

Abstract
Pain is a key component of most rheumatologic diseases. In fibromyalgia, the importance of central nervous system pain mechanisms (for example, loss of descending analgesic activity and central sensitization) is well documented. A few studies have also noted alterations in central pain processing in osteoarthritis, and some data, including the observation of widespread pain sensitivity, suggest that central pain-processing defects may alter the pain response in rheumatoid arthritis patients. When central pain is identified, different classes of analgesics (for example, serotonin-norepinephrine reuptake inhibitors, α2δ ligands) may be more effective than drugs that treat peripheral or nociceptive pain (for example, nonsteroidal anti-inflammatory drugs and opioids).
Importance of chronic pain in the rheumatic diseases
Although pain is commonly the patients' utmost priority and the reason most patients seek rheumatologic consultation, the medical community has historically had a poor understanding of the etiology, mechanisms and treatment of pain. Rheumatologists often consider pain a peripheral entity, but there is great discordance between pain severity and purported peripheral causes of pain, such as inflammation and structural joint damage (for example, cartilage degradation, erosions).
In recognition of the importance of pain in the rheumatic diseases, the American College of Rheumatology Pain Management Task Force established an initiative to increase awareness and call for organized research and education [1]. This initiative emphasizes the need for high-quality, quantitative research to understand the mechanisms underlying individual differences in pain among patients with rheumatic disease. Currently, most advances in the study of pain mechanisms have been in non-inflammatory diseases, such as fibromyalgia [2]. These studies have highlighted the role of central pain-processing mechanisms, such as loss of descending analgesic activity and central pain augmentation or sensitization. Some pain researchers also believe that these mechanisms may have a significant impact on pain severity among patients with osteoarthritis (OA) and rheumatoid arthritis (RA), diseases that have historically been associated with peripheral pain due to joint damage and inflammation.
In the present review we give a brief overview of the basic biology of acute and chronic pain, including the role of central pain-processing defects. We discuss the role of these mechanisms in diseases commonly seen in rheumatology practices (for example, fibromyalgia, OA and RA) and consider potential treatments that may correct deficits in central pain processing.
Basic biology of pain in healthy individuals
To determine the cause of pain, rheumatologists frequently categorize pain into acute pain and chronic pain. Acute pain typically lasts from seconds to weeks or months. Acute pain is often sudden in onset, as it is usually the direct result of a noxious stimulus. In contrast, chronic pain is, by definition, present for at least 3 months. Chronic pain may persist because the original inciting stimulus is still present and/or because changes to the nervous system have occurred, making it more sensitive to pain.
Acute pain
Acute pain develops when a stimulus, such as pressure, heat or inflammation, is presented to the body. Specialized receptors sense these stimuli and transport the signals to the central nervous system (CNS) via nerve fibers that extend into the dorsal horn of the spinal cord. The specialized receptors include low-threshold receptors that respond to non-noxious levels of stimuli, and high-threshold receptors that sense noxious stimuli (nociceptors). Both nerve fibers reside in soft tissue throughout the body, including the muscle, skin and internal organs.
Two types of nociceptors, the Aδ afferent and the C afferent, are responsible for the sensation and differentiation of mechanical, chemical and heat stimuli. The Aδ nerve fiber has two classes, Type I and Type II, which respond to mechanical and heat stimuli. Type I fibers have higher heat thresholds than Type II fibers, while Type II fibers have higher mechanical thresholds than Type I fibers [3]. Consequently, the Type I Aδ afferents usually transmit noxious mechanical stimuli while the Type II Aδ afferents often transmit noxious heat stimuli. The C nerve fibers detect mechanical and heat stimuli, as well as chemical stimuli. Compared with pain mediated by Aδ fibers, pain mediated by unmyelinated C fibers tends to be poorly localized [4].
Chronic pain
Chronic pain is associated with many rheumatologic conditions, varying from non-inflammatory syndromes, such as fibromyalgia, to systemic inflammatory diseases, such as RA. Depending on the condition, as well as individual factors, differing pain mechanisms are involved. Mechanisms of chronic pain can be divided into peripheral mechanisms and central mechanisms.
Peripheral pain mechanisms stem from abnormalities in the peripheral nerves, leading to local areas of enhanced pain sensitivity. The most commonly cited peripheral pain mechanism besides direct nociceptive input is peripheral sensitization, which probably plays important roles in chronic pain mediated by OA and RA. This topic is covered in depth by Schaible and colleagues in an earlier manuscript in this Biology of Pain review series [5].
Central pain mechanisms operate at the level of the CNS, leading to enhanced widespread pain sensitivity. Individuals with augmented central pain processing will display diffuse hyperalgesia (increased pain in response to normally painful stimuli) and allodynia (pain in response to normally nonpainful stimuli).
Abnormalities in central pain processing are divided into abnormalities in the descending facilitatory and inhibitory pain pathways, and central sensitization (Figure 1). The descending pain pathways descend from the brainstem, hypothalamus and cortical structures, and modulate sensory input from primary afferent fibers and projection neurons in the dorsal horn of the spinal cord [6]. The best characterized descending analgesic path-ways are the serotonergic-noradrenergic pathway and the opioidergic pathway. These pathways lead to the release of serotonin, norepinephrine and endogenous opioids, which inhibit the release of excitatory neurotransmitters such as glutamate. These pathways are activated in response to noxious stimuli, leading to a widespread decrease in pain sensitivity after exposure to an acutely painful stimulus. In chronic pain syndromes, descending analgesic activity is often impaired or absent - hence the term loss of descending analgesia.
Figure 1.
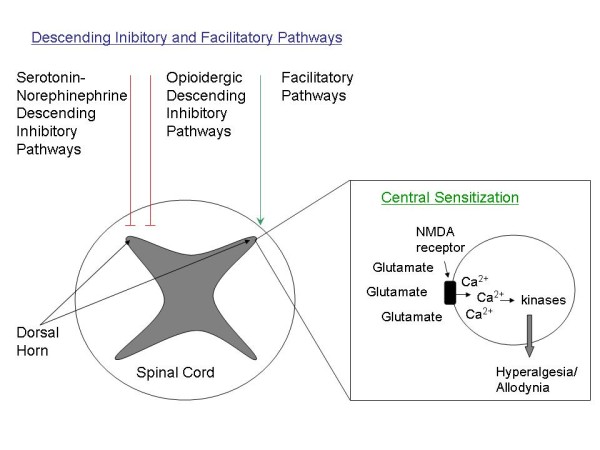
Descending pain pathways and central sensitization. Descending pain pathways and central sensitization modulate the pain response in the dorsal horn of the spinal cord. Descending analgesic pathways include the serotonin-norepinephrine and opioidergic descending pathways, which dampen pain sensitivity response. Loss of descending analgesia leads to hyperalgesia and allodynia. Central sensitization occurs through the action of glutamate on the N-methyl-D-aspartate (NMDA) receptor, resulting in an increase in intracellular calcium levels and kinase activation, leading to hyperalgesia and allodynia.
In the present review, loss of descending analgesia is used synonymously with the term loss of diffuse noxious inhibitory controls. Experimentally, diffuse noxious inhibitory control is commonly assessed by exposing subjects to two types of stimuli: the conditioning stimulus, and the test stimulus. The conditioning stimulus is an acute noxious stimulus that activates descending analgesic pathways, leading to a diffuse decrease in pain sensitivity throughout the body [7]. In healthy controls, a wide range of noxious stimuli - including ice-cold water, contact heat and tourniquet ischemia - are all effective conditioning stimuli, producing increased pain thresh-olds throughout the body [7]. The test stimulus is a painful stimulus that is applied at baseline and during/after exposure to the conditioning stimulus. The magnitude of the descending analgesic response is the difference between the pain rating of the test stimulus before exposure to the conditioning stimulus and the pain rating of the test stimulus after exposure to the conditioning stimulus [7].
When evaluating these studies, it is important to understand that, although commonly used to assess descending analgesia, these studies do not specifically localize the areas of pain modulation to the descending spinal tracts. Changes in pain threshold after noxious pain stimulation may also partly reflect changes in attention (for example, distraction) or other processes that influence pain perception. To directly assess the descending spinal pathways, electrophysiologic assessments of the spinal nociceptive flexion reflex must be performed.
While descending analgesic pathways are typically tonically active and inhibit the upward transmission of pain signals, other descending pain-processing mechanisms involve enhanced activity down the descending facilitatory pain pathways that lead to generalized increases in sensory sensitivity [8]. The role of these facilitatory pathways, however, has not been well established in human studies.
In addition to descending inhibitory and facilitatory pathways, central sensitization also leads to enhanced CNS neuron excitability and increased transmission of pain signals. In the literature, the term central sensitization may be used in two ways: to describe general abnormalities in central pain processing (which, in the present review, we will refer to as central augmentation); and to describe a specific defect in central pain processing associated with activation of N-methyl-D-aspartate (NMDA) receptor channels (which we will refer to as central sensitization).
Central sensitization occurs largely as a result of enhanced release of glutamate and substance P at the level of the spinal cord. Glutamate is the major excitatory neurotransmitter in the nervous system, and it acts on three receptor subsets: the α-amino-3-hydroxy-5-methyl-4-isoxazeloproprionic acid receptor, the NMDA receptor and the G-protein-coupled metabotropic family of receptors. While the α-amino-3-hydroxy-5-methyl-4-isoxazeloproprionic acid receptor is responsible for the baseline response to noxious stimuli, the NMDA receptor enhances and extends the pain response [9]. NMDA receptor activation results in calcium influx, stimulating calcium/calmodulin-dependent kinases and extracellular signal-regulated kinases. These changes modulate CNS plasticity, resulting in the hyperalgesia and allodynia that characterize central sensitization [9].
Experimentally, central sensitization is characterized by di use pain sensitivity and increased pain severity during and after repeated stimuli. Individuals with central sensitization have low thermal and mechanical thresholds in a diffuse pattern, reflecting enlargement of the spinal cord neuron receptive fields [4]. Repeated stimulation results in painful after-sensations that persist after a stimulus is withdrawn; and results in enhanced temporal summation of pain such that the pain rating for the last stimulus is higher than the pain rating for the first stimulus, even though the stimuli are exactly the same. NMDA receptor antagonists, such as dextromethorphan and ketamine, inhibit temporal summation [10-12].
Studies suggest that maintenance of central augmentation requires persistent noxious peripheral input, even in syndromes such as fibromyalgia, which is characterized by the absence of well-defined, localized, pain-causing lesions [13,14]. A recent study of 68 fibromyalgia patients with myofascial pain syndromes and 56 fibromyalgia patients with regional joint pain showed that peripheral trigger point injections and hydroelectrophoresis ameliorate fibromyalgia pain and increase pain thresholds at sites distant from the therapeutic interventions [15], providing further evidence that painful peripheral stimuli contribute to the perpetuation of central augmentation.
Fibromyalgia
Fibromyalgia is the prototypical non-inflammatory chronic pain syndrome. The disease is characterized by chronic widespread pain and associated symptoms, including sleep problems, fatigue, cognitive dysfunction and depression. Quantitative sensory testing methods have consistently identified abnormalities in pain perception among fibromyalgia patients (Table 1). Most notably, patients with fibromyalgia have diffusely lower pressure pain thresholds than healthy controls [16]. This diffuse hyperalgesic state of central augmentation of pain processing has been repeatedly identified using functional neuroimaging techniques [17,18] and may partly be due to specific defects such as loss of descending analgesic activity and central sensitization.
Table 1.
Peripheral and central quantitative sensory testing findings in fibromyalgia, osteoarthritis and rheumatoid arthritis
| Peripheral findings | Central findings | ||||
|---|---|---|---|---|---|
| Low pain thresholds at affected sites | Low pain thresholds in a widespread distribution | Loss of descending analgesia (conditioned pain modulation) | Temporal summation | Expanded areas of hyperalgesia | |
| Fibromyalgia | X | X | X | X | X |
| Osteoarthritis | X | X | X | X | X |
| Rheumatoid arthritis | X | X | X | X | |
Evidence for the role of defects in descending analgesic activity in fibromyalgia comes from studies of conditioned pain modulation [19-21]. In a study of 26 healthy controls and 25 fibromyalgia patients, heat stimulation of the foot increased pain thresholds to electric stimulation of the forearm among healthy controls but not among fibromyalgia patients [19]. Similarly, tourniquet ischemic pain increased pressure pain threshold in 10 healthy controls but not in 10 fibromyalgia patients [20], and a noxious cold stimulus reduced heat pain ratings among 20 healthy controls but not among 45 fibromyalgia patients [21].
These defects in inhibitory pain responses may be due to blunted activity of the descending serotonergic-noradrenergic system. Fibromyalgia patients have reduced serum levels of serotonin and its precursor, L-tryptophan, as well as reduced levels of the principal serotonin metabolite, 5-hydroxyindoleacetic acid, in their cerebral spinal fluid [22]. Levels of 3-methoxy-4-hydroxyphen-ethylene, the principal metabolite of norepinephrine, are also lower in the cerebral spinal fluid of fibromyalgia patients compared with healthy controls [22]. In contrast, biochemical and imaging findings suggest that fibromyalgia patients actually have increased activity of endogenous opioidergic systems, which is consistent with anecdotal experience that opioids are ineffective analgesics in patients with fibromyalgia and related conditions [23,24].
The evidence for central sensitization in fibromyalgia predominantly consists of studies comparing the magnitude of temporal summation in fibromyalgia patients with healthy controls. Although both fibromyalgia patients and healthy controls experience temporal summation, the magnitude of temporal summation may be slightly greater in fibromyalgia patients [25]. The magnitude of temporal summation is decreased by treatment with either fentanyl injections or ketamine, an NMDA antagonist [10,12].
In addition to heightened sensitivity to pain, fibromyalgia patients are also more sensitive to a variety of other sensory stimuli [26,27]. This polysensory augmentation may partly be due to enhanced neural activity that has been consistently observed in brain regions such as the insula, a region known to code for the intensity of all sensory information [17]. Previous studies suggest that the anterior insula is involved in the affective/emotional modulation of pain processing, while the posterior insula is involved in the sensory/discriminative processing of pain [28]. Compared with controls, fibromyalgia patients have higher levels of glutamate in the posterior insula, and changes in glutamate levels in the posterior insula are correlated with changes in pain and tenderness after acupuncture [29,30]. These studies suggest that at least a component of pain in fibromyalgia is a result of sensory amplification, rather than just affective processing.
Genetic studies also support an association between the serotonergic-noradrenergic system and fibromyalgia. In candidate gene studies, polymorphisms in the metabolism and transport of monoamines (for example, catecholamine-o-methyltransferase, serotonin 5-hydroxytryp-tamine type 2a receptor, serotonin transporter) have been associated with the diagnosis or severity of fibromyalgia [31-35]. Most of these studies were small, however, and conflicting data exist - with some studies reporting no association between these genes and fibromyalgia [31,36-38]. Future studies, incorporating a larger number of fibromyalgia patients and/or utilizing meta-analysis techniques, are needed.
In addition to genetic studies, a recent surge of interest has surrounded the use of functional magnetic resonance imaging (fMRI) to study pain in a more quantitative, objective manner. This area of research, however, is still relatively new. As such, we present the following results as preliminary evidence for the role of the CNS in pain modulation, rather than as well-established facts.
In one of the early studies of fMRI in fibromyalgia, Gracely and colleagues reported that fibromyalgia patients, compared with controls, exhibit enhanced activation in the contralateral primary somatosensory cortex (SI), inferior parietal lobe, insula, anterior cingulate cortex, posterior cingulate cortex, ipsilateral secondary somatosensory cortex (SII) cortex, bilateral superior temporal gyrus and cerebellum when exposed to experimental pain of the same magnitude (for example, same pressure) [17]. When exposed to experimental pain stimuli rated of similar intensity (moderate), however, fibromyalgia patients exhibited activation in the same neural structures (contralateral SI, SII, contralateral superior temporal gyrus, inferior parietal lobe, contralateral putamen, ipsilateral cerebellum and contralateral insula) as controls. These observations provided the first fMRI-based evidence for central augmentation of pain sensitivity in fibromyalgia.
Cook and colleagues noted similar findings in a study examining responses to heat stimuli [39]. In addition, their study reported post hoc analyses showing no neural activation in the periaqueductal gray region of fibromyalgia patients exposed to painful heat stimuli but significant activity in the periaqueductal gray region of healthy controls exposed to painful heat stimuli. Because previous studies have suggested that the periaqueductal gray region is involved in descending pain modulation, these findings were interpreted as possible evidence for loss of descending analgesia among fibromyalgia patients. A more recent article by Jensen and colleagues showed similar decreases in neuronal activation in the anterior cingulate cortex, a region also involved in pain modulation [40].
The fMRI techniques examining resting-state functional connectivity have also identified the default mode network as a potential modulator of spontaneous clinical pain in fibromyalgia patients. The default mode network consists of neural regions (medial frontal gyri, hippocampus, lateral temporal cortex, posterior cingulate cortex, precuneus, inferior parietal lobe) that are active at rest and may be involved in self-referential thought. In a study of 18 fibromyalgia patients and 18 age-matched and sex-matched controls, Napadow and colleagues noted that connectivity between the default mode network and the insula was positively correlated with clinical pain severity [41].
Osteoarthritis
OA is a common degenerative joint disease, characterized by damage to cartilage and bone, which affects approximately 27 million people in the United States [42]. Individuals with OA often suffer from chronic pain, ultimately leading to significant disability and healthcare costs. Despite the significant impact of pain in OA patients, little is known about the causes of the pain associated with OA.
On a population level, pain intensity (via patient self-report) correlates poorly with peripheral joint damage assessed by the Kellgren-Lawrence radiologic classification criteria [43]. Within individuals, however, pain severity is strongly associated with radiographic damage [44]. Taken together, these studies suggest that other mechanisms of pain that are not knee specific (for example, enhanced pain sensitivity due to alterations in central pain processing) may play a role in the variability in pain severity across individuals.
Studies utilizing quantitative sensory testing indicate that OA patients are more sensitive to experimental pain stimuli than healthy controls (Table 1). Most studies have focused on pain sensitivity at sites close to affected joints, showing that OA patients have lower mechanical and thermal pain thresholds (for example, higher pain sensitivity) than healthy controls [45-49]. Intriguingly, O'Driscoll and Jayson also reported low-pressure pain thresholds at the forehead, a clinically nonpainful site, unaffected by OA [50]. Similarly, among 15 patients with OA of the hip, Kosek and Ordeberg noted increased sensitivity to pressure, ischemia and innocuous warm stimuli at the affected hip and at the contralateral hip, indicating a diffuse process extending beyond just the affected joint. These studies suggest that OA pain, historically considered a peripheral entity, may also be modulated via widespread mechanisms controlled by the CNS.
Assessments of the widespread nature of pain sensitivity in OA have provided further support for the role of central pain mechanisms in OA. Bajaj and colleagues infused hypertonic saline into the tibialis anterior muscles of 14 OA patients and of 14 age-matched and sex-matched controls. OA patients reported increased pain intensity and larger pain areas, extending to the toes, whereas healthy controls reported lower pain intensity with the distribution of pain ending near the ankle. The authors attributed these findings to changes in central pain mechanisms [51]. In a larger study of 62 female knee OA patients and 22 age-matched healthy controls, Imamura and colleagues highlighted the widespread distribution of pain sensitivity, showing subcutaneous hyperalgesia to pressure stimuli at seven dermatome levels, myotomal hyperalgesia at nine lower extremity muscle groups, and sclerotomal hyperalgesia at eight sites across the lower back and legs. The authors speculated that both peripheral and central mechanisms contribute to the chronic pain state, with peripheral mechanisms being more important in the early stages, and central mechanisms dominating in the later stages [52].
Additional evidence for defects in central pain processing comes from studies assessing specific pain-processing mechanisms, such as loss of descending analgesic activity. In a study of 48 knee OA patients and 24 age-matched and sex-matched controls, OA patients exhibited greater loss of descending analgesic activity than healthy controls [49] - a finding similar to the previous study by Kosek and Ordeberg of 15 hip OA patients [47]. The study by Kosek and Ordeberg was particularly interesting because it showed that loss of descending analgesic activity is contingent upon the chronic pain state and that loss of descending analgesic activity can be reversed [47]. After the initial evaluation, 13 out of 15 hip OA patients underwent surgery, resulting in significant clinical pain relief. When the patients were reassessed 6 to 14 months after surgery (when pain-free), they exhibited significant increases in pain thresholds compared with pre surgery. Postsurgery pain thresholds were similar to pain thresholds among healthy controls. Furthermore, modulation of pain through descending analgesic pathways was restored. These results suggest that dysfunctional central pain mechanisms are associated with the chronic pain state, and removal of the inciting pain stimulus may lead to normalization of central pain processing [47].
In addition to loss of descending analgesic activity, central sensitization may also alter pain processing among OA patients. In a study examining the effects of repeated pressure stimulation on pain sensitivity, temporal summation at the knee and tibialis anterior muscle was significantly greater among patients with knee OA compared with controls [49].
Studies utilizing fMRI during quantitative sensory testing have also shown enhanced activity in the periaqueductal gray matter of OA patients compared with healthy controls [48]. This finding was interpreted as an increase in activity of the descending facilitatory path-ways, a mechanism that would have the same net effect as a decrease in descending analgesic activity. Notably, this finding is the opposite of that found by Cook and colleagues in fibromyalgia patients [39]. Cook and colleagues reported lower levels of activity in the periaqueductal gray matter of fibromyalgia patients compared with pain-free controls, which the authors interpreted as an impairment in descending analgesic pathways. Other studies using fMRI have suggested that OA-related knee pain is modulated by the medial pain system, a network of brain structures associated with the affective dimension of pain processing [53].
Rheumatoid arthritis
In contrast to fibromyalgia and OA, RA is characterized by systemic inflammation. Although inflammation contributes to pain in RA, it may not be the only factor. For some patients, pain does not improve despite treatment with anti-inflammatory disease-modifying anti-rheumatic drugs. In a cross-sectional analysis of 12,090 RA patients recruited from rheumatology practices, pain levels were almost constant over RA duration, even though most participants were treated with a disease-modifying anti-rheumatic drug, an anti-TNF agent or both [54]. A large longitudinal study, consisting of 882 RA patients, reported that pain initially decreased during the first 3 years after diagnosis but subsequently increased over time. The authors speculated that the initial decrease in pain was due to control of inflammation while the later rise in pain was attributed to other pain pathways [55].
Although few studies have specifically examined the role of central pain-processing mechanisms in RA, studies utilizing dolorimetry to assess pain thresholds suggest that these other pathways may include deficits in central pain processing. Deficits in central pain processing are characterized by enhanced pain sensitivity in a widespread distribution, and studies have consistently shown that RA patients have lower pressure pain thresholds (higher pain sensitivity) than healthy controls at joint and nonjoint sites [56-58].
Only one study has directly examined the role of descending analgesic activity in RA patients [59]. The study compared the magnitude of descending analgesic activity in 11 patients with RA of short duration to 11 healthy controls and in 10 patients with RA of long duration to 10 healthy controls. The magnitude of descending analgesic activity in both groups of RA patients was less than the magnitude of descending analgesic activity in healthy controls. These differences were not statistically significant [59], but given the small samples sizes it was difficult to determine whether there really was no difference between the two groups or whether the study was underpowered to detect an effect.
A few small studies have provided support for the role of central sensitization in pain augmentation among RA patients. Wendler and colleagues demonstrated using electroencephalography that, compared with age-matched and sex-matched controls, RA patients had enhanced cortical responses to repeated noxious stimulation, suggesting changes in CNS modulation of pain [60]. Morris and colleagues showed that capsaicin induces a larger area of hyperalgesia among RA patients compared with healthy controls [61]. This area of enhanced hyperalgesia may correspond to the enlargement of spinal cord neuron receptive fields, characteristic of central sensitization.
In addition to central augmentation of pain through central sensitization and/or loss of descending analgesia, functional neuroimaging studies suggest that structures in the medial pain system may modulate pain processing in RA. Using positron emission tomography, Jones and Derbyshire observed that regional cerebral blood flow in the dorsolateral prefrontal cortex, anterior cingulate cortex and cingulofrontal transition cortex was lower in RA patients compared with healthy controls exposed to heat pain [62]. More recently, Schwienhardt and colleagues showed that fMRI signal intensity in the medial pre-frontal cortex was significantly associated with depression severity among 20 RA patients with provoked joint pain [63]. These differences in cortical activity may reflect enhanced cortical opioid peptide release in patients with RA [64].
The relationships between inflammation, psychosocial factors and peripheral and central pain processing are intricately entwined. In a recent study of 59 female RA patients, we showed that C-reactive protein levels were inversely associated with pain thresholds at joint sites but not nonjoint sites, consistent with peripheral sensitization [65]. Sleep disturbance, on the other hand, was associated with pain thresholds at both joint and nonjoint sites, indicating a central mechanism linking pain sensitivity and sleep problems. Recent studies in healthy women [66] and in patients with temperomandibular joint disorder [67] support this hypothesis, showing that short sleep duration and forced awakenings are associated with loss of descending analgesic activity.
Mechanism-based treatment
The rheumatologist's approach to pain management has historically focused on treatment of the underlying disease process. With recent advances in the study of pain mechanisms, it has become clear that pain is multifactorial in origin, and successful treatment may require a combination of medications with different mechanisms of action. Although most rheumatologists are familiar with the use of nonsteroidal anti-inflammatory drugs for pain, few are experienced with newer classes of medications, such as antidepressants and anti-convulsants, that target central pain-processing mechanisms. Current treatments for central pain have mainly been used in the fibromyalgia population, although a few studies have examined these agents in OA patients and RA patients. In the remainder of the present review we give an overview of the medications that are likely to play an increasing role in pain management among patients with rheumatic disease.
Tricyclic antidepressants
Tricyclic antidepressants (TCAs) work by inhibiting serotonin and norepinephrine reuptake. The most commonly used TCA is amitriptyline. Other TCAs include dothiepin and imipramine.
Ten randomized, double-blinded, placebo-controlled trials have examined the efficacy of amitriptyline in fibromyalgia [68]. A meta-analysis of these studies revealed poor to moderate evidence for the efficacy of amitriptyline 25 mg daily over 6 to 8 weeks but no evidence for the efficacy of amitriptyline at higher doses or longer treatment durations. Outcome measures included patient and physician global assessment of disease, the visual analog pain scale and the tender point count [68]. Although these studies were classified as of high methodological quality by Jadad's score, other quality issues (for example, sample size, duration of follow-up and retention rates) were not considered and may limit the strength of these results.
Studies of TCAs in OA and RA have been limited. To our knowledge, no studies have specifically assessed the role of TCAs in the treatment of pain in OA - although one study examined the efficacy of imipramine in the treatment of pain in a mixed population of 66 OA, RA and ankylosing spondylitis patients, showing significant pain relief in patients treated with imipramine compared with placebo [69]. In RA, four out of six studies reported significant improvements in pain among RA patients taking TCAs compared with RA patients on placebo [70-73]. The largest study, including 184 RA patients, showed a decrease in pain among patients treated with dothiepin, but the change in pain scores was not statistically different from the change in pain scores among patients treated with placebo [74]. Studies examining the effects of TCAs on depression and pain showed that improvements in pain were independent of improvements in depression [70,73].
In clinical practice, the use of TCAs is often problematic because TCAs are associated with substantial adverse effects, and compliance with these medications is low. In addition to inhibiting serotonin and norepinephrine reuptake, TCAs also block cholinergic, histaminic and α-adrenergic receptors. As a result, many patients taking TCAs experience side effects such as sedation, dizziness, blurred vision, constipation and dry mouth. Dry mouth is particularly problematic in the RA population because many patients also have secondary Sjogren's syndrome, an inflammatory disorder characterized by decreased salivary gland function.
Serotonin norepinephrine reuptake inhibitors
Serotonin norepinephrine reuptake inhibitors (SNRIs) have similar noradrenergic/serotonergic reuptake ratios compared with TCAs. While TCAs have many effects other than inhibiting serotonin and norepinephrine reuptake, however, SNRIs are selective. A selective SNRI, such as duloxetine or milnacipran, could thus show greater overall benefit by enhancing the serotonergic and noradrenergic effects that lead to drug efficacy, while minimizing the dose-limiting effects of toxicity.
SNRIs modulate the descending serotonin-norepinephrine pathways involved in central pain-inhibiting mechanisms and are effective in the treatment of conditions characterized by defects in central pain processing (for example, fibromyalgia). In a group of 40 healthy individuals with low descending analgesic activity at baseline, treatment with duloxetine 60 mg daily resulted in an increase in descending analgesic activity from 0.15 to 19.35 within 1 week [75].
Two SNRIs, duloxetine and milnacipran, are approved by the Food and Drug Administration for the treatment of fibromyalgia. In three large, randomized placebo-controlled trials of fibromyalgia patients, duloxetine was associated with significant improvements in clinical pain [76-78]. Similar results have been reported in studies examining the effects of milnacipran on fibromyalgia pain [79-81]. The pain-relieving effects of these agents have been observed in depressed patients and in non-depressed patients [79].
Recent studies have expanded the potential use of SNRIs to other chronic painful conditions, including OA. In a 13-week, randomized, double-blind, placebo-controlled trial of 231 patients with knee OA, duloxetine 60 to 120 mg daily significantly reduced mean 24-hour pain scores [82]. Duloxetine was also associated with significant improvements in the Western Ontario and McMasters physical function scores. To date, no studies have examined the effect of SNRIs on pain in RA.
The α2δ ligands
The α2δ ligands, pregabalin and gabapentin, are anticonvulsants used to treat chronic pain conditions such as postherpetic neuralgia and diabetic neuropathy. Pregabalin and gabapentin bind to the α2δ subunit of calcium channels, inhibiting the release of neurotransmitters, including glutamate, noradrenaline, serotonin, and substance P. These compounds could thus work in individuals with central sensitization, as well as decreased descending analgesic response due to low serotonergic-noradrenergic activity.
Among fibromyalgia patients, pregabalin has consistently been associated with improvements in pain severity [83,84]. A Cochrane systematic review including 1,376 fibromyalgia patients treated with pregabalin 300 to 450 mg daily reported a relative benefit between 1.5 (95% confidence interval 1.2 to 1.9) and 1.7 (95% confidence interval 1.4 to 2.1) for a 50% decrease in pain [85]. The authors concluded that although some patients will experience moderate pain relief from pregabalin, few will experience a large effect [85]. No studies have examined the effect of pregabalin on pain in OA or RA patients, although a recent animal study suggested that pregabalin decreased pain sensitivity in a rat model of OA [86].
Conclusions
Central pain mechanisms play important roles in wide-spread pain syndromes, including fibromyalgia. The role of these mechanisms in rheumatologic diseases such as OA and RA is not well understood. A few small studies, utilizing quantitative sensory testing and fMRI, have documented loss of descending analgesic activity and alterations in CNS activity among OA patients, and a couple of small studies suggest a role for central sensitization in RA (Table 1). The data regarding loss of descending analgesic activity in RA, however, remain inconclusive.
Larger studies involving extensive pain phenotyping and comprehensive information about disease characteristics are necessary to better understand the impact of central pain mechanisms in OA and RA. Studies are also necessary to determine whether these patients, or a sub-group of these patients, may benefit from treatment with drugs such as SNRIs and α2δ ligands that target central pain mechanisms. If central pain mechanisms do play a significant role in pain processing among OA and RA patients, these medications may be attractive adjunctive treatments to manage pain in patients with rheumato-logic disease.
Abbreviations
CNS: central nervous system; fMRI: functional magnetic resonance imaging; NMDA: N-methyl-D-aspartate; OA: osteoarthritis; RA: rheumatoid arthritis; SI: primary somatosensory cortex; SII: secondary somatosensory cortex; SNRI: serotonin norepinephrine reuptake inhibitor; TCA: tricyclic antidepressant; TNF: tumor necrosis factor.
Competing interests
YCL receives grant support from Forest Laboratories and holds stocks in Merck and Company, Inc., Novartis, and Elan Corporation. DJC has acted as a consultant for Pfizer, Lilly, Forest Laboratories, Cypress Biosciences, Pierre Fabre, UCB, Johnson and Johnson, Nuvo, Merck and Company, Inc. and Wyeth. DJC has also received grant support from Pfizer, Cypress Bioscience, and Forest. NJN declares that he has no competing interests.
Note
This article is part of the series Evolving understanding of the biology of pain and its application to patient care, edited by Daniel Clauw and Anthony Jones. Other articles in this series can be found at http://arthritis-research.com/series/pain
Contributor Information
Yvonne C Lee, Email: ylee9@partners.org.
Nicholas J Nassikas, Email: nnassikas@partners.org.
Daniel J Clauw, Email: dclauw@med.umich.edu.
Acknowledgements
Grant support was received from NIH/NIAMS grant AR057578. The funding body played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
References
- Borenstein D, Altman R, Bello A, Chatham W, Clauw DJ, Crofford LJ, Croft J, Hassett A, Kozin F, Pisetsky D, Richardson J, Schanberg LE, Starz T, Witter J. Report of the American College of Rheumatology Pain Management Task Force. Arthritis Care Res. 2010;62:590–599. doi: 10.1002/acr.20005. [DOI] [PubMed] [Google Scholar]
- Buskila D. Developments in the scientific and clinical understanding of fibromyalgia. Arthritis Res Ther. 2009;11:242. doi: 10.1186/ar2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible HG, Ebersberger A, Von Banchet GS. Mechanisms of pain in arthritis. Ann N Y Acad Sci. 2002;966:343–354. doi: 10.1111/j.1749-6632.2002.tb04234.x. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Ebersberger A, Natura G. Update on peripheral mechanisms of pain: beyond prostaglandins and cytokines. Arthritis Res Ther. 2011;13:210. doi: 10.1186/ar3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/S0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Pud D, Granovsky Y, Yarnitsky D. The methodology of experimentally induced diffuse noxious inhibitory control (DNIC)-like effect in humans. Pain. 2009;144:16–19. doi: 10.1016/j.pain.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG. Descending control of persistent pain: inhibitory or facilitatory? Brain Res Brain Res Rev. 2004;46:295–309. doi: 10.1016/j.brainresrev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140:441–451. doi: 10.7326/0003-4819-140-8-200404200-00010. [DOI] [PubMed] [Google Scholar]
- Price DD, Staud R, Robinson ME, Mauderli AP, Cannon R, Vierck CJ. Enhanced temporal summation of second pain and its central modulation in fibromyalgia patients. Pain. 2002;99:49–59. doi: 10.1016/S0304-3959(02)00053-2. [DOI] [PubMed] [Google Scholar]
- Arendt-Nielsen L, Nielsen J, Petersen-Felix S, Schnider TW, Zbinden AM. Effect of racemic mixture and the (S+)-isomer of ketamine on temporal and spatial summation of pain. Br J Anaesth. 1996;77:625–631. doi: 10.1093/bja/77.5.625. [DOI] [PubMed] [Google Scholar]
- Graven-Nielsen T, Aspegren Kendall S, Henriksson KG, Bengtsson M, Sorensen J, Johnson A, Gerdle B, Arendt-Nielsen L. Ketamine reduces muscle pain, temporal summation, and referred pain in fibromyalgia patients. Pain. 2000;85:483–491. doi: 10.1016/S0304-3959(99)00308-5. [DOI] [PubMed] [Google Scholar]
- Staud R, Nagel S, Robinson ME, Price DD. Enhanced central pain processing of fibromyalgia patients is maintained by muscle afferent input: a randomized, double-blind, placebo-controlled study. Pain. 2009;145:96–104. doi: 10.1016/j.pain.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge HY, Nie H, Madeleine P, Danneskiold-Samsoe B, Graven-Nielsen T, Arendt-Nielsen L. Contribution of the local and referred pain from active myofascial trigger points in fibromyalgia syndrome. Pain. 2009;147:233–240. doi: 10.1016/j.pain.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Affaitati G, Costantini R, Fabrizio A, Lapenna D, Tafuri E, Giamberardino MA. Effects of treatment of peripheral pain generators in fibromyalgia patients. Eur J Pain. 2011;15:61–69. doi: 10.1016/j.ejpain.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Geisser ME, Casey KL, Brucksch CB, Ribbens CM, Appleton BB, Crofford LJ. Perception of noxious and innocuous heat stimulation among healthy women and women with fibromyalgia: association with mood, somatic focus, and catastrophizing. Pain. 2003;102:243–250. doi: 10.1016/S0304-3959(02)00417-7. [DOI] [PubMed] [Google Scholar]
- Gracely RH, Petzke F, Wolf JM, Clauw DJ. Functional magnetic resonance imaging evidence of augmented pain processing in fibromyalgia. Arthritis Rheum. 2002;46:1333–1343. doi: 10.1002/art.10225. [DOI] [PubMed] [Google Scholar]
- Giesecke T, Gracely RH, Grant MA, Nachemson A, Petzke F, Williams DA, Clauw DJ. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004;50:613–623. doi: 10.1002/art.20063. [DOI] [PubMed] [Google Scholar]
- Lautenbacher S, Rollman GB. Possible deficiencies of pain modulation in fibromyalgia. Clin J Pain. 1997;13:189–196. doi: 10.1097/00002508-199709000-00003. [DOI] [PubMed] [Google Scholar]
- Kosek E, Hansson P. Modulatory influence on somatosensory perception from vibration and heterotopic noxious conditioning stimulation (HNCS) in fibromyalgia patients and healthy subjects. Pain. 1997;70:41–51. doi: 10.1016/S0304-3959(96)03295-2. [DOI] [PubMed] [Google Scholar]
- Julien N, Goffaux P, Arsenault P, Marchand S. Widespread pain in fibromyalgia is related to a deficit of endogenous pain inhibition. Pain. 2005;114:295–302. doi: 10.1016/j.pain.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Russell IJ, Vaeroy H, Javors M, Nyberg F. Cerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritis. Arthritis Rheum. 1992;35:550–556. doi: 10.1002/art.1780350509. [DOI] [PubMed] [Google Scholar]
- Baraniuk JN, Whalen G, Cunningham J, Clauw DJ. Cerebrospinal fluid levels of opioid peptides in fibromyalgia and chronic low back pain. BMC Musculoskelet Disord. 2004;5:48–54. doi: 10.1186/1471-2474-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RE, Clauw DJ, Scott DJ, McLean SA, Gracely RH, Zubieta JK. Decreased central mu-opioid receptor availability in fibromyalgia. J Neurosci. 2007;27:10000–10006. doi: 10.1523/JNEUROSCI.2849-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staud R, Cannon RC, Mauderli AP, Robinson ME, Price DD, Vierck CJ Jr. Temporal summation of pain from mechanical stimulation of muscle tissue in normal controls and subjects with fibromyalgia syndrome. Pain. 2003;102:87–95. doi: 10.1016/s0304-3959(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Geisser ME, Glass JM, Rajcevska LD, Clauw DJ, Williams DA, Kileny PR, Gracely RH. A psychophysical study of auditory and pressure sensitivity in patients with fibromyalgia and healthy controls. J Pain. 2008;9:417–422. doi: 10.1016/j.jpain.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Geisser ME, Strader Donnell C, Petzke F, Gracely RH, Clauw DJ, Williams DA. Comorbid somatic symptoms and functional status in patients with fibromyalgia and chronic fatigue syndrome: sensory amplification as a common mechanism. Psychosomatics. 2008;49:235–242. doi: 10.1176/appi.psy.49.3.235. [DOI] [PubMed] [Google Scholar]
- Singer T, Seymour B, O'Doherty J, Kaube H, Dolan RJ, Frith CD. Empathy for pain involves the affective but not sensory components of pain. Science. 2004;303:1157–1162. doi: 10.1126/science.1093535. [DOI] [PubMed] [Google Scholar]
- Harris RE, Sundgren PC, Craig AD, Kirshenbaum E, Sen A, Napadow V, Clauw DJ. Elevated insular glutamate in fibromyalgia is associated with experimental pain. Arthritis Rheum. 2009;60:3146–3152. doi: 10.1002/art.24849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RE, Sundgren PC, Pang Y, Hsu M, Petrou M, Kim SH, McLean SA, Gracely RH, Clauw DJ. Dynamic levels of glutamate within the insula are associated with improvements in multiple pain domains in fibromyalgia. Arthritis Rheum. 2008;58:903–907. doi: 10.1002/art.23223. [DOI] [PubMed] [Google Scholar]
- Vargas-Alarcon G, Fragoso JM, Cruz-Robles D, Vargas A, Vargas A, Lao-Villadoniga JI, Garcia-Fructuoso F, Ramos-Kuri M, Hernandez F, Springall R, Bojalil R, Vallejo M, Martinez-Lavin M. Catechol-O-methyltransferase gene haplotypes in Mexican and Spanish patients with fibromyalgia. Arthritis Res Ther. 2007;9:R110. doi: 10.1186/ar2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Neumann L, Glazer Y, Ebstein RP, Buskila D. The relationship between a common catechol-O-methyltransferase (COMT) polymorphism val(158) met and fibromyalgia. Clin Exp Rheumatol. 2009;27:S51–S56. [PubMed] [Google Scholar]
- Bondy B, Spaeth M, Offenbaecher M, Glatzeder K, Stratz T, Schwarz M, de Jonge S, Kruger M, Engel RR, Farber L, Pongratz DE, Ackenheil M. The T102C polymorphism of the 5-HT2A-receptor gene in fibromyalgia. Neurobiol Dis. 1999;6:433–439. doi: 10.1006/nbdi.1999.0262. [DOI] [PubMed] [Google Scholar]
- Offenbaecher M, Bondy B, de Jonge S, Glatzeder K, Kruger M, Schoeps P, Ackenheil M. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis Rheum. 1999;42:2482–2488. doi: 10.1002/1529-0131(199911)42:11<2482::AID-ANR27>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Buskila D, Sarzi-Puttini P. Biology and therapy of fibromyalgia. Genetic aspects of fibromyalgia syndrome. Arthritis Res Ther. 2006;8:218–222. doi: 10.1186/ar2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tander B, Gunes S, Boke O, Alayli G, Kara N, Bagci H, Canturk F. Polymorphisms of the serotonin-2A receptor and catechol-O-methyltransferase genes: a study on fibromyalgia susceptibility. Rheumatol Int. 2008;28:685–691. doi: 10.1007/s00296-008-0525-8. [DOI] [PubMed] [Google Scholar]
- Potvin S, Larouche A, Normand E, Souza JB, Gaumond I, Marchand S, Grignon S. No relationship between the ins del polymorphism of the serotonin transporter promoter and pain perception in fibromyalgia patients and healthy controls. Eur J Pain. 2010;14:742–746. doi: 10.1016/j.ejpain.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Gursoy S. Absence of association of the serotonin transporter gene polymorphism with the mentally healthy subset of fibromyalgia patients. Clin Rheumatol. 2002;21:194–197. doi: 10.1007/s10067-002-8284-5. [DOI] [PubMed] [Google Scholar]
- Cook DB, Lange G, Ciccone DS, Liu WC, Steffener J, Natelson BH. Functional imaging of pain in patients with primary fibromyalgia. J Rheumatol. 2004;31:364–378. [PubMed] [Google Scholar]
- Jensen KB, Kosek E, Petzke F, Carville S, Fransson P, Marcus H, Williams SC, Choy E, Giesecke T, Mainguy Y, Gracely R, Ingvar M. Evidence of dysfunctional pain inhibition in fibromyalgia reflected in rACC during provoked pain. Pain. 2009;144:95–100. doi: 10.1016/j.pain.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Napadow V, LaCount L, Park K, As-Sanie S, Clauw DJ, Harris RE. Intrinsic brain connectivity in fibromyalgia is associated with chronic pain intensity. Arthritis Rheum. 2010;62:2545–2555. doi: 10.1002/art.27497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, Jordan JM, Katz JN, Kremers HM, Wolfe F. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedson J, Croft PR. The discordance between clinical and radiographic knee osteoarthritis: a systematic search and summary of the literature. BMC Musculoskelet Disord. 2008;9:116–126. doi: 10.1186/1471-2474-9-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neogi T, Felson D, Niu J, Nevitt M, Lewis CE, Aliabadi P, Sack B, Torner J, Bradley L, Zhang Y. Association between radiographic features of knee osteoarthritis and pain: results from two cohort studies. BMJ. 2009;339:b2844–b2850. doi: 10.1136/bmj.b2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel J. The reliability and validity of pain threshold measurements in osteoarthritis of the knee. Scand J Rheumatol. 1995;24:238–242. doi: 10.3109/03009749509100881. [DOI] [PubMed] [Google Scholar]
- Farrell M, Gibson S, McMeeken J, Helme R. Pain and hyperalgesia in osteoarthritis of the hands. J Rheumatol. 2000;27:441–447. [PubMed] [Google Scholar]
- Kosek E, Ordeberg G. Lack of pressure pain modulation by heterotopic noxious conditioning stimulation in patients with painful osteoarthritis before, but not following, surgical pain relief. Pain. 2000;88:69–78. doi: 10.1016/S0304-3959(00)00310-9. [DOI] [PubMed] [Google Scholar]
- Gwilym SE, Keltner JR, Warnaby CE, Carr AJ, Chizh B, Chessell I, Tracey I. Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis Rheum. 2009;61:1226–1234. doi: 10.1002/art.24837. [DOI] [PubMed] [Google Scholar]
- Arendt-Nielsen L, Nie H, Laursen MB, Laursen BS, Madeleine P, Simonsen OH, Graven-Nielsen T. Sensitization in patients with painful knee osteoarthritis. Pain. 2010;149:573–581. doi: 10.1016/j.pain.2010.04.003. [DOI] [PubMed] [Google Scholar]
- O'Driscoll SL, Jayson MI. Pain threshold analysis in patients with osteoarthrosis of hip. Br Med J. 1974;3:714–715. doi: 10.1136/bmj.3.5933.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj P, Bajaj P, Graven-Nielsen T, Arendt-Nielsen L. Osteoarthritis and its association with muscle hyperalgesia: an experimental controlled study. Pain. 2001;93:107–114. doi: 10.1016/S0304-3959(01)00300-1. [DOI] [PubMed] [Google Scholar]
- Imamura M, Imamura ST, Kaziyama HH, Targino RA, Hsing WT, de Souza LP, Cutait MM, Fregni F, Camanho GL. Impact of nervous system hyperalgesia on pain, disability, and quality of life in patients with knee osteoarthritis: a controlled analysis. Arthritis Rheum. 2008;59:1424–1431. doi: 10.1002/art.24120. [DOI] [PubMed] [Google Scholar]
- Kulkarni B, Bentley DE, Elliott R, Julyan PJ, Boger E, Watson A, Boyle Y, El-Deredy W, Jones AK. Arthritic pain is processed in brain areas concerned with emotions and fear. Arthritis Rheum. 2007;56:1345–1354. doi: 10.1002/art.22460. [DOI] [PubMed] [Google Scholar]
- Wolfe F, Michaud K. Assessment of pain in rheumatoid arthritis: minimal clinically significant difference, predictors, and the effect of anti-tumor necrosis factor therapy. J Rheumatol. 2007;34:1674–1683. [PubMed] [Google Scholar]
- Rupp I, Boshuizen HC, Roorda LD, Dinant HJ, Jacobi CE, van den Bos G. Course of patient-reported health outcomes in rheumatoid arthritis: comparison of longitudinal and cross-sectional approaches. J Rheumatol. 2006;33:228–233. [PubMed] [Google Scholar]
- Huskisson EC, Hart FD. Pain threshold and arthritis. Br Med J. 1972;4:193–195. doi: 10.1136/bmj.4.5834.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerecz-Simon EM, Tunks ER, Heale JA, Kean WF, Buchanan WW. Measurement of pain threshold in patients with rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, and healthy controls. Clin Rheumatol. 1989;8:467–474. doi: 10.1007/BF02032098. [DOI] [PubMed] [Google Scholar]
- Konttinen YT, Honkanen VE, Gronblad M, Keinonen M, Santavirta N, Santavirta S. The relation of extraarticular tenderness to inflammatory joint disease and personality in patients with rheumatoid arthritis. J Rheumatol. 1992;19:851–855. [PubMed] [Google Scholar]
- Leffler AS, Kosek E, Lerndal T, Nordmark B, Hansson P. Somatosensory perception and function of diffuse noxious inhibitory controls (DNIC) in patients suffering from rheumatoid arthritis. Eur J Pain. 2002;6:161–176. doi: 10.1053/eujp.2001.0313. [DOI] [PubMed] [Google Scholar]
- Wendler J, Hummel T, Reissinger M, Manger B, Pauli E, Kalden JR, Kobal G. Patients with rheumatoid arthritis adapt differently to repetitive painful stimuli compared to healthy controls. J Clin Neurosci. 2001;8:272–277. doi: 10.1054/jocn.1999.0775. [DOI] [PubMed] [Google Scholar]
- Morris VH, Cruwys SC, Kidd BL. Characterisation of capsaicin-induced mechanical hyperalgesia as a marker for altered nociceptive processing in patients with rheumatoid arthritis. Pain. 1997;71:179–186. doi: 10.1016/S0304-3959(97)03361-7. [DOI] [PubMed] [Google Scholar]
- Jones AK, Derbyshire SW. Reduced cortical responses to noxious heat in patients with rheumatoid arthritis. Ann Rheum Dis. 1997;56:601–607. doi: 10.1136/ard.56.10.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwienhardt P, Kalk N, Wartolowska K, Chessel I, Wordsworth P, Tracey I. Investigation into the neural correlates of emotional augmentation of clinical pain. Neuroimage. 2008;40:759–766. doi: 10.1016/j.neuroimage.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Jones AK, Cunningham VJ, Ha-Kawa S, Fujiwara T, Luthra SK, Silva S, Derbyshire S, Jones T. Changes in central opioid receptor binding in relation to inflammation and pain in patients with rheumatoid arthritis. Br J Rheumatol. 1994;33:909–916. doi: 10.1093/rheumatology/33.10.909. [DOI] [PubMed] [Google Scholar]
- Lee YC, Chibnik LB, Lu B, Wasan AD, Edwards RR, Fossel AH, Helfgott SM, Solomon DH, Clauw DJ, Karlson EW. The relationship between disease activity, sleep, psychiatric distress and pain sensitivity in rheumatoid arthritis: a cross-sectional study. Arthritis Res Ther. 2009;11:R160. doi: 10.1186/ar2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Edwards RR, McCann UD, Haythornthwaite JA. The effects of sleep deprivation on pain inhibition and spontaneous pain in women. Sleep. 2007;30:494–505. doi: 10.1093/sleep/30.4.494. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Grace E, Peterson S, Klick B, Haythornthwaite JA, Smith MT. Sleep continuity and architecture: associations with pain-inhibitory processes in patients with temporomandibular joint disorder. Eur J Pain. 2009;13:1043–1047. doi: 10.1016/j.ejpain.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishishinya B, Urrutia G, Walitt B, Rodriguez A, Bonfill X, Alegre C, Darko G. Amitriptyline in the treatment of fibromyalgia: a systematic review of its efficacy. Rheumatology (Oxford) 2008;47:1741–1746. doi: 10.1093/rheumatology/ken317. [DOI] [PubMed] [Google Scholar]
- Gringras M. A clinical trial of Tofranil in rheumatic pain in general practice. J Int Med Res. 1976;4:41–49. [PubMed] [Google Scholar]
- Macfarlane JG, Jalali S, Grace EM. Trimipramine in rheumatoid arthritis: a randomized double-blind trial in relieving pain and joint tenderness. Curr Med Res Opin. 1986;10:89–93. doi: 10.1185/03007998609110424. [DOI] [PubMed] [Google Scholar]
- Sarzi Puttini P, Cazzola M, Boccassini L, Ciniselli G, Santandrea S, Caruso I, Benvenuti C. A comparison of dothiepin versus placebo in the treatment of pain in rheumatoid arthritis and the association of pain with depression. J Int Med Res. 1988;16:331–337. doi: 10.1177/030006058801600502. [DOI] [PubMed] [Google Scholar]
- Frank RG, Kashani JH, Parker JC, Beck NC, Brownlee-Duffeck M, Elliott TR, Haut AE, Atwood C, Smith E, Kay DR. Antidepressant analgesia in rheumatoid arthritis. J Rheumatol. 1988;15:1632–1638. [PubMed] [Google Scholar]
- Ash G, Dickens CM, Creed FH, Jayson MI, Tomenson B. The effects of dothiepin on subjects with rheumatoid arthritis and depression. Rheumatology (Oxford) 1999;38:959–967. doi: 10.1093/rheumatology/38.10.959. [DOI] [PubMed] [Google Scholar]
- Chuck AJ, Swannell AJ, House AO, Pownall R. The effects of dothiepin on subjects with rheumatoid arthritis and depression. Rheumatology (Oxford) 2000;39:1425–1427. doi: 10.1093/rheumatology/39.12.1425. [DOI] [PubMed] [Google Scholar]
- Yarnitsky D, Granovsky Y, Fadel S, Sprecher E, Granot M. Proceedings of the 12th World Congress on Pain: 2008; Glasgow, Scotland. Seattle, WA: IASP Press; 2008. Endogenous analgesia and SSNRI towards individually tailored pain therapy. [Google Scholar]
- Arnold LM, Lu Y, Crofford LJ, Wohlreich M, Detke MJ, Iyengar S, Goldstein DJ. A double-blind, multicenter trial comparing duloxetine with placebo in the treatment of fibromyalgia patients with or without major depressive disorder. Arthritis Rheum. 2004;50:2974–2984. doi: 10.1002/art.20485. [DOI] [PubMed] [Google Scholar]
- Arnold LM, Rosen A, Pritchett YL, D'Souza DN, Goldstein DJ, Iyengar S, Wernicke JF. A randomized, double-blind, placebo-controlled trial of duloxetine in the treatment of women with fibromyalgia with or without major depressive disorder. Pain. 2005;119:5–15. doi: 10.1016/j.pain.2005.06.031. [DOI] [PubMed] [Google Scholar]
- Russell IJ, Mease PJ, Smith TR, Kajdasz DK, Wohlreich MM, Detke MJ, Walker DJ, Chappell AS, Arnold LM. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: results from a 6-month, randomized, double-blind, placebo-controlled, fixeddose trial. Pain. 2008;136:432–444. doi: 10.1016/j.pain.2008.02.024. [DOI] [PubMed] [Google Scholar]
- Gendreau RM, Thorn MD, Gendreau JF, Kranzler JD, Ribeiro S, Gracely RH, Williams DA, Mease PJ, McLean SA, Clauw DJ. Efficacy of milnacipran in patients with fibromyalgia. J Rheumatol. 2005;32:1975–1985. [PubMed] [Google Scholar]
- Mease PJ, Clauw DJ, Gendreau RM, Rao SG, Kranzier J, Chen W, Palmer RH. The efficacy and safety of milnacipran for treatment of fibromyalgia: a randomized, double-blind, placebo-controlled trial. J Rheumatol. 2009;36:398–409. doi: 10.3899/jrheum.080734. [DOI] [PubMed] [Google Scholar]
- Clauw DJ, Mease P, Palmer RH, Gendreau RM, Wang Y. Milnacipran for the treatment of fibromyalgia in adults: a 15-week, multicenter, randomized, double-blind, placebo-controlled, multiple-dose clinical trial. Clin Ther. 2008;30:1988–2004. doi: 10.1016/j.clinthera.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Chappell AS, Ossanna MJ, Liu-Seifert H, Iyengar S, Skljarevski V, Li LC, Bennett RM, Collins H. Duloxetine, a centrally acting analgesic, in the treatment of patients with osteoarthritis knee pain: a 13-week, randomized, placebocontrolled trial. Pain. 2009;146:253–260. doi: 10.1016/j.pain.2009.06.024. [DOI] [PubMed] [Google Scholar]
- Crofford LJ, Rowbotham MC, Mease PJ, Russell IJ, Dworkin RH, Corbin AE, Young JP, LaMoreaux LK, Martin SA, Sharma U. Pregabalin for the treatment of fibromyalgia syndrome: results of a randomized, doubleblind, placebo-controlled trial. Arthritis Rheum. 2005;52:1264–1273. doi: 10.1002/art.20983. [DOI] [PubMed] [Google Scholar]
- Crofford LJ, Mease PJ, Simpson SL, Young JP, Martin SA, Haig GM, Sharma U. Fibromyalgia relapse evaluation and efficacy for durability of meaningful relief (FREEDOM): a 6-month, double-blind, placebo-controlled trial with pregabalin. Pain. 2008;136:419–431. doi: 10.1016/j.pain.2008.02.027. [DOI] [PubMed] [Google Scholar]
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst Rev. 2009;3:CD007076. doi: 10.1002/14651858.CD007076.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman W, Bauer CS, Bannister K, Vonsy JL, Dolphin AC, Dickenson AH. Descending serotonergic facilitation and the antinociceptive effects of pregabalin in a rat model of osteoarthritic pain. Mol Pain. 2009;5:45–61. doi: 10.1186/1744-8069-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]