


Abstract
Detection of similarity is particularly difficult for small proteins and thus connections between many of them remain unnoticed. Structure and sequence analysis of several metal-binding proteins reveals unexpected similarities in structural domains classified as different protein folds in SCOP and suggests unification of seven folds that belong to two protein classes. The common motif, termed treble clef finger in this study, forms the protein structural core and is 25–45 residues long. The treble clef motif is assembled around the central zinc ion and consists of a zinc knuckle, loop, β-hairpin and an α-helix. The knuckle and the first turn of the helix each incorporate two zinc ligands. Treble clef domains constitute the core of many structures such as ribosomal proteins L24E and S14, RING fingers, protein kinase cysteine-rich domains, nuclear receptor-like fingers, LIM domains, phosphatidylinositol-3-phosphate-binding domains and His-Me finger endonucleases. The treble clef finger is a uniquely versatile motif adaptable for various functions. This small domain with a 25 residue structural core can accommodate eight different metal-binding sites and can have many types of functions from binding of nucleic acids, proteins and small molecules, to catalysis of phosphodiester bond hydrolysis. Treble clef motifs are frequently incorporated in larger structures or occur in doublets. Present analysis suggests that the treble clef motif defines a distinct structural fold found in proteins with diverse functional properties and forms one of the major zinc finger groups.
INTRODUCTION
The natural diversity provided by 20 amino acids is not sufficient to ensure all the functional and structural flexibility required in nature. Proteins frequently recruit other compounds (co-factors) as helpers to perform chemical functions and to achieve structural integrity and stability. The smallest but probably most widely used co-factors are metal ions. Zinc is particularly important and is used for diverse functions, from catalytic to structural (1). A great variety of small zinc-containing domains have been cataloged (2–4). These 20–50 amino acid polypeptides use zinc in their structural cores. Zinc ions facilitate folding and increase protein stability, which is not easily achievable for small polypeptide chains. Zinc utilization is analogous to the employment of Ca2+ by larger proteins to increase thermostability (5–7), or disulfides by small domains (8).
Small zinc-containing domains are termed ‘fingers’ and typically function as interaction modules. They bind to nucleic acids or proteins. The classical zinc finger domain, the C2H2 finger, is a DNA-binding unit (9). Structurally, it is composed of a β-hairpin and an α-helix with Zn2+ sandwiched between them. The α-helix binds in the DNA major groove. More than 6500 different sequences with this motif are known (4). Another large group of fingers covers the rubredoxin-like zinc ribbon domain (10–12). Its structure contains two zinc knuckles (non-canonical turns with consensus CPxCG) (13) and typically a three-stranded β-sheet. Each knuckle carries two zinc ligands. This domain is known for being ‘mobile’ and is frequently found inserted in larger proteins. Many other zinc fingers have been classified into small families (10–100 members) and the relationships between them remain largely uncertain.
Here, a zinc-binding structural motif that we term the ‘treble clef finger’ is described. The motif is composed of a zinc knuckle followed by a loop, a β-hairpin and an α-helix, and is characterized by the distinct structural arrangement of these elements. The zinc knuckle and the first turn of the helix typically donate two Zn2+ ligands each. A projection of the domain Cα trace on one of the planes resembles the treble clef sign, which suggested the name (Table 1). Treble clef finger structures are present in several groups of proteins that display virtually undetectable sequence similarity to each other and have been classified into seven different folds in the SCOP (structural classification of proteins) database (v.1.53) (3). The major groups encompass RING fingers, nuclear receptor-like fingers, protein kinase cysteine-rich domains and His-Me finger endonucleases. We argue that the treble clef finger motif constitutes the structural core of all of these proteins, suggesting their unification into a single group.
Table 1. Pairwise comparison of treble clef finger structures.
 |
PDB entries of 42 treble clef-containing proteins with sequences shown in the alignment from Figure 2 are numbered and listed in the left column. Top row contains only protein numbers. The values in the upper right half of the table (above the diagonal) are r.m.s.d. in Å between each pair of proteins. Each pair of proteins was superimposed by main chain atoms with the Insight II package (MSI): see Materials and Methods for the definition of superimposed regions. The lower left half of the table (diagonal and below) contains BLOSUM62 (36) scores of sequences aligned on Figure 2. For each pair of sequences, only sites without gaps were used in score calculation. Each raw BLOSUM62 score (the sum of pairwise scores for each site) is multiplied by 10 and divided by the number of sites used in the score calculation. The families are divided by lines. The picture above the table illustrates the origin of the domain name. The structure on the right is a RING finger domain of signal transduction protein Cbl (PDB entry 1fbv, residues A381–A403) depicted as a coil with no smoothing (29,30). N- and C-termini are labeled and Zn2+ is shown as a ball. The same structure (smoothed) is shown in the middle.
MATERIALS AND METHODS
Structure analysis
Structure similarity searches against the Protein Data Bank (PDB) (14) maintained at the Research Collaboratory for Structural Bioinformatics were performed using DALI (15), VAST (16) and CE (17) programs with default parameters. The ribosomal proteins L24E (18,19) (PDB entry 1ffk, chain K) and S14 (20,21) (PDB entry 1fjf, chain N) were taken to initiate the searches that were continued using each detected protein as a query. The pairwise structure-based alignments generated by the above-mentioned programs were used as a basis for the multiple structure-based alignment shown in Figure 2. The SCOP database (v.1.53; 11 410 PDB entries, July 1, 2000) (3) was used as a source of protein classification. Protein structures were visualized and superimposed using the InsightII package (MSI) and SwissPDB viewer (22), and the multiple structure-based alignment was built on the basis of the superpositions made in InsightII. Detection of six principal positions (marked with red asterisks in Figure 2), four zinc ligands and one position in each of the β-strands of the β-hairpin, aided in construction of the multiple alignment. Residues used for the superpositions cover regions marked by asterisks in the multiple alignment (Fig. 2). For each pair of proteins, the maximum possible number of residues within the above-defined region (from 14 to 25 residues) was used for superposition. Structure diagrams were rendered using Bobscript (23), a modified version of Molscript (24).
Figure 2.

Structure-based sequence alignment of treble clef fingers. For each sequence, gene identification (gi) number of the NCBI/GenBank protein sequence database, PDB entry name, chain ID (if any), fragment number (if more than one fragment is shown), and starting and ending residue numbers are given. The numbers correspond to the numbering in the PDB file. Sequences are divided into families with the family numbers shown on the left. Families are separated from each other by a larger spacing between the sequences. The families are: I, ribosomal protein L24E; II, ribosomal protein S14; III, RING finger; IV, Pyk2-associated protein β-ARF-GAP domain; V, protein kinase cysteine-rich domain; VI, phosphatidylinositol-3-phosphate-binding domain; VII, GATA-1, LIM and DNA repair factor XPA zinc-binding domains; VIII, nuclear receptor DNA-binding domain; IX, His-Me finger endonucleases/MH1 domain of Smad. Zinc ligands in the signature of the treble clef motif are boxed in black, non-zinc-binding residues in corresponding positions are shown in blue, Zn2+ ligands in other sites are boxed in dark gray, Mg2+ ligands are boxed in olive, active site histidine in endonucleases is shown in green, uncharged residues (all amino acids except D,E,K,R) in mostly hydrophobic sites are highlighted in yellow, non-hydrophobic residues (all amino acids except W,F,Y,M,L,I,V) at mostly hydrophilic sites are highlighted in light gray, small residues (G,P,A,S,C,T,V) at positions occupied by mostly small residues are shown in red letters. Secondary structure consensus is shown below the alignment. β-Strands are displayed as arrows, α-helix is shown as a cylinder. Colors and labels are according to the scheme from Figure 1. Arcs connect hydrogen-bonded residues in the β-hairpins. The sites used in r.m.s.d. minimization are marked with asterisks above the alignment. Red asterisks are for the crucial six sites used in the alignment construction.
Sequence analysis
Sequence similarity searches against the non-redundant protein database (nr) maintained at the National Center for Biotechnology Information (Bethesda, MD) were performed using the PSI-BLAST program (25). The non-redundant database (nr, October 17, 2000 release; 574 979 sequences, 180 825 488 total letters) sequences were filtered for low-complexity regions using the SEG program (26,27) with the following parameters: window 40, trigger 2.7 and extension 3.2. The query sequences were not subjected to SEG filtering. The BLOSUM62 matrix (28) was used for scoring, E-value thresholds were set to 0.01 or 0.001 (25) for inclusion in the profile calculation. All sequences of treble clef domains in proteins with known structure were used as initial PSI-BLAST queries. Since the results of searches strongly depend on the query sequence, all significantly different (BLAST score <1.0 bits per site, which corresponds to ∼50% identity) sequences found in the course of iterations were taken as queries for additional PSI-BLAST searches. If a protein of determined structure was detected in the searches, PSI-BLAST alignment was verified by the structure-based alignment to ensure the validity of the match. Sequence analysis protocols were carried out using SEALS (29).
RESULTS AND DISCUSSION
Structural description of the treble clef domain
The treble clef structural motif is defined by the unique spatial arrangement of the following consecutive elements: zinc knuckle, loop, β-hairpin and α-helix, which are folded around a zinc ion (Fig. 1). Zinc knuckles are unique turns, having the consensus sequence CPxCG, where the two cysteines function as zinc ligands. The short 2–4 residue sequence segments preceding and following the knuckle turn usually adopt an extended conformation. These segments typically form two to three hydrogen bonds with each other and comprise a short β-hairpin (β-strands a and b, shown in purple in Fig. 1A). The loop following this knuckle β-hairpin varies in length (0–9 residues) and conformation (Figs 1A and B and 2). It can be structured as a tight turn [in ribosomal protein S14 (20,21); Fig. 1A and B, 1fjf_N], as a long and possibly flexible loop [in Cys2 activator-binding domain of protein kinase Cδ (30); Fig. 1A and B, 1ptq] or fold into a single turn–helix [in the FYVE domain of Vps27p (31) protein second treble clef finger, 1vfy_A(2), Fig. 1A and B]. The axis and plane of the primary β-hairpin of the treble clef (β-strands c and d, shown in yellow in Fig. 1A) is almost perpendicular to those of the knuckle β-hairpin. The four β-strands of the two hairpins (abcd) form a left-handed ββββ unit. The length of the β-strands in the primary β-hairpin is typically between 4 and 8 residues, and the turn between them can bear long insertions that might be structured as subdomains (e.g. in 1mdh_A, Figs 1A and B and 2). The second β-strand (d) of the primary β-hairpin is followed by an α-helix (A, shown in blue in Fig. 1A). The first turn of the α-helix incorporates the remaining two zinc ligands. One of the ligands occupies the position of the N-terminal cap in the α-helix. The length of the α-helix is variable and ranges from one to six turns. The β-hairpin and the α-helix (cdA) are folded as a left-handed ββα unit (32); thus, the treble clef motif can be described as a left-handed superhelix of the five secondary structural elements (abcdA). The features described above are unique to the treble clef motif and allow for its easy detection in protein structures.
Figure 1.

Structural comparisons of representative treble clef fingers. (A) Structural diagrams of ribosomal protein S14 (1fjf, chain N, residues 21–53), ribosomal protein L24E (1ffk, chain R, residues 3–45), RING finger of RAG1 (dimerization domain) (1rmd, residues 23–60), ARF-GAP domain of Pyk2-associated protein β (1dcq, chain A, residues 262–297), Cys2 activator-binding domain of protein kinase Cδ (1ptq, residues 241–280), FYVE domain of Vps27p protein, first treble clef finger (1vfy, chain A, residues 172–207), FYVE domain of Vps27p protein, second treble clef finger (1vfy, chain A, residues 190–235), retinoid X receptor α DNA-binding domain (2nll, chain B, residues 300–336), recombination endonuclease VII (1en7, chain A, residues 19–79), MH1 domain of Smad (1mhd, chain A, residues 93–132) showing the treble clef finger domains from each protein. In each protein, N- and C-termini are labeled with N and C. β-Strands and α-helices are labeled in lower and upper case letters, respectively. The color of the letter corresponds to the color of the element. The short β-strands in the zinc knuckle region are shown in purple (a and b) with the zinc knuckle turn colored red. Side-chains of zinc ligands and corresponding residues in 1mhd are shown in ball-and-stick representation. Zinc ions are shown as orange balls. The ribbon diagrams were rendered by Bobscript (23), a modified version of Molscript (24). (B) Stereo diagram of superimposed Cα-traces of the 10 structures from (A) shown in the same orientation. The Cα-traces of proteins, side chains of zinc ligands and Zn2+ are shown. Superpositions were made using Insight II package (MSI). The regions used in r.m.s.d. minimization are outlined in thicker lines. Color coding of structures corresponds to the dot color scheme (in front of each PDB entry) in (C). (C) Structure-based sequence alignment of treble clef motif regions of the 10 proteins illustrated in panel (A). For each sequence, the PDB entry name and chain ID, starting and ending residue numbers are given. Zinc ligands are boxed in black. Color shading and labels of secondary structure elements correspond to those in (A). Long insertions are not displayed: the number of omitted residues is specified in brackets.
Unlike most globular proteins, the treble clef domain does not have a pronounced hydrophobic core and is apparently stabilized by the zinc ion. Consequently, the most structurally conserved part of the treble clef motif is the Zn2+-binding site (Fig. 1B). The zinc knuckle and the first turn of the α-helix display the smallest r.m.s.d. between different proteins (usually 0.2–0.7 Å) and thus constitute the structural core of the domain. The primary β-hairpin, in particular its first β-strand (c), which is remote from the core, shows greater structural variation.
Sequence description of the treble clef domain
Structure-based sequence alignment of treble clef domains revealed rather limited sequence conservation (Fig. 2). Most of the sequences contain 25 positions in common (marked with asterisks above the alignment in Fig. 2). Sequence conservation is restricted mainly to the zinc ligands. Zinc ligands are arranged in two pairs that form the two ‘halves’, or sub-sites, of the zinc-binding site (zinc knuckle, N-terminal CXXC pair; and first turn of an α-helix, C-terminal CXXC pair). Most of the ligands are cysteines; however, histidines are also present, contributed by the C-terminal sub-site. It is clear that the two halves of the zinc-binding site display distinct patterns of sequence conservation. The sequence signal in the N-terminal sub-site is characteristic of a classical zinc knuckle (CPXCG consensus) with small residues immediately following the cysteines. Glycine followed by a primarily hydrophilic residue typically occurs after the second cysteine. There is no sequence conservation before the first cysteine of the motif. In the C-terminal sub-site, a small residue precedes the second cysteine. A pair of uncharged residues, the second of which is mostly hydrophobic, is usually present before the first cysteine. Little sequence conservation is noticeable after the C-terminal sub-site. The absence of structural and sequence similarity between the two Zn2+-binding half-sites is a distinguishing feature of treble clef motifs that can be explored in their detection. The two Zn2+ half sites in zinc ribbon motifs, for example, are similar. The distance between the second cysteine of the N-terminal sub-site and the first cysteine of the C-terminal sub-site ranges from 12 to ∼50 residues. This region incorporates three β-strands with two loops between them. A conserved small residue that starts the last β-strand (d) and a single usually uncharged residue in each of the β-strands b and c are characteristic of this segment (colored yellow in Fig. 2). Sequence conservation is lowest in β-strand c (Fig. 2), which correlates with the large variations in the structure of this β-strand (Fig. 1B).
Zinc fingers of different classes are susceptible to replacements in zinc-binding sites and consequent loss of zinc-binding properties (10,11). This feature is also manifested in treble clef fingers. Among the proteins with known structure, loss of zinc-binding sites occurs in His-Me finger endonucleases (33–36) and the MH1 domain of Smad protein (37) (Fig. 2). Substitution of Zn2+ ligands usually makes motif detection more challenging (11,38). However, in His-Me finger endonucleases, the presence of the conserved active site histidine residue combined with the local sequence and structural conservation leave little doubt about their homology (36,39–41) despite the absence of a zinc-binding site in some of the sequences (Fig. 2). Zinc-binding site deterioration, from partial to complete loss of ligands, occurs in ribosomal protein S14 as well. For example, the following S14 proteins from different organisms contain four, three, two, one and no cysteines, respectively (results not shown): gi|10835576, gi|3790138, gi|7388136, gi|6094132 and gi|7388131 (gene identification numbers from NCBI protein sequence database are given).
Treble clef finger: a versatile metal-binding motif
Analysis of treble clef domains in various proteins revealed that, in addition to the primary zinc-binding site between the zinc knuckle and the helix (site 1 in Fig. 3), treble clefs can accommodate seven other different metal-binding sites (Fig. 3, eight different sites in total). Two metal-binding sites are defined as different if at least some of the ligands are contributed by different positions in the multiple sequence alignment (Fig. 2). Virtually every turn, loop or cavity in the treble clef structure can serve as a metal-binding site. The primary zinc-binding site is the most buried (1 in Fig. 3). Three different sites (2, 3 and 8) are placed between the β-hairpin (cd) and the α-helix. Other sites (4, 5, 6 and 7) utilize additional ligands that are contributed by structures outside the treble clef motif. Site 5 is mostly hosted by a long insertion in the β-hairpin (cd). Site 6 shares a ligand with the primary site 1. It is likely that most of these metal-binding sites serve a structural role. However, at least one of them, namely site 8 in Figure 3, incorporates a catalytically important ion (Mg, Ni or Zn) in His-Me finger endonucleases (42–48).
Figure 3.
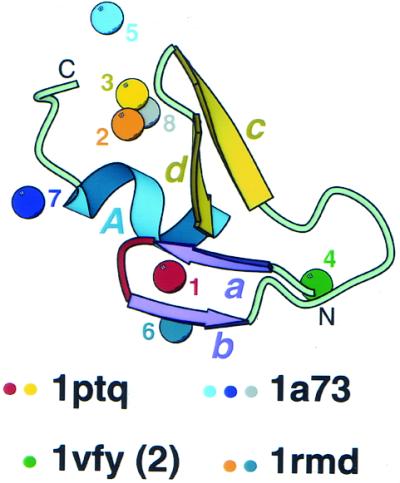
Metal-binding sites in treble clef fingers. All 42 treble clef finger structures (Fig. 2) are superimposed and metal ions bound at different sites in the structural motif (having zinc ligands from different sites in sequence alignment) are displayed. Metal ions are shown as balls and are numbered from 1 to 8. Protein ribbon corresponds to Cys2 activator-binding domain of protein kinase Cδ (1ptq, residues 241–280). Color coding and labeling of secondary structural elements correspond to Figure 1. PDB codes of representative structures that cover all distinct zinc-binding sites are shown below and colored dots indicate the sites that are present in the structures.
It is not understood why the treble clef is particularly suited to accommodate a variety of metal ion sites. Other frequently occurring zinc finger motifs, such as the classical C2H2 finger or zinc ribbon, rarely incorporate more than a single metal-binding site. It is probable that the conformationally unique treble clef, the structure of which is composed of three layers and can incorporate long loops between secondary structural elements, offers a particularly versatile metal-binding template with at least eight different places suitable for zinc-binding.
Treble clef domains in larger protein structures
In many treble-clef-containing proteins, such as S14 (20,21) and L24E (18,19), the finger is the only domain present; these proteins contain only five secondary structural elements of the motif (abcdA) (Fig. 1). However, incorporation of treble clef fingers in multidomain proteins is more typical. Duplications are among the most frequent events in protein evolution, and in several proteins, treble clef domains were detected in tandem, suggesting possible duplication events. LIM domains (49–52) (Fig. 4A) are made of two consecutive treble clef fingers. The two fingers are arranged almost in parallel, so that the two α-helices (A1 and A2 in Fig. 4A) are nearly co-axial. The loops of the β-hairpins (c1d1 and c2d2 in Fig. 4A) are close together and side chains between the hairpins form a hydrophobic core between the two treble clef domains.
Figure 4.
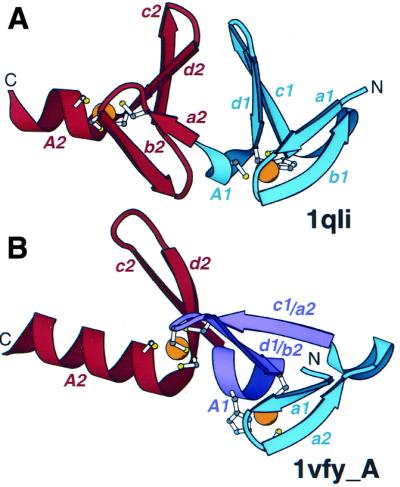
Arrangement of treble clef pairs. (A) A pair of tandem treble clef motifs in the LIM domain of cysteine-rich protein CRIP (1qli, residues 117–145). (B) Two zinc-binding motifs of the FYVE domain of vps27p protein (1vfy, chain A, residues 173–235). One treble clef finger is colored in blue, the other is colored in red. Secondary structural elements are named according to Figure 1. Indices 1 and 2 refer to the first and second treble clef finger, respectively. The segment that belongs to both treble clef fingers is colored in purple. Side chains of zinc ligands are shown in ball-and-stick representation. Zn2+ is shown as an orange ball.
A more unusual doublet of treble clefs is present in phosphatidylinositol-3-phosphate-binding domain (31,53,54) (Fig. 4B). This domain contains two zinc-binding sites, such that each site is formed by a zinc knuckle and the first turn of an α-helix. Thus, each site constitutes the core of a treble clef domain. Each of the two motifs, when considered separately, corresponds perfectly to one treble clef template. However, they overlap over half of the motif length. It turns out that the primary β-hairpin (c1d1) of the first treble clef domain is also the knuckle β-hairpin of the second treble clef domain (a2b2). The turn in the latter hairpin accommodates two zinc ligands of the second treble clef, and the hairpin is placed between the two sub-sites of the first zinc-binding site. The α-helix of the first treble clef motif (A1) is situated between the two β-hairpins of the second treble clef motif. Thus the C-terminal segment of the first treble clef motif is simultaneously the N-terminal segment of the second treble clef with the zinc sub-sites being ‘interleaved’ (first sub-site of the second treble clef is between the two sub-sites of the first treble clef; Figs 2 and 4B). Again, as in the case of a simple duplication (Fig. 4A), the axes of the two α-helices (A1 and A2 in Fig. 4B) almost coincide. How could these overlapping motifs have evolved? It is possible that one of the motifs (for example, the second) represents the ‘true’ treble clef motif that is homologous to others, and the other motif originated independently by convergence from the N- (or C-)terminal extension. It is also possible that the structure is formed by a triplet of homologous ββα motifs (colored blue, purple and red in Fig. 4B). If this is true, a single treble clef motif might be formed by a duplication of such a ββα unit. In any event, the overlapping treble clefs in the phosphatidylinositol-3-phosphate-binding domain provide an evolutionary puzzle.
Since the treble clef motif is very short, typically approximately 30 residues, additional secondary structure elements are frequently incorporated into treble-clef-containing domains. These elements can occur as N- or C-terminal extensions or insertions with the treble clef secondary structures (abcdA) and a zinc ion constituting the core of the domains. The representative structures illustrating incorporation of additional elements into the treble clef finger are shown in Figure 5.
Figure 5.

Treble clef fingers inside larger structures. Structural diagrams of ARF-GAP domain of Pyk2-associated protein β (1dcq, chain A, residues 248–365), RING finger of RAG1 (dimerization domain) (1rmd, residues 1–87), retinoid X receptor α DNA-binding domain (2nll, chain B, residues 300–369), Cys2 activator-binding domain of protein kinase Cδ (1ptq, residues 243–280), S.marcescens endonuclease (1ql0, chain A, residues 6–245) and intron-encoded homing endonuclease I-PpoI (1a73, chain A, residues 21–139). The treble clef finger motif is outlined in red. β-Strands and α-helices that are not part of the motif are shown in yellow and blue, respectively. Side-chains of zinc ligands, residues in sites corresponding to ligands of zinc #1, and active site residues in 1a73 are shown in ball-and-stick representation. Active site residues are colored in green. Zn2+ and Mg2+ are displayed as orange and green balls, respectively. In each protein, N- and C-termini are labeled with N and C. β-Strands and α-helices are labeled in lower and upper case letters, respectively. The color of the letter corresponds to the color of the element. Zinc ions are labeled with numbers corresponding to those in Figure 3.
Most typically, the extensions are structured as additional β-strands that form hydrogen bonds with either of the two treble clef β-hairpins. For example, the third β-strand that H-bonds to the β-strand c of the principal β-hairpin is present in several treble clef motif proteins, such as RING finger domains (55) (Fig. 5, 1rmd, β-strand g), the ADP ribosylation factor (ARF)-GTPase-activating protein (GAP) domain of Pyk2-associated protein β (56) (Fig. 5, 1dcq, β-strand e) and the Cys2 activator-binding domain of protein kinase Cδ (30) (Fig. 5, 1ptq, β-strand a′). In the first two proteins, the additional β-strand is C-terminal from the treble clef motif. In Cys2 activator-binding domain, a spatially equivalent β-strand is contributed by the N-terminal part of the protein. Thus, the Cys2 activator-binding domain has a circularly permuted RING finger topology (Fig. 5, 1ptq and 1rmd). The third β-strand in these proteins gives rise to the ββαβ structural motif (or βββα in a circularly permuted version), where βαβ is right-handed, and the ββα unit is a part of the treble clef domain. Such ββαβ motifs are commonly present in many unrelated protein structures (3); however, in treble clef fingers the axis of the α-helix usually forms a larger angle with the plane of the β-sheet. The zinc knuckle β-hairpin is much shorter but can be extended by an additional β-strand in some treble clef structures, such as in the ARF-GAP domain of Pyk2-associated protein β (Fig. 5, 1dcq, β-strand g) and in some nuclear receptors (Fig. 5, 2nll, β-strand e).
The principal β-hairpin of the treble clef domain interacts with the α-helix A on one side and is exposed to the solvent on the other side. However, in many treble clef domains additional structures interact with this solvent-exposed side. Typically, an α-helix contributed either by N-terminal (Fig. 5, 1dcq and 1a73, α-helix A′) or C-terminal (Fig. 5, 1rmd, α-helix B) parts of the molecule packs across the β-hairpin cd. Alternatively, a loop can occupy this position (Fig. 5, 1ptq, a loop between a′ and a). Additional elements can be packed on the zinc ion side of the treble clef finger (near the zinc knuckle and α-helix). An α-helix interacts with the α-helix A proximally to the zinc-binding site in the ARF-GAP domain (Fig. 5, 1dcq, α-helix D) and in nuclear receptors (57) (Fig. 5, 2nll, α-helix B). A seven-stranded β-meander occupies this position in Serratia marcescens endonuclease (44,45) (Fig. 5, 1ql0, β-strands b1–h1).
In summary, it appears that additional secondary structural elements incorporated into a treble clef finger can interact with almost any surface of its structure, except the distal surface of the α-helix A (Fig. 5). This surface remains free from intra-molecular interactions and is used to mediate inter-molecular contacts by many treble clef finger domains.
Treble clef domains in different protein superfamilies
Treble clef structural motifs can be identified in proteins from seven different SCOP folds (3). The structural core of ribosomal proteins L24E and S14, not yet in SCOP v.1.53, is also a treble clef. Here, the results of structure similarity searches with computer programs, such as DALI (15), VAST (16) and CE (17) are discussed. Alignment-based (Fig. 2) sequence similarity scores and r.m.s.d. of structural superpositions for each pair of treble clefs are presented in Table 1. The results show that most of the treble clef fingers from different families are structurally very similar (r.m.s.d. of 1.5–2.5 Å) despite significant sequence differences among some of them (negative scores). Sequence similarity is typically pronounced within the families (Table 1).
Ribosomal proteins L24E and S14: prototypes for treble clef finger. Ribosomal proteins are likely to represent some of the most ancient proteins. Crystal structures of both ribosomal subunits solved recently confirmed that ∼10% of ribosomal protein domains are structured as zinc-binding modules. Two of these, one from each ribosomal subunit, L24E (18,19) (Fig. 1A, PDB entry 1ffk, chain R) and S14 (20,21) (Fig. 1A, PDB entry 1fjf, chain N) are treble clef fingers. S14 is the smallest treble clef representative with only 12 residues between the second and third cysteines. Structure superpositions (Table 1) reveal the close resemblance of S14 to some nuclear receptor domains (r.m.s.d. of 1.7–1.8 Å), which are also characterized by a short β-hairpin cd. The L24E domain, which is surprisingly very similar structurally (r.m.s.d. of 1.3 Å) to T4 recombination endonuclease VII (1en7), possesses a longer hairpin cd. DALI searches initiated with either of these two ribosomal proteins do not find any matches with Z-scores higher than 2. The VAST program detects T4 recombination endonuclease VII (1en7) with L24E as a query (P-value 0.06). Phosphatidylinositol-3-phosphate-binding domain (1zbd, P-value 0.04) and RING finger (1rmd, P-value 0.08) are retrieved in a VAST search using the S14 structure as a query. Thus, the VAST algorithm detects the links between ribosomal protein structures and treble clef domains.
RING finger. RING fingers participate in a variety of cellular processes such as development, apoptosis, cell-cycle control, ubiquitination, etc., and function as protein interaction modules (58). The RING finger domain is characterized by the presence of the second zinc-binding site (Fig. 5, 1rmd, site 2) and the third β-strand bonded to the primary β-hairpin (Fig. 5, β-strand g). The surrounding structures in the second zinc site resemble a circularly permuted zinc ribbon motif (β-strands e, f, g, c and d) (12). Five different RING finger structures have been reported (55,59–62) [1chc, 1bor, 1rmd (Figs 1 and 5), 1fbv and 1g25]. Surprisingly, despite pronounced sequence similarity which can be detected by PSI-BLAST (results not shown), RING finger structures are rather dissimilar. For example, r.m.s.d. between the acute promyelocytic leukaemia proto-onkoprotein PML RING finger domain (60) (1bor) and the N-terminal domain of the TFIIH MAT1 subunit (62) (1g25) is 3.9 Å, despite the positive sequence similarity scores (Table 1). These differences might be caused by real structural variation among these small modules or may indicate problems in the structure determination. In any case, homology among the members of RING family is hard to question, and the presence of a common secondary structural core assembled around the two zinc-binding sites is apparent. However, neither DALI nor VAST detect structural similarities between 1bor and other RING fingers. RING finger domains illustrate the difficulties of structure similarity searches with small proteins, for which sequence analysis often offers a more powerful method of homology detection (11). The VAST match between RAG1 RING finger (1rmd) and ribosomal protein L24E (1ffk chain R) with a P-value of 0.08 links RING finger domains with the other treble clef fingers.
Pyk2-associated protein β ARF-GAP domain. The ARF-GAP domain facilitates hydrolysis of GTP by ARF. ARFs are RAS-related GTPases that are crucial for vesicular trafficking and stimulation of phospholipase D (56). The structure of ARF-GAP is formed around the treble clef motif with a single zinc-binding site (56,63) (Figs 1 and 5, 1dcq). Similar to the RING finger domain, ARF-GAP contains an additional β-strand (e in Fig. 5) but lacks the second zinc-binding site. Mandiyan et al. (56) reported that a DALI search of PDB with ARF-GAP Zn2+-binding module detects the phorbol-binding domain of protein kinase C (30) (Fig. 5, 1ptq).
Protein kinase cysteine-rich domain. Protein kinases C and RAF are activated by various small molecule ligands that bind to their cysteine-rich domains. Structures of cysteine-rich domains from several kinases have been determined (30,64–66) [1ptq (Figs 1 and 5), 1tbo and 1far]. Cysteines or histidines were found to form two zinc-binding sites (Fig. 5, 1ptq) placed at similar locations to the Zn2+ sites in RING fingers (Fig. 5, 1rmd). This similarity combined with the presence of a third β-strand (Fig. 5, a′ in 1ptq and g in 1rmd) interacting with the primary β-hairpin cd might indicate homology between the two variants of the treble clef finger. The topological difference between the protein kinase cysteine-rich domain and RING fingers can be explained by a circular permutation: the third β-strand is placed at the N-terminus of the protein kinase cysteine-rich domain before its treble clef segment (Fig. 5, a′ in 1ptq), and the third β-strand follows the treble clef motif in the RING finger domain (Fig. 5, g in 1rmd).
Besides other kinase cysteine-rich domains, a DALI search initiated with the protein kinase Cδ phorbol-binding domain (30) (1ptq) finds representatives of other SCOP folds, such as the phosphatidylinositol-3-phosphate-binding FYVE domain (1vfy: Z-score 2.4, r.m.s.d. 2.3 Å for 35 residues), the effector domain of rabphilin (1zbd: Z-score 2.1, r.m.s.d. 2.9 Å for 37 residues) and LIM domains of cysteine-rich intestinal protein (1iml: Z-score 2.1, r.m.s.d. 2.6 Å for 38 residues; 1b8t: Z-score 2.0, r.m.s.d. 2.0 Å for 34 residues). VAST detects glucocorticoid receptor and erythroid transcription factor GATA-1 DNA-binding domains (1glu and 1gnf, P-values 0.05). PSI-BLAST sequence similarity searches are consistent with DALI results. Due to the short length of treble clef domains and limited sequence similarity, PSI-BLAST iterations converge before retrieving proteins found by DALI with statistically significant E-values. However, rabphilin domains (e.g. gi|1350829) and LIM domains (e.g. gi|8393153) are consistently found with marginal scores (25–30 bits) below the E-value thresholds of 0.01–0.001. Thus, sequence and structure similarity searches suggest that protein kinase cysteine-rich domains are homologous to phosphatidylinositol-3-phosphate-binding domains and LIM domains.
Phosphatidylinositol-3-phosphate-binding domain. Phosphatidylinositol-3-phosphate, an essential regulator of membrane trafficking in eukaryotes (67), binds to the FYVE domain. This domain represents an unusual overlapping doublet of treble clefs in which the C-terminal half of the first finger serves as the N-terminal half of the second one. The overlapping treble clef finger domains such as FYVE (31,54) [1vfy (Figs 1 and 4B) and 1dvp] and the effector domain of rabphilin-3a (53) (1zbd) are likely to interact with the membranes. DALI searches with the FYVE domain as a query (1vfy) find the protein kinase C cysteine-rich domain (1ptq: Z-score 2.4, r.m.s.d. 2.3 Å for 35 residues) and LIM domains of cysteine-rich intestinal protein (1b8t: Z-score 2.2, r.m.s.d. 4.0 Å for 44 residues). VAST detects essentially the same proteins (1tbo, P-value 0.02; 1b8t, P-value 0.01). These similarities as well as an additional similarity of the FYVE domain with GATA-1 DNA-binding domain are discussed by Misra and Hurley (31).
Nuclear receptor-like finger. This group contains the structures of GATA-1 (68,69) (1gat, 1gnf), the nuclear receptor DNA-binding domain (57,70–75) [2nll (Figs 1A and 5), 1cit, 1a6y, 1dsz, 1glu and 1hcq], the LIM domain (49–52) [1b8t, 1qli (Fig. 4A), 1iml and 1zfo] and the DNA repair factor XPA zinc-binding domain (70) (1xpa). SCOP unifies all these structures in a single superfamily termed the glucocorticoid receptor-like domain. These treble clef domains are rather diverse both in structure (Table 1) and function. They include both DNA- and protein-binding domains.
Nuclear receptors form a tight cluster of treble clef domains with r.m.s.d. values ranging from 0.3 to 1.4 Å between them (Table 1, brown). Nuclear receptor DNA-binding domains accommodate two zinc-binding sites (Fig. 5, 2nll). The N-terminal site (site 1 in Fig. 5) is situated within the clearly defined treble clef domain. The C-terminal subdomain also shows some resemblance to the treble clef finger. Notably, the second pair of cysteines is located in the N-terminal turn of an α-helix. However, there is no clearly defined β-hairpin preceding that α-helix, and the first pair of zinc ligands displays different structural arrangement compared to that found in treble clef fingers. Since duplications are very common in protein evolution, it is possible that nuclear receptor DNA-binding domains originated by duplication of an ancestral treble clef domain and the C-terminal treble clef structure changed substantially afterwards. However, because the structure of the C-terminal subdomain differs significantly from that of typical treble clef domains, it is not considered here.
The most divergent structure is the zinc-binding domain of DNA repair factor XPA (70) (1xpa), which has an α-helix in place of β-strand c. As a result, r.m.s.d. values of 1xpa with other treble clef structures are elevated to 2.5–3.5 Å. LIM domains function as protein interaction modules and are formed by a doublet of treble clefs (49–52) (Fig. 4A). Similarity among LIM domains is less pronounced than that among the nuclear receptor DNA-binding domains (Table 1). However, the PSI-BLAST program can detect LIM structures 1b8t, 1qli, 1iml and 1zfo as being homologous with E-values <0.01. The GATA-1 domain (1gat) appears to be more similar to the LIM domain. A DALI search with 1gat detects the LIM domain structure (1b8t) with Z-score 2.2 (r.m.s.d. 2.1 Å from 37 residues). The DALI program also finds links between the glucocorticoid receptor-like superfamily proteins and other treble clef domains classified in different SCOP folds. For example, the LIM domain structure (1b8t) retrieves the following representative proteins: rabphilin-3a (1zbd: Z-score 2.4, r.m.s.d. 5.5 Å from 55 residues), the phosphatidylinositol-3-phosphate-binding FYVE domain (1vfy: Z-score 2.2, r.m.s.d. 4.0 Å from 44 residues) and the protein kinase C cysteine-rich domain (1ptq: Z-score 2.0, r.m.s.d. 2.0 Å from 34 residues). A VAST search produces similar results and additionally finds the glucocorticoid receptor DNA-binding domain (1glu) with a P-value of 0.01.
His-Me finger endonucleases and the MH1 domain of Smad. His-Me finger endonucleases are likely to possess the same catalytic mechanism, which utilizes a metal ion (Mg2+, Ni2+, Zn2+) and an invariant histidine residue (48). The structures of five representatives of this superfamily have been determined: DNase domains of colicins E7 and E9 (42,43) (7cei, 1bxi), S.marcescens endonuclease (44,45) (Fig. 5, 1ql0), intron-encoded endonuclease I-PpoI (46) (Fig. 5, 1a73) and T4 recombination endonuclease VII (47) (Fig. 1, 1en7). Detection of structural similarity between the members of this superfamily has been challenging and resulted in several studies (36,39–41). Although His-Me finger endonuclease structures share only a short ββα supersecondary structural segment in common, homology offers the most likely scenario for their relationship due to the similar active site arrangements and a common catalytic mechanism. The ββα segment covers a part of the treble clef domain (Fig. 1A, cdA unit).
One of the five His-Me finger endonuclease structures, namely T4 recombination endonuclease VII (47) (Fig. 1A, 1en7), firmly establishes the presence of a treble clef in the members of this superfamily. Only this protein contains a zinc-binding site characteristic of the treble clef motif. Other structures do not bind zinc at a homologous site. Analysis of available sequences of HNH-motif endonucleases demonstrates that some members of this family possess zinc-binding residues at corresponding treble clef sites, while others (e.g. colicins 7cei and 1bxi) do not (33–35). It appears that deterioration of the treble clef zinc-binding site is a frequent trend in the His-Me finger endonuclease superfamily and obscures detection of the treble clef motif in them. Despite this trend, VAST detects structural similarities between the three families of His-Me finger endonucleases and only fails to find a S.marcescens endonuclease structure. Specifically, DNase of colicin E7 (7cei) finds endonuclease I-PpoI (1a73) with a P-value of 0.01, and T4 endonuclease VII (1en7) retrieves DNase of colicin E9 (1bxi) with a P-value of 0.05. However, only the T4 endonuclease VII structure with the treble clef zinc-binding site can link His-Me finger endonucleases with other treble clef proteins. For example, a DALI search reveals structural similarity between T4 endonuclease VII (1en7) and the effector domain of rabphilin (1zbd: Z-score 2.1, r.m.s.d. 2.9 Å for 50 residues), and VAST finds T4 endonuclease VII (1en7) and the GATA-1 DNA-binding domain (1gnf) to be structural neighbors (P-value 0.01). Structure superposition and sequence similarity score analyses reveal strong tendencies due to zinc site deterioration (Table 1). r.m.s.d. values between the His-Me finger endonuclease structures with deteriorated zinc-binding modules and treble clef finger domains (3.4–6.0 Å) are much larger that those between T4 endonuclease VII (1en7) and treble clef fingers (1.4–3.2 Å), and are comparable to those between T4 endonuclease VII and other His-Me finger endonucleases. Corresponding results are also apparent for sequence similarity scores (Table 1).
Smad proteins are eukaryotic transcription regulators in the TGF-β signaling cascade (76). The MH1 domain of Smad is a DNA interaction module. Its structure has been determined (37) (Fig. 1, 1mhd) and it has been classified in a fold of its own in the SCOP database. However, DALI finds pronounced similarity between the Smad MH1 domain (1mhd) and I-PpoI endonuclease (1a73: Z-score 2.7, r.m.s.d. 3.3 Å for 78 residues) which spans over the entire domain length. In addition to this global fold resemblance, both proteins possess a conserved motif of three cysteines and one histidine, which form a zinc-binding site in I-PpoI (Fig. 2, between β-strands c and d; Figs 3 and 5, site 5). MH1 of Smad3 and I-PpoI exhibit similar nucleic acid-binding modes and interact with the DNA major groove through an anti-parallel β-sheet (37,46). Sequence conservation in the zinc-binding region, structural similarity and similar functional properties strongly suggest that the MH1 domain of Smad and I-PpoI endonuclease are homologous (77). Therefore, the region structurally equivalent to the treble clef domain in I-PpoI endonuclease is treated as a rudiment of a treble clef motif here. This structural segment does not include the α-helix A, its treble clef zinc-binding site is missing (as in I-PpoI endonuclease), and only the primary β-hairpin cd is present (Fig. 1A, 1mhd). The C-terminal segment of the Smad MH1 domain represents an extreme case of deterioration of the treble clef motif, detection of which was only possible due to extensive similarities in the regions outside the motif.
Treble clef—protein fold or supersecondary structural element
The treble clef motif has a unique appearance due to unusual packing of the two β-hairpins (knuckle hairpin ab and primary hairpin cd, Fig. 1), which are situated in almost orthogonal planes, and an α-helix that follows them. This makes treble clef motifs easy to recognize. The usual presence of a zinc-binding site with ligands contributed by a zinc knuckle and the first turn in an α-helix facilitates detection of treble clef fingers as well. Thus, manual inspection of the structure offers a reliable way to identify the motif. Traditionally used generic structure similarity search programs such as DALI (15), VAST (16) and CE (17) are not consistent in detection of treble clef structures. This phenomenon is well known for small protein domains, whose size does not allow for convincing statistical support to be obtained (11). However, as discussed above, transitive DALI and VAST searches make it possible to establish connections between all classes of treble clef fingers starting from the ribosomal protein structures L24E and S14 as the search queries.
In addition to the ribosomal proteins that are not yet entered in SCOP (v.1.53) (3), treble clef fingers are detected in proteins from seven different SCOP folds. Moreover, these folds are attributed to two different protein classes, α+β and small proteins. The question arises, does the presence of the treble clef motif in all these proteins warrant their unification in a single fold, or is treble clef merely a supersecondary structural element, similar, for example, to a βαβ right-handed unit, that can be used to build up different folds? More importantly, do all treble clefs have a common evolutionary origin (homology), or are they diversified from several evolutionarily unrelated proteins (convergence, parallelism)? The answer to the first question depends largely on definitions and personal preferences. Clearly, treble clef fingers are very small and contain only five secondary structural elements. Furthermore, many of them recruit additional secondary structures that are packed into a single domain (Fig. 5). However, in several proteins, the most notable example being ribosomal proteins L24E and S14 (Fig. 1), the treble clef is the only globular domain. In others, such as His-Me finger endonucleases, treble clefs incorporated in different structural contexts are the only structurally similar segment, among homologous enzymes and thus constitute the core. Additionally, structural differences between treble clefs within a SCOP fold are often larger than those in different folds. For example, the treble clefs in the structures of the zinc-binding domain of DNA repair factor XPA (1xpa) and the LIM domain (1qli) which belong to the same superfamily within the glucocorticoid receptor-like SCOP fold can be superimposed with r.m.s.d. of only 3.4 Å, while the treble clefs of T4 recombination endonuclease VII (1en7) and the FYVE domain (1dvp) which belong not only to two different SCOP folds, but also to two different SCOP protein classes (α+β and small proteins), superimpose with r.m.s.d. of 1.1 Å (Table 1). Thus, it can be argued that, due to its distinct appearance, ability to form a structural and/or functional core, and presence as a sole domain in some proteins, the treble clef deserves the status of a protein fold. In this case, all domains described here should be classified together.
On the basis of the results presented here, we cannot argue convincingly that all of the treble clef fingers are homologous. It is clear that homology offers an easy explanation for sequence and structural similarity in this unique motif. However, the motif is very short. Additionally, the existence of overlapping motifs (Fig. 4) might suggest independent origins for different treble clef domains. Thus, it is possible that some of the treble clef fingers do not share a common ancestor with others. Despite this evolutionary difficulty, the treble clef is a clearly defined unique structural motif that deserves recognition.
Treble clef fingers: functional implications
One of the most remarkable features of the treble clef domain is its ability to adopt various functions. Despite the small size of the domain with the core of 25 residues (Fig. 2), treble clefs are more diverse functionally than many larger domains. Most of the treble clef domains bind one or several metal ions (Figs 1, 3 and 5). However, it is likely that these ions serve a structural role (with one exception), and metal-binding is not considered a functional property here. Treble clefs can perform many classes of function from binding to catalytic, which include protein-binding, small ligand-binding, nucleic acid (DNA and RNA)-binding and enzymatic properties. As far as cellular functions are concerned, treble clef fingers are largely involved in regulation through molecular recognition and participate in different signal transduction pathways (2,31,74,76,78).
Functional properties of treble clef fingers are exemplified in Figure 6. Treble clefs such as RING fingers and LIM domains are known to be protein-binding modules (51,78). The structure of the RING finger domain of the signal transduction protein Cbl in complex with ubiquitin-conjugating enzyme Ubch7 (61) (1fbv chains A and C) reveals that the latter is bound near the zinc-binding site and most contacts are formed with the treble clef α-helix and zinc knuckle (Fig. 6A).
Figure 6.

Functional properties of treble clef fingers. Stereo diagrams of (A) RING finger domain of signal transduction protein Cbl (black) in complex with ubiquitin-conjugating enzyme Ubch7 (blue) (1fbv, chain A, residues 376–431 in black, segments of the chain C in blue); (B) Cys2 activator-binding domain of protein kinase Cδ (black) in complex with phorbol ester (orange) (1ptr, residues 231–280); (C) retinoid X receptor α DNA-binding domain (black) in complex with DNA (red) (2nll, chain B residues 300–336 in black, chains C and D in red); (D) intron-encoded homing endonuclease I-PpoI (black) in complex with DNA (red) (1a73, chain A, residues 49–125 in black, chains C and D in red). Cα traces of treble-clef-containing proteins are displayed in black with N- and C-termini labeled. The treble clef motif is shown in thicker lines. Zinc ions are represented by a gray ball. Side chains of zinc ligands or residues in corresponding sites are shown in black. Side chains of active site residues and an active site Mg2+ ion are shown in green. Cα traces of the polypeptide chains interacting with the treble clef domain are dark blue, small molecules are in orange, DNA chains are in red.
Binding of small molecules to treble clefs is elucidated by the structure of the cysteine-rich domain of protein kinase Cδ in complex with phorbol ester (30) (Fig. 6B, 1ptr). Phorbol binds between two loops: the one before and the one after the knuckle β-hairpin. It was hypothesized that a similar lipid-binding mode should be present in the FYVE domain and rabphilin-3a (31).
Most treble clefs, however, interact with nucleic acids. RNA-binding might have been the function of ancestral treble clef fingers such as ribosomal protein L24E and S14–like domains. The DNA-binding mode of the treble clef motif is illustrated by the structure of retinoid X receptor α in complex with DNA (57) (Fig. 6C, 2nll, chains B, C and D). The mode of nucleic acid-binding is similar in all known complexes that involve treble clef motifs. The treble clef α-helix is situated in the DNA major groove and the outer β-strand c of the primary β-hairpin interacts with the phosphate backbone.
Intron-encoded homing endonuclease I-PpoI binds to DNA (46) (Fig. 6D, 1a73, chains A, C and D) in a similar manner. However, in addition to its DNA-binding property, the treble clef finger carries out an enzymatic function. First, it binds a metal ion (Mg2+) that participates in catalysis; secondly, it incorporates an invariant histidine residue. It has been suggested that His-Me finger endonucleases employ a single-metal catalytic mechanism in which the metal ion stabilizes the transition state, and a water molecule is activated by the histidine for an attack on the scissile phosphate (48,79).
Due to the similarities in both structure and DNA-binding mechanism between His-Me finger endonucleases and non-catalytic treble clef fingers, it is likely that these endonucleases originated from the broader class of nucleic acid-binding domains that served as transcription regulators. A possible gain of enzymatic activity by RING fingers has also been described (80). The eukaryotic transcriptional regulator MH1 domain of Smad, which is more similar to endonuclease I-PpoI than to other endonucleases or other treble clef fingers, probably descended from His-Cys box endonucleases (81) through the loss of catalytic activity. Thus, treble clef proteins provide a unique example of domains that were able to gain enzymatic activity in the course of evolution from transcription regulators to endonucleases and then lose it again on the path from endonucleases to Smad transcription regulators.
Despite some differences in the location of binding sites for proteins, small molecules and nucleic acids on the treble clef surface, it appears that all binding sites are confined to a particular side of the structure. If a sphere is drawn around a treble clef domain with the poles placed at the tip of the primary β-hairpin and near the N-terminus of the α-helix, functional sites appear to be confined to the hemisphere with the pole in the α-helix area (Fig. 6). The α-helix and loops near the zinc-binding site provide surfaces and grooves suitable for placing binding and catalytic sites. In summary, the treble clef finger, a short 25–45 residue motif defined by a unique and unusual structure of two orthogonally placed β-hairpins and an α-helix assembled around a zinc ion, offers a particularly versatile template adaptable for a variety of functions, both in catalysis or in binding to proteins, small molecules and nucleic acids.
Acknowledgments
ACKNOWLEDGEMENTS
I am grateful to Steve Sprang, Hong Zhang and Sara Cheek for critical reading of the manuscript and helpful comments, and Tammiko Jones for excellent technical assistance.
References
- 1.Alberts I.L., Nadassy,K. and Wodak,S.J. (1998) Analysis of zinc binding sites in protein crystal structures. Protein Sci., 7, 1700–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mackay J.P. and Crossley,M. (1998) Zinc fingers are sticking together. Trends Biochem. Sci., 23, 1–4. [DOI] [PubMed] [Google Scholar]
- 3.Murzin A.G., Brenner,S.E., Hubbard,T. and Chothia,C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol., 247, 536–540. [DOI] [PubMed] [Google Scholar]
- 4.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Finn,R.D. and Sonnhammer,E.L. (1999) Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res., 27, 260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teplyakov A., Polyakov,K., Obmolova,G., Strokopytov,B., Kuranova,I., Osterman,A., Grishin,N., Smulevitch,S., Zagnitko,O., Galperina,O. et al. (1992) Crystal structure of carboxypeptidase T from Thermoactinomyces vulgaris. Eur. J. Biochem., 208, 281–288. [DOI] [PubMed] [Google Scholar]
- 6.Burmeister W.P., Cusack,S. and Ruigrok,R.W. (1994) Calcium is needed for the thermostability of influenza B virus neuraminidase. J. Gen. Virol., 75, 381–388. [DOI] [PubMed] [Google Scholar]
- 7.Smith C.A., Toogood,H.S., Baker,H.M., Daniel,R.M. and Baker,E.N. (1999) Calcium-mediated thermostability in the subtilisin superfamily: the crystal structure of Bacillus Ak.1 protease at 1.8 A resolution. J. Mol. Biol., 294, 1027–1040. [DOI] [PubMed] [Google Scholar]
- 8.Wedemeyer W.J., Welker,E., Narayan,M. and Scheraga,H.A. (2000) Disulfide bonds and protein folding. Biochemistry, 39, 4207–4216. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe S.A., Nekludova,L. and Pabo,C.O. (2000) DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct., 29, 183–212. [DOI] [PubMed] [Google Scholar]
- 10.Aravind L. and Koonin,E.V. (1999) DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res., 27, 4658–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grishin N.V. (2000) C-terminal domains of Escherichia coli topoisomerase I belong to the zinc-ribbon superfamily. J. Mol. Biol., 299, 1165–1177. [DOI] [PubMed] [Google Scholar]
- 12.Chen H.T., Legault,P., Glushka,J., Omichinski,J.G. and Scott,R.A. (2000) Structure of a (Cys3His) zinc ribbon, a ubiquitous motif in archaeal and eucaryal transcription. Protein Sci., 9, 1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B., Jones,D.N., Kaine,B.P. and Weiss,M.A. (1998) High-resolution structure of an archaeal zinc ribbon defines a general architectural motif in eukaryotic RNA polymerases. Structure, 6, 555–569. [DOI] [PubMed] [Google Scholar]
- 14.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- 16.Gibrat J.F., Madej,T. and Bryant,S.H. (1996) Surprising similarities in structure comparison. Curr. Opin. Struct. Biol., 6, 377–385. [DOI] [PubMed] [Google Scholar]
- 17.Shindyalov I.N. and Bourne,P.E. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng., 11, 739–747. [DOI] [PubMed] [Google Scholar]
- 18.Nissen P., Hansen,J., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 19.Ban N., Nissen,P., Hansen,J., Moore,P.B. and Steitz,T.A. (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science, 289, 905–920. [DOI] [PubMed] [Google Scholar]
- 20.Carter A.P., Clemons,W.M., Brodersen,D.E., Morgan-Warren,R.J., Wimberly,B.T. and Ramakrishnan,V. (2000) Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature, 407, 340–348. [DOI] [PubMed] [Google Scholar]
- 21.Wimberly B.T., Brodersen,D.E., Clemons,W.M.,Jr, Morgan-Warren,R.J., Carter,A.P., Vonrhein,C., Hartsch,T. and Ramakrishnan,V. (2000) Structure of the 30S ribosomal subunit. Nature, 407, 327–339. [DOI] [PubMed] [Google Scholar]
- 22.Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 23.Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 133–138. [DOI] [PubMed] [Google Scholar]
- 24.Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- 25.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wootton J.C. (1994) Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput. Chem., 18, 269–285. [DOI] [PubMed] [Google Scholar]
- 27.Wootton J.C. and Federhen,S. (1996) Analysis of compositionally biased regions in sequence databases. Methods Enzymol., 266, 554–571. [DOI] [PubMed] [Google Scholar]
- 28.Henikoff S. and Henikoff,J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker D.R. and Koonin,E.V. (1997) SEALS: a system for easy analysis of lots of sequences. Ismb, 5, 333–339. [PubMed] [Google Scholar]
- 30.Zhang G., Kazanietz,M.G., Blumberg,P.M. and Hurley,J.H. (1995) Crystal structure of the cys2 activator-binding domain of protein kinase C δ in complex with phorbol ester. Cell, 81, 917–924. [DOI] [PubMed] [Google Scholar]
- 31.Misra S. and Hurley,J.H. (1999) Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting motif, the FYVE domain of Vps27p. Cell, 97, 657–666. [DOI] [PubMed] [Google Scholar]
- 32.Kajava A.V. (1992) Left-handed topology of super-secondary structure formed by aligned α-helix and β-hairpin. FEBS Lett., 302, 8–10. [DOI] [PubMed] [Google Scholar]
- 33.Shub D.A., Goodrich-Blair,H. and Eddy,S.R. (1994) Amino acid sequence motif of group I intron endonucleases is conserved in open reading frames of group II introns. Trends Biochem. Sci., 19, 402–404. [DOI] [PubMed] [Google Scholar]
- 34.Gorbalenya A.E. (1994) Self-splicing group I and group II introns encode homologous (putative) DNA endonucleases of a new family. Protein Sci., 3, 1117–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalgaard J.Z., Klar,A.J., Moser,M.J., Holley,W.R., Chatterjee,A. and Mian,I.S. (1997) Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the HNH family. Nucleic Acids Res., 25, 4626–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aravind L., Makarova,K.S. and Koonin,E.V. (2000) Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res., 28, 3417–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y., Wang,Y.F., Jayaraman,L., Yang,H., Massague,J. and Pavletich,N.P. (1998) Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell, 94, 585–594. [DOI] [PubMed] [Google Scholar]
- 38.Aravind L. and Koonin,E.V. (2000) The U box is a modified RING finger—a common domain in ubiquitination. Curr. Biol., 10, R132–R134. [DOI] [PubMed] [Google Scholar]
- 39.Friedhoff P., Franke,I., Meiss,G., Wende,W., Krause,K.L. and Pingoud,A. (1999) A similar active site for non-specific and specific endonucleases. Nat. Struct. Biol., 6, 112–113. [DOI] [PubMed] [Google Scholar]
- 40.Friedhoff P., Franke,I., Krause,K.L. and Pingoud,A. (1999) Cleavage experiments with deoxythymidine 3′,5′-bis-(p-nitrophenyl phosphate) suggest that the homing endonuclease I-PpoI follows the same mechanism of phosphodiester bond hydrolysis as the non-specific Serratia nuclease. FEBS Lett., 443, 209–214. [DOI] [PubMed] [Google Scholar]
- 41.Kuhlmann U.C., Moore,G.R., James,R., Kleanthous,C. and Hemmings,A.M. (1999) Structural parsimony in endonuclease active sites: should the number of homing endonuclease families be redefined? FEBS Lett., 463, 1–2. [DOI] [PubMed] [Google Scholar]
- 42.Ko T.P., Liao,C.C., Ku,W.Y., Chak,K.F. and Yuan,H.S. (1999) The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure Fold. Des., 7, 91–102. [DOI] [PubMed] [Google Scholar]
- 43.Kleanthous C., Kuhlmann,U.C., Pommer,A.J., Ferguson,N., Radford,S.E., Moore,G.R., James,R. and Hemmings,A.M. (1999) Structural and mechanistic basis of immunity toward endonuclease colicins. Nat. Struct. Biol., 6, 243–252. [DOI] [PubMed] [Google Scholar]
- 44.Miller M.D., Tanner,J., Alpaugh,M., Benedik,M.J. and Krause,K.L. (1994) 2.1 Å structure of Serratia endonuclease suggests a mechanism for binding to double-stranded DNA. Nat. Struct. Biol., 1, 461–468. [DOI] [PubMed] [Google Scholar]
- 45.Shlyapnikov S.V., Lunin,V.V., Perbandt,M., Polyakov,K.M., Lunin,V.Y., Levdikov,V.M., Betzel,C. and Mikhailov,A.M. (2000) Atomic structure of the Serratia marcescens endonuclease at 1.1 Å resolution and the enzyme reaction mechanism. Acta Crystallogr. D, 56, 567–572. [DOI] [PubMed] [Google Scholar]
- 46.Flick K.E., Jurica,M.S., Monnat,R.J.,Jr and Stoddard,B.L. (1998) DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature, 394, 96–101. [DOI] [PubMed] [Google Scholar]
- 47.Raaijmakers H., Vix,O., Toro,I., Golz,S., Kemper,B. and Suck,D. (1999) X-ray structure of T4 endonuclease VII: a DNA junction resolvase with a novel fold and unusual domain-swapped dimer architecture. EMBO J., 18, 1447–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galburt E.A., Chevalier,B., Tang,W., Jurica,M.S., Flick,K.E., Monnat,R.J.,Jr and Stoddard,B.L. (1999) A novel endonuclease mechanism directly visualized for I-PpoI. Nat. Struct. Biol., 6, 1096–1099. [DOI] [PubMed] [Google Scholar]
- 49.Yao X., Perez-Alvarado,G.C., Louis,H.A., Pomies,P., Hatt,C., Summers,M.F. and Beckerle,M.C. (1999) Solution structure of the chicken cysteine-rich protein, CRP1, a double-LIM protein implicated in muscle differentiation. Biochemistry, 38, 5701–5713. [DOI] [PubMed] [Google Scholar]
- 50.Konrat R., Weiskirchen,R., Krautler,B. and Bister,K. (1997) Solution structure of the carboxyl-terminal LIM domain from quail cysteine-rich protein CRP2. J. Biol. Chem., 272, 12001–12007. [DOI] [PubMed] [Google Scholar]
- 51.Perez-Alvarado G.C., Kosa,J.L., Louis,H.A., Beckerle,M.C., Winge,D.R. and Summers,M.F. (1996) Structure of the cysteine-rich intestinal protein, CRIP. J. Mol. Biol., 257, 153–174. [DOI] [PubMed] [Google Scholar]
- 52.Hammarstrom A., Berndt,K.D., Sillard,R., Adermann,K. and Otting,G. (1996) Solution structure of a naturally-occurring zinc–peptide complex demonstrates that the N-terminal zinc-binding module of the Lasp-1 LIM domain is an independent folding unit. Biochemistry, 35, 12723–12732. [DOI] [PubMed] [Google Scholar]
- 53.Ostermeier C. and Brunger,A.T. (1999) Structural basis of Rab effector specificity: crystal structure of the small G protein Rab3A complexed with the effector domain of rabphilin-3A. Cell, 96, 363–374. [DOI] [PubMed] [Google Scholar]
- 54.Mao Y., Nickitenko,A., Duan,X., Lloyd,T.E., Wu,M.N., Bellen,H. and Quiocho,F.A. (2000) Crystal structure of the VHS and FYVE tandem domains of Hrs, a protein involved in membrane trafficking and signal transduction. Cell, 100, 447–456. [DOI] [PubMed] [Google Scholar]
- 55.Bellon S.F., Rodgers,K.K., Schatz,D.G., Coleman,J.E. and Steitz,T.A. (1997) Crystal structure of the RAG1 dimerization domain reveals multiple zinc-binding motifs including a novel zinc binuclear cluster. Nat. Struct. Biol., 4, 586–591. [DOI] [PubMed] [Google Scholar]
- 56.Mandiyan V., Andreev,J., Schlessinger,J. and Hubbard,S.R. (1999) Crystal structure of the ARF-GAP domain and ankyrin repeats of PYK2-associated protein β. EMBO J., 18, 6890–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rastinejad F., Perlmann,T., Evans,R.M. and Sigler,P.B. (1995) Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature, 375, 203–211. [DOI] [PubMed] [Google Scholar]
- 58.Borden K.L. (2000) RING domains: master builders of molecular scaffolds? J. Mol. Biol., 295, 1103–1112. [DOI] [PubMed] [Google Scholar]
- 59.Barlow P.N., Luisi,B., Milner,A., Elliott,M. and Everett,R. (1994) Structure of the C3HC4 domain by 1H-nuclear magnetic resonance spectroscopy. A new structural class of zinc-finger. J. Mol. Biol., 237, 201–211. [DOI] [PubMed] [Google Scholar]
- 60.Borden K.L., Boddy,M.N., Lally,J., O’Reilly,N.J., Martin,S., Howe,K., Solomon,E. and Freemont,P.S. (1995) The solution structure of the RING finger domain from the acute promyelocytic leukaemia proto-oncoprotein PML. EMBO J., 14, 1532–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng N., Wang,P., Jeffrey,P.D. and Pavletich,N.P. (2000) Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell, 102, 533–539. [DOI] [PubMed] [Google Scholar]
- 62.Gervais V., Busso,D., Wasielewski,E., Poterszman,A., Egly,J.-M., Thierry,J.-C. and Kieffer,B. (2001) Solution structure of the N-terminal domain of the human TFIIH MAT1 subunit: new insights into the RING finger family. J. Biol. Chem., 276, 7457–7464. [DOI] [PubMed] [Google Scholar]
- 63.Goldberg J. (1999) Structural and functional analysis of the ARF1–ARFGAP complex reveals a role for coatomer in GTP hydrolysis. Cell, 96, 893–902. [DOI] [PubMed] [Google Scholar]
- 64.Ichikawa S., Hatanaka,H., Takeuchi,Y., Ohno,S. and Inagaki,F. (1995) Solution structure of cysteine-rich domain of protein kinase C α. J. Biochem. (Tokyo), 117, 566–574. [DOI] [PubMed] [Google Scholar]
- 65.Mott H.R., Carpenter,J.W., Zhong,S., Ghosh,S., Bell,R.M. and Campbell,S.L. (1996) The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc. Natl Acad. Sci. USA, 93, 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu R.X., Pawelczyk,T., Xia,T.H. and Brown,S.C. (1997) NMR structure of a protein kinase C-γ phorbol-binding domain and study of protein–lipid micelle interactions. Biochemistry, 36, 10709–10717. [DOI] [PubMed] [Google Scholar]
- 67.De Camilli P., Emr,S.D., McPherson,P.S. and Novick,P. (1996) Phosphoinositides as regulators in membrane traffic. Science, 271, 1533–1539. [DOI] [PubMed] [Google Scholar]
- 68.Omichinski J.G., Clore,G.M., Schaad,O., Felsenfeld,G., Trainor,C., Appella,E., Stahl,S.J. and Gronenborn,A.M. (1993) NMR structure of a specific DNA complex of Zn-containing DNA binding domain of GATA-1. Science, 261, 438–446. [DOI] [PubMed] [Google Scholar]
- 69.Kowalski K., Czolij,R., King,G.F., Crossley,M. and Mackay,J.P. (1999) The solution structure of the N-terminal zinc finger of GATA-1 reveals a specific binding face for the transcriptional co-factor FOG. J. Biomol. NMR, 13, 249–262. [DOI] [PubMed] [Google Scholar]
- 70.Ikegami T., Kuraoka,I., Saijo,M., Kodo,N., Kyogoku,Y., Morikawa,K., Tanaka,K. and Shirakawa,M. (1998) Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat. Struct. Biol., 5, 701–706. [DOI] [PubMed] [Google Scholar]
- 71.Meinke G. and Sigler,P.B. (1999) DNA-binding mechanism of the monomeric orphan nuclear receptor NGFI-B. Nat. Struct. Biol., 6, 471–477. [DOI] [PubMed] [Google Scholar]
- 72.Zhao Q., Khorasanizadeh,S., Miyoshi,Y., Lazar,M.A. and Rastinejad,F. (1998) Structural elements of an orphan nuclear receptor–DNA complex. Mol. Cell ., 1, 849–861. [DOI] [PubMed] [Google Scholar]
- 73.Rastinejad F., Wagner,T., Zhao,Q. and Khorasanizadeh,S. (2000) Structure of the RXR–RAR DNA-binding complex on the retinoic acid response element DR1. EMBO J., 19, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luisi B.F., Xu,W.X., Otwinowski,Z., Freedman,L.P., Yamamoto,K.R. and Sigler,P.B. (1991) Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature, 352, 497–505. [DOI] [PubMed] [Google Scholar]
- 75.Schwabe J.W., Chapman,L., Finch,J.T. and Rhodes,D. (1993) The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell, 75, 567–578. [DOI] [PubMed] [Google Scholar]
- 76.Massague J. and Wotton,D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J., 19, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grishin N.V. (2001) MH1 domain of Smad is a degraded homing endonuclease. J. Mol. Biol., 301, 31–37. [DOI] [PubMed] [Google Scholar]
- 78.Borden K.L. (1998) RING fingers and B-boxes: zinc-binding protein–protein interaction domains. Biochem. Cell Biol., 76, 351–358. [DOI] [PubMed] [Google Scholar]
- 79.Mannino S.J., Jenkins,C.L. and Raines,R.T. (1999) Chemical mechanism of DNA cleavage by the homing endonuclease I-PpoI. Biochemistry, 38, 16178–16186. [DOI] [PubMed] [Google Scholar]
- 80.Lorick K.L., Jensen,J.P., Fang,S., Ong,A.M., Hatakeyama,S. and Weissman,A.M. (1999) RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl Acad. Sci. USA, 96, 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johansen S., Embley,T.M. and Willassen,N.P. (1993) A family of nuclear homing endonucleases. Nucleic Acids Res., 21, 4405. [DOI] [PMC free article] [PubMed] [Google Scholar]