

Summary
Apolipoprotein E (ApoE) is a 299 amino acid protein encoded by the APOE gene. Three common polymorphisms in the APOE gene, ε2, ε3, and ε4, result in single amino changes in the ApoE protein. The APOEε2, ε3, and ε4 alleles strongly and dose-dependently alter the likelihood of developing Alzheimer’s disease (AD) and cerebral amyloid angiopathy (CAA). In particular, APOE ε4 is associated with increased risk for AD, whereas APOEε2 is associated with decreased risk. The effects of APOE genotype on AD and CAA risk are likely mediated, in large part, by differential effects of the ApoE protein on amyloid-β (Aβ) accumulation in the brain and cerebrovasculature. Recent data indicate that responses to AD treatments may differ according to APOE genotype. The APOE ε4 allele is also associated with poor outcome following traumatic brain injury and brain hemorrhage, though the mechanisms underlying these associations are unclear. Given the convincing body of literature tying APOE genotype to AD and CAA risk, APOE has also been studied in relation to other neurological diseases. While the possibility that APOE plays a role in these diseases is of great interest, convincing associations have not yet emerged.
Introduction
With few efficacious treatment options available, the management of many neurological disorders presents clinicians with significant challenges, while healthcare systems are burdened with enormous strain. Understanding environmental and/or genetic components that modulate neurological disease risk and outcome may provide useful information for managing these devastating diseases. Following a series of landmark studies identifying the strong association of the APOE ε4 allele with increased AD risk and decreased age of onset and the protective role of the APOE ε2 allele,1–5 numerous studies have investigated putative associations of APOE genotype with risk or progression for a wide variety of neurological disorders. Given the various proposed roles of apoE in influencing Aβ metabolism, CNS lipid homeostasis, synaptic activity, response to cellular injury, and neuroinflammation, these investigations were hypothesized to reveal strong associations with several diseases. To date, however, AD and CAA are the only neurological diseases for which the level of evidence for an association between APOE genotype and disease risk and age of onset is compelling. APOE is believed to be linked to AD and CAA risk through isoform-dependent modulation of Aβ accumulation. Associations of APOE with outcome following traumatic brain injury (TBI) and risk of Down’s syndrome-associated dementia (DAD) are also hypothesized to be, in part, mediated through isoform-dependent modulation of Aβ accumulation. To date, roles for APOE genotype in stroke, vascular dementia (VaD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and others have been suggested, but strong consensus has been lacking. This Review evaluates the level of evidence for associations of APOE genotype with risk and outcome for various neurological diseases with particular attention to AD and CAA, for which evidence of an association is strong (Table 1). The many proposed roles of apoE in the nervous system present a unique challenge to understanding its roles in neurological disease pathogenesis as well as in treatment strategies. Differences in treatment response according to APOE status in recent AD clinical trials will also be evaluated.
Table 1.
Levels of evidence for an association of APOE with occurrence and progression for neurological disorders. Levels of evidence are adapted from the Categories of Association established and used by the Institute of Medicine for association between a factor and a specific health outcome (Committee on Health Effects Associated with Exposures During the Gulf War. Institute of Medicine, 2000) [also see Tarawneh et al., 2010].172
| Disease | Disease Occurrence and Levels of Evidence | Disease Progression and Levels of Evidence | Possible Mechanisms of ApoE In Disease | References |
|---|---|---|---|---|
| AD | ε4>ε3>ε2 Sufficient evidence of a direct relationship (A) |
Inadequate/insufficient evidence to determine whether an association exists (C) |
|
1–5,13–24,35–40, 41, 45, 47–57,160–162, 164–170 |
| CAA | ε4>ε3 Sufficient evidence of a direct relationship (A) ε2 and ε4 risk for hemorrnage Suggestive evidence of an association (B) |
ε4>ε3 sufficient evidence of a direct relationship (A) |
Aβ metabolism | 25–34, 38–40, 43, 46, 160–162, 164–170 |
| TBI | Not Applicable | ε4>ε3 Sufficient evidence of a direct relationship (A) |
Aβ and Tau accumulation | 82–101,160–162, 164–170 |
| DAD | ε4> Non-carriers Suggestive evidence of an association (B) |
ε4> Non-carriers Suggestive evidence of an association (B) |
Aβ metabolism | 102–105, 160–162, 164–170 |
| Stroke (IS, SAH, ICH) | Inadequate/insufficient evidence to determine whether an association exists (C) | IS: Inadequate/insufficient evidence to determine whether an association exists (C) SAH: ε4 > Non-carriers ICH: ε >Non-carriers suggestive evidence of an association (B) |
Unclear | 106–111, 160–170 |
| VaD | ε4> Non-carriers Suggestive evidence of an association (B) |
Inadequate/sufficient evidence to determine whether an association exists (C) | Unclear | 112–120, 160–162, 164–170 |
| CJD | Suggestive evidence of no association (D) | Suggestive evidence of no association (D) | Not applicable | 2, 121–124, 160–162, 164–170 |
| MS | Suggestive evidence of no association (D) | ε4> Non-carriers Suggestive evidence of an association (B) |
Unclear | 125–128, 160–162, 164–170 |
| ALS | Suggestive evidence of no association (D) | ε4>ε2 Suggestive evidence of an association (B) |
Unclear | 129–132, 160–162, 162–170 |
| IBM | Inadequate/insufficient evidence to determine whether an association exists (C) | Inadequate/insufficient evidence to determine whether an association exists (C) | Not applicable | 133–139, 160–164–170 |
| PD | ε2> Non-carriers Suggestive evidence of an association (B) |
ε2> Non-carriers Suggestive evidence of an association (B) |
Unclear | 140–145, 160–162, 164–170 |
| DLB | ε4> Non-carriers Suggestive evidence of an association (B) |
Inadequate/insufficient evidence to determine whether an association exists (C) | Aβ metabolism | 146–152, 160–162, 164–170 |
| CP, HD, FTD, TL-E | Inadequate/insufficient evidence to determine whether an association exists (C) | Inadequate/insufficient evidence to determine whether an association exists (C) | None proposed | 153–159, 162–164, 164–170 |
Sufficient evidence of a direct relationship: Evidence fulfills the guidelines for sufficient evidence of an association, is supported by experimental data in humans and animals, and satisfies several of the guidelines used to assess causality: strength of association, dose response relationship, and consistency of association.
Suggestive evidence of an association: Evidence is suggestive of an association between APOE and the neurological disorder in humans, but the body of evidence is limited by the inability to exclude chance and bias, and confounding factors with confidence.
Inadequate/insufficient evidence to determine whether an association exists: Evidence is of insufficient quantity, quality, or consistency to permit a conclusion regarding the existence of an association between APOE and the neurological disorder in humans.
Suggestive evidence of no association. There are several adequate studies that are consistent in not showing a positive association between APOE and the neurological disorder in humans.
Table Abbreviations: AD: Alzheimer’s disease; CAA: cerebral amyloid angiopathy; TBI: traumatic brain injury; IS: ischemic stroke, ICH: intracerebral hemorrhage; SAH: subarachnoid hemorrhage; DAD: Down’s syndrome-associated dementia; CJD: Creutzfeldt-Jakob disease; MS: multiple sclerosis; ALS: amyotrophic lateral sclerosis; IBM: Inclusion-body myositis; PD: Parkinson’s disease; VaD: Vascular dementia; DLB: Dementia with Lewy bodies; CP: cerebral palsy; HD: Huntington’s disease; TL-E: temporal lobe-epilepsy; FTD: Frontotemporal dementia
Physiological function of apoE
Human apolipoprotein E (apoE) is a lipoprotein of 299 amino acids expressed in multiple organs with the highest expression in liver followed by the brain.6 ApoE exists mainly as a component of lipoprotein complexes along with other apolipoproteins and proteins in plasma and cerebrospinal fluid (CSF).6 In humans, there are three polymorphic forms of apoE: apoE2 (Cys-112, Cys-158), apoE3 (Cys-112, Arg-158), and apoE4 (Arg-112, Arg-158).7 The amino acid differences at these positions are suggested to be critical as they alter the charge and structural properties of the protein, ultimately influencing the functional properties of apoE isoforms.6
ApoE is one of the key lipoproteins of lipoprotein complexes that regulate the metabolism of lipids by directing their transport, delivery, and distribution from one tissue or cell type to another through apoE receptors and proteins associated with lipid transfer and lipolysis.6, 8 The apoE isoform-specific associations with lipoprotein complexes in the plasma and uptake of apoE lipoprotein complexes by LDL receptors have significant effects on peripheral lipid metabolism, having important implications in diseases like Type III Hyperlipoproteinemia (HLP) and atherosclerosis. In brain, apoE is mostly produced by astrocytes, followed by microglial cells and, under certain conditions, by neurons.9 In the CSF, apoE is associated predominantly with cholesterol and phospholipid-rich, high-density lipoprotein (HDL)-like complexes. Unlike plasma, CSF exclusively contains HDL-like lipoproteins and no LDL or VLDL. ApoE’s association with HDL-like particles in the CSF occurs without any known isoform specificity.10–12 Unlike in the periphery, where apoE isoforms differentially alter lipoprotein metabolism, there is no convincing evidence that apoE isoforms differentially influence general measures of CNS cholesterol or phospholipid metabolism, suggesting that structural differences in apoE isoforms may influence neurological disorders via mechanisms not directly linked to isoform-specific effects on lipid metabolism.
APOE and Alzheimer’s disease (AD)
AD is a devastating neurodegenerative disease affecting approximately 26 million people worldwide. AD is pathologically defined by the extracellular accumulation of Aβ, intracellular accumulation of tau, neuronal and synaptic loss, brain atrophy, and inflammation.13 Mutations in three genes, APP, PS1, and PS2 cause rare forms of autosomal-dominant familial AD with clinical onset typically between the ages of 30–60.13 Several susceptibility genes have also been implicated in influencing AD risk, one of which, APOE, has been confirmed to confer risk for sporadic, late-onset AD (age > 60), as well as autosomal-dominant familial AD.1, 14 The APOE ε3 allele is the most frequent in all populations, with a frequency range of 50–90%, whereas APOE ε4 and APOE ε2 allele frequencies range from 5–35% and 1–5%, respectively.6 Risk for AD is associated with APOE allele (ε4 >ε3>ε2), with the APOE ε4 allele present in ~50% of patients who develop late-onset AD compared to 20–25% in controls.1–3 Having one or two copies of the APOE ε4 allele increases late-onset AD risk approximately 3- or 12-fold, respectively. Moreover, having one or two copies of APOE ε4 shifts the age of onset earlier by approximately one to two decades relative to non-carriers in late-onset AD.1 APOE ε4 also results in earlier onset of dementia in individuals with a PS1 mutation in a large Colombian kindred 14. APOE ε2 individuals have reduced risk for developing late-onset AD.1–3, 14–15 Multiple epidemiological studies from various populations have confirmed the increased frequency of the APOE ε4 allele in late-onset AD patients compared to non-carriers, though the frequency varies in different ethnicities.4 It is important to note that APOE ε4 is neither necessary nor sufficient for the development of AD so that apoE polymorphism cannot be utilized alone for the diagnosis of AD.16–17
There is controversy as to whether APOE polymorphism associates with the rate of progression of cognitive decline in AD after its onset.3, 18 In particular, there is discrepancy as to the role of APOE ε4 in the rate of cognitive and functional decline after dementia onset in AD. Several reports suggest that homozygous APOE ε4 patients experience more rapid cognitive and functional decline following clinical disease onset,3 suggesting that the factors determining disease onset may also have a role in the rate of progression and clinical outcome. Others have also reported that disease onset and rate of progression as factors are different in the context of APOE ε4.3, 19–20 An MRI study from a large, cognitively normal population suggested that APOE ε4-carriers have decreased entorhinal cortex volume in children and adolescents, suggesting a potential developmental effect.21 Longitudinal MRI studies of subjects already diagnosed with AD revealed that the rate of volume decrease of entorhinal cortex and hippocampus is greater in those who are APOE ε4-positive.22 Conversely, a recent study reported that cognitively normal APOE ε2-carriers had slower hippocampal atrophy measured over two years compared to non-carriers.23 A recent large study of cognitively normal individuals found that age-related memory decline starting in the late fifties was greater in APOE ε4-carriers vs. non-carriers,24 suggesting that the consequences of AD pathology may manifest in the brain as early as the sixth decade of life. The role of APOE in the predisposition of AD is well established, though further studies are needed to understand the possible association of APOE with rate of AD progression.
APOE and cerebral amyloid angiopathy (CAA)
CAA co-occurs in patients with late-onset AD, though a substantial number of individuals develop symptomatic CAA in the absence of dementia. CAA is pathologically characterized by the accumulation of Aβ, mostly Aβ40, in the adventitia and media of penetrating arterioles and in arterioles of the leptomeninges in the CNS.25–26 CAA is a frequent cause of lobar hemorrhage in individuals over the age of 60. The fact that Aβ deposits are present in brain parenchyma in AD and in vessels in CAA has motivated several investigations to understand the role of apoE isoforms in CAA. APOE ε4 is associated with sporadic CAA risk and risk for brain hemorrhage, the severity of which is APOE ε4 dose-dependent.25, 27–31 Interestingly, while APOE ε4 is associated with an increase in ICH, the APOE ε2 allele appears to predispose individuals to ICH if CAA is present,32 possibly due to the CAA-related vasculopathic abnormalities associated with this allele.25 Though the association of APOE ε4 with CAA is well established in several studies, 25, 27–31 there are a few conflicting reports.32–34 Possible reasons for discrepancies include diversity in genetic background of the populations studied, difference in the proportion of AD and non-AD cases with CAA, and the high mortality rate associated with APOE ε4 after CAA-related ICH in post-mortem studies.
Aβ-dependent roles for apoE in AD and CAA
A variety of in vitro and animal studies have demonstrated that apoE plays an important role in determining whether and when Aβ converts from a monomeric, non-toxic molecule into higher molecular weight forms, such as oligomers and fibrils, of which certain forms likely mediate toxicity.35–37 For example, in mice engineered to develop Aβ plaque and CAA pathology, apoE is required for the formation of these pathologies in addition to Aβ-associated toxicity, e.g., dystrophic neurite formation and CAA-associated hemorrhages.38–40 Furthermore, expressing human apoE in these models results in apoE isoform-dependent differences in Aβ accumulation (E4>E3>E2).41–44 Whether Aβ toxicity in the brain is due to small Aβ oligomers or larger aggregates such as fibrils, or both, is unclear, though there is evidence that apoE isoforms can influence both Aβ fibril formation and the toxicity of Aβ oligomers.45 Though it is likely that CAA and AD share common mechanistic pathways with regard to apoE and Aβ, specific mechanisms have also been suggested in animal studies. For example, in mice, APOE ε4 was found to promote the development of CAA probably via increasing the ratio of Aβ40 to Aβ42 in the brain, which results in a shift of Aβ accumulation from the brain parenchyma to vessels43. Further, it has been suggested that apoE may influence Aβ drainage through perivascular channels.46
In vitro, animal, and human studies suggest that the accumulation of Aβ in the brain is a driving force for AD pathogenesis.13 While the mechanism by which apoE isoforms affect AD risk is not entirely understood, there is strong evidence that apoE isoforms differentially modulate Aβ metabolism and accumulation. In postmortem tissue from AD patients, apoE is present in Aβ plaques, a major hallmark of AD pathology.47 Several studies have observed an increase in senile and neuritic plaques in APOE ε4 homozygous AD patients compared to APOE ε4/ε3 or APOE ε3 homozygous AD patients and increased plaques in APOE ε4-positive vs. ε4-negative patients.48–52 However, one group found no significant effect on plaque density or number.9 In a large cohort study in autopsy-confirmed AD patients, the presence of both APOE ε4 alleles was found to be a crucial factor in increasing neuritic plaque accumulation in all neocortical areas of brain. APOE ε2 AD patients had reduced plaque accumulation, though the sample size was very small.17 Perhaps most relevant in understanding how APOE genotype influences AD risk is whether it influences AD pathology in relation to the time course of disease onset. Converging evidence suggests that the initial pathological feature of AD is Aβ deposition in the brain, which is estimated to begin 10–15 years prior to the onset of any clinical signs and symptoms of cognitive decline.53 Various events appear downstream of Aβ deposition in the AD pathological process, including neurofibrillary tangle formation, neuroinflammation, and neuronal/synaptic loss. The period of AD pathological changes in the absence of clinically detectable disease has been termed “preclinical” or “presymptomatic” AD. If APOE genotype is linked to AD risk by influencing the probability of onset of Aβ accumulation, it would be expected that cognitively normal individuals at a given age would have greater brain Aβ burden in the order, ε4 >ε3>ε2. In fact, in both CSF biomarker and amyloid imaging studies, isoform-dependent brain Aβ pathology (ε4 >ε3>ε2) has been reported in cognitively normal individuals aged 45–90.23, 54–56 These data suggest that APOE genotype modulates AD risk by affecting the likelihood that Aβ begins to deposit, such that the timing of Aβ accumulation is shifted earlier or later in the preclinical phase depending on APOE genotype (Figure 1). Given the clear effect of APOE in modulating AD risk and Aβ pathology, a major hypothesis for which there is accumulating evidence is that apoE4 increases Aβ aggregation and/or impairs Aβ clearance relative to other apoE isoforms.
Figure 1.
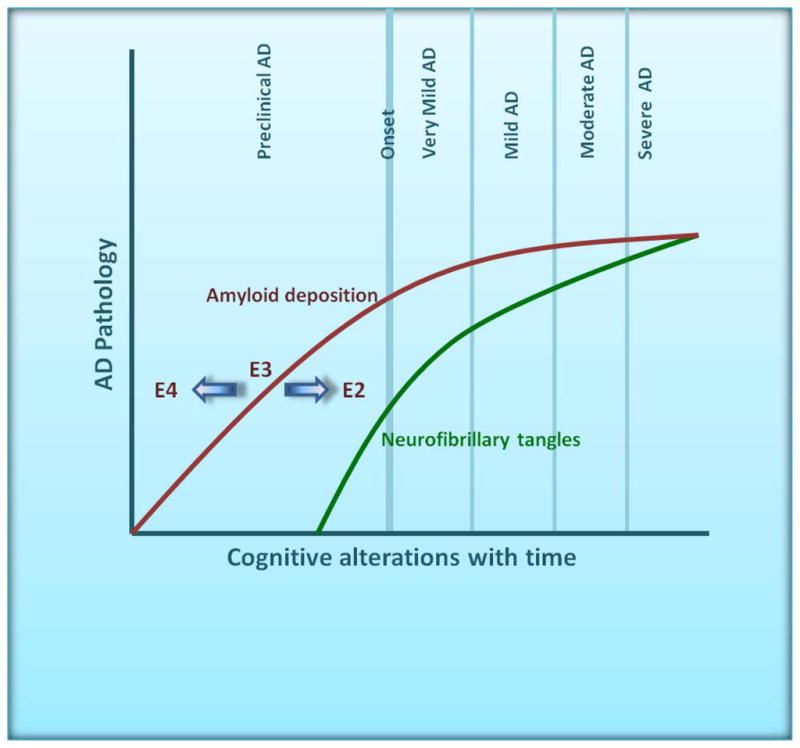
A large body of evidence suggests that the dominant effect of human apoE isoforms on AD risk is to shift the onset of disease via alterations in the probability of amyloid deposition, likely during the preclinical, asymptomatic phase. Consistent with this hypothesis, individuals appear to accumulate amyloid in the order ε4 > ε3 > ε2, well before AD clinical symptomology becomes manifest (Figure adapted from Holtzman, DM 2008 Nature, 454(7203):418–20)171.
Aβ-independent roles for apoE in AD
While the differential effects of apoE isoforms on lipid metabolism are well-characterized in the periphery, it remains unclear whether apoE isoforms have isoform-specific effects via lipoprotein receptors and transporter binding in the human brain leading to disturbances in lipid metabolism or neuronal signaling in the CNS. Such effects, if present, are proposed to have significant consequences on synaptic plasticity and membrane remodeling following neuronal injury. Consistent with this possibility, in vitro and animal studies have indicated that apoE3 appears to enhance synaptic plasticity and decrease neurotoxicity compared to apoE4, though there are a few conflicting reports.9, 57
In humans, PET studies have revealed that the brains of cognitively normal APOE ε4 individuals display reduced cerebral glucose metabolism two decades earlier than the expected mean age of onset for APOE ε3 homozygousindividuals, 58 consistent with several studies demonstrating that young and elderly APOE ε4-carriers exhibit regional glucose hypometabolism compared to non-carriers.58–59 Interestingly, cerebral glucose hypometabolism is correlated with increase in mid-life serum cholesterol in cognitively normal APOE ε4-carriers.60 Recent fMRI studies reported that brain activity in the default mode network, a brain network spatially overlapping with that of amyloid deposition,61 is increased in young, middle-aged, and elderly carriers of APOE ε4, both with and without amyloid deposition.62–64 In AD patients, it is unknown whether apoE isoform-specific lipid dysregulation or cellular signaling contributes to cerebral hypometabolism specifically or whether the hypometabolism is secondary to brain injury, altered Aβ metabolism, or other AD-related dysfunction.
ApoE isoform-specific roles in neurotoxicity, independent of interactions with Aβ, may have implications in mechanisms of neurodegeneration in AD.65 Though most apoE is secreted by glia, neuronal apoE4 secreted in the context of certain models of cellular stress is more sensitive to chymotrypsin-like serine proteolytic cleavage than apoE3,66 leading to the generation of potentially neurotoxic C-terminal truncated apoE4 fragments. Mouse studies suggest that these fragments lead to neurodegeneration and behavioral deficits through effects on tau phosphorylation and cytoskeletal disruption.66 In AD patients, C-terminal truncated fragments of apoE are increased relative to normal individuals; however, there are presently no reports of increased C-terminal truncated fragments in APOE ε4-positive AD patients. It will be important to determine the overall impact of apoE produced by neurons in neurotoxicity. Since the potential effects of apoE isoforms on lipid metabolism, cell signaling, and neurotoxicity in the CNS have the potential to influence both AD and other neurological disorders, further investigation on this topic is warranted.
Effects of APOE genotype on AD therapeutic response
Several AD treatments have been proposed and have been or are being tested in clinical trials. While not all trials have assessed the effect of APOE genotype in regard to efficacy and safety, several reports suggest that this could be an important factor in trial design. Three acetylcholinesterase inhibitors (AChE-I), donepezil (Aricept ®), rivastigmine (Exelon®), and galantamine (Reminyl®) are widely used for symptomatic treatment of AD. A recent prospective study in a cohort of mild to moderate AD patients treated with Aricept, Exelon, and Reminyl for 36 weeks found that APOE ε4 status did not influence drug efficacy;67–68 however, previous studies showed that APOE ε4-carriers benefit less than non-carriers from treatment with tacrine (Cognex®), an AChE-I.69–70 A large study of mild cognitive impairment showed that donepezil had a significant effect on delaying progression from MCI to AD in the first 12 months of the study, but this effect was not observed after 3 years. Secondary analysis showed that the observed effect was more profound among APOE ε4-positive than ε4-negative carriers throughout the entire length of the study.68 However, the benefit observed in APOE ε4 carriers may be due not to a biological link between APOE and donepezil, but rather the ease in demonstrating the conversion and progression from MCI to AD in APOE ε4 carriers compared to non-carriers.
Preliminary clinical trials with Rosiglitazone (RSG), a PPARγ agonist that modulates energy/lipid metabolism and inflammation, demonstrated a significant improvement in cognition in RSG-treated mild to moderate AD patients not carrying copies of APOE ε4; however, a decline in cognition in APOE ε4-carriers was observed.71 However, a phase III study later found no significant improvement in cognition in RSG-treated mild to moderate AD patients regardless of APOE ε4 genotype.72
Active and passive immunizations against soluble or insoluble Aβ in several mouse models of amyloid deposition resulted in reduced amyloid pathology in brain tissue and cerebral vasculature with behavioral improvements in various memory-related tasks.73–77 A recent phase II passive immunization clinical trial in mild to moderate AD patients with bapineuzumab, a humanized anti-Aβ monoclonal antibody, did not find significant treatment differences in pre-specified within-dose cohort analyses.78 However, secondary analysis revealed significantly improved clinical outcomes among bapineuzumab-treated patients. By some clinical measures, treatment efficacy was greater in non-carriers of APOE ε4 relative to carriers. Importantly, reversible vasogenic edema (VE) was observed in approximately 10% of bapineuzumab-treated patients, the incidence of which was greater in higher dose groups and APOE ε4-carriers. Increased vascular amyloid burden, differences in vascular permeability, and increased neuroinflammation in APOE ε4-carriers have all been proposed to account for the VE observed in bapineuzumab-treated APOE ε4-carriers.78–80 A recent animal study suggests that the murine version of bapineuzumab can result in rapid inflammatory changes in the brain in areas of amyloid deposition including around CAA.81 These studies emphasize that drugs designed to alter amyloid load in AD or other neurological disorders may interact with APOE genotype, resulting in differential efficacy and outcome. This interaction is an important factor to consider in trial design.
APOE and traumatic brain injury (TBI)
More than a decade of literature has suggested a link between traumatic brain injury (TBI) and AD risk.82–84 Several groups have reported increased risk for AD in TBI patients carrying an APOE ε4 allele,85–86 an association for which the level of evidence is strong (Table 1). One hypothesis for the increased development of dementia in TBI patients posits that brain trauma precipitates Aβ deposition, a hypothesis substantiated by the increased incidence of Aβ deposition identified in postmortem cortical extracts from acute brain injury patients.84 The likelihood of identifying Aβ deposits or CAA in postmortem tissue from TBI patients was elevated in APOE ε4-carriers relative to non-carriers.87–88 Given that amyloid deposition is higher in APOE ε4-carriers, the interpretation that the brains of APOE ε4-carriers may harbor Aβ deposits prior to their acute brain injury remains a formal possibility. Studies using an experimental swine model of diffuse TBI, however, demonstrated the deposition of Aβ following controlled impact.89–90 A series of studies has also revealed that possession of the APOE ε4 allele renders TBI patients more susceptible to poor neurological outcome following brain injury.91–96 In particular, one prospective cohort study found that TBI patients carrying the APOE ε4 allele were twice as likely to suffer poor neurological outcome as non-carriers,93 an association later found to be most striking in younger patients in a larger cohort.97 Some discrepancy exists among studies with regard to whether possession of APOEε4 is associated with initial severity of injury versus increased risk of poor outcome following TBI. However, a recent meta-analysis analyzed 14 prospective TBI studies, concluding that the major effect of APOE ε4 was on poor neurological outcome following TBI.98 Repetitive subacute brain injury commonly results in a progressive disorder known as chronic traumatic encephalopathy (CTE) or the variant, dementia pugilistica (DP), both of which share many of the clinical features of common dementias.99 Strong associations between APOE ε4 and increased risk for CTE and DP have been reported in high exposure football players and boxers.100–101 Interestingly, in spite of the link between brain injury and increased Aβ accumulation, the brains of patients with these disorders at autopsy also have a significant amount of tau pathology with denser accumulations of tau in more superficial cortical layers than in AD and with not all cases exhibiting increased Aβ deposition.99 Taken together, these studies strongly suggest a link between APOEε 4 and aberrant Aβ metabolism in the wake of brain injury. In addition, an Aβ-independent process involving tau and other pathways may also be involved in the APOE ε4 and TBI connection. Further studies are needed to assess the relative contribution of Aβ, tau, and other mechanisms, in negative outcome following brain injury and the extent to which APOE ε4 aggravates pathology.
APOE and Down’s syndrome-associated dementia (DAD)
By the age of 40, all individuals with Down’s syndrome (DS) develop neuropathological features of AD.102 Several studies have suggested that the APOE ε4 allele predisposes and reduces the age of onset for DAD, while APOE ε2 alleles may be protective.103 One case-control study in a Dutch population reported no effect of APOE genotype on incidence or onset of DAD,103–104 though an early meta-analysis of association studies found that within DS patients, the incidence of dementia was higher in patients carrying APOE ε4. A major limitation of these studies has been the relatively small number of patients analyzed. However, a recent longitudinal, large cohort study of DS patients reported that APOE ε4-carriers are at higher risk for DAD with earlier onset and accelerated progression compared to non-carriers.105 Available data suggest that the level of evidence for an association between APOE ε4 and DAD is highly suggestive (Table 1).
APOE and outcome after stroke
The observation that APOE ε4 associates with poor neurological outcome after TBI has provided the impetus to investigate a possible association between APOE and three major pathological types of acute stroke, ischemic stroke (IS), intracerebral hemorrhage (ICH), and subarachnoid hemorrhage (SAH). Multiple studies assessing large cohorts of different ethnicities suggest that the outcome of IS in patients carrying the APOE ε4 allele is similar to that of non-carriers.106–108 A recent meta-analysis and several subsequent studies observed an association between APOE ε4 and poor neuropsychological outcome after SAH, though there are conflicting reports.106, 108–110 A similar association was identified in small cohort studies of ICH patients, though APOE ε4 also appeared to associate with poor survival in these patients.111 At present, the level of evidence is suggestive for an association between APOE ε4 and ICH and SAH, but not IS (Table 1). It will be critical to employ large cohort studies with standardized follow-up methods to understand whether apoE isoforms play a role in outcome following acute stroke.
APOE and Vascular dementia (VaD)
VaD is the second most common contributor to dementia, accounting for 17–20% of dementia patients in the United States.112–113 Several studies in multiple ethnicities have shown that possession of APOE ε4 increases risk for VaD,114–116 though an equal number of studies report that APOE ε4 does not confer risk.117–118 Inconsistent results among these studies are likely due to small cohort sizes, misdiagnosis of VaD with AD and mixed dementia, and lack of population-based, prospective study design. A recent population-based, prospective study following 3424 elderly individuals found that APOE ε4 is associated with increased risk for VaD in an allele dose-dependent fashion, with 1.5-fold and 4-fold increased risk for homozygous and heterozygous APOE ε4 subjects, respectively (Table 1).113 While it has been suggested that vascular risk factors like hypertension, diabetes and dyslipidemia are important factors in VaD development,119 multiple studies suggest VaD risk of APOE ε4 is independent of the other vascular risk factors.113, 120 This may be due to the effect of APOE ε4 on concomitant AD pathology contributing to cognitive decline in patients with vascular dementia. Further studies are needed to understand how vascular factors and APOE status converge to modulate VaD risk.
APOE and Creutzfeldt-Jakob disease (CJD)
Following a report that apoE immunoreactivity was found in kuru amyloid plaques from CJD patients, an initial study found that the APOE ε4 allele frequency of CJD patients did not differ from that of controls.2 A contradictory report found that the APOE ε4 allele associated with definite or probable CJD while APOE ε2 decreased CJD-related mortality.121 Unlike several subsequent studies reporting no such association of APOE genotype in several different ethnic populations,122–124 the Amouyel et al. study analyzed the frequency of allele bearers rather than alleles in addition to including both familial and sporadic CJD patients that were not age-matched with controls. Given the available data, it appears that apoE associates with plaques in CJD patients with no definite role in the disease process (Table 1).
APOE and multiple sclerosis (MS)
Another neurological disorder for which the association of APOE genotype and disease is unclear is MS. Though an initial study reported a higher frequency of MS in APOE ε4 homozygous individuals from a Danish population,125 a number of studies have found no association with APOE ε4 allele frequency and MS susceptibility.126 In particular, a meta-analysis of many association studies did not support a role of APOE in modulating MS risk, though the effect of population admixture may have limited effect size in the analysis.126–127 A number of studies support a role of APOE ε4 as a progression modifier of MS, having a negative effect on brain pathology, cognitive dysfunction, and severity, though these conclusions are not without controversy.128 More uniformity in cognitive measures as well as larger studies with longer follow-up in MS patients is needed before conclusions can be drawn regarding the association of APOEε4 and MS progression (Table 1).
APOE and Amyotrophic lateral sclerosis (ALS)
There is a strong consensus among studies that APOE genotype is not associated with sporadic or familial ALS risk.129 However, ample debate exists as to the role of apoE isoforms in differentially modulating age at onset, progression rate, and site of onset in ALS patients. One survival analysis study reported that APOE status is not associated with age of onset or site of onset of ALS; however, the progression of the disease appeared to be more rapid in APOE ε4-carriers.130 A family-based association analysis found that APOE ε2 was protective, resulting in a later disease onset relative to non-carriers; the presence of APOE ε4 had no effect on onset despite its suggested role in progression.131 However, a recent study found an APOE ε4 gene dose-dependent association with lower age of onset of sporadic ALS with APOE ε4-carriers.132 Most studies only identify APOE as a weak modifier of ALS onset and progression, though additional studies may be needed to clarify the relative effects of APOE ε4 and APOE ε2 (Table 1).
APOE and Inclusion-body Myositis (IBM)
Several neuropathological features are shared among sporadic and hereditary IBM (s-IBM/h-IBM) and AD.133 Along with many other proteins, apoE is present in vacuolated muscle fibers of patients with s-IBM and h-IBM,134–135 prompting groups to assess whether APOE genotype is associated with s-IBM/h-IBM. An early study performed in a small cohort of 14 patients reported increased frequency of APOE ε4 in s-IBM,136 but several studies later found no association.135, 137–138 A recentstudy in a moderately sized cohort of s-IBM patients and a meta-analysis (studies prior to 2007) demonstrated no evidence of risk for s-IBM associated with APOE status139 (Table 1).
APOE and Parkinson’s disease (PD)
Clinical and pathological features of PD and AD frequently overlap, leading several groups to investigate the association of APOE and PD onset or PD dementia (PDD). The majority of studies have not reported associations between APOE ε4 and susceptibility to PD/PDD or age at onset,140–141 though there are conflicting reports.142–143 A meta-analysis of 22 studies reported a positive association between the APOE ε2 allele frequency and PD risk, while no such association was found in APOE ε3 or APOE ε4 allele carriers.144 Several reports suggest that APOE plays no role in modulating the clinical features of PDD, suggesting that the underlying cause of dementia in PD differs from that of AD.140, 145 Given that APOE ε2 appears to increase PD risk in some studies, it is likely that the role of apoE in PD may be mechanistically distinct from that in other neurological disorders associated with APOE ε4 (Table 1).
APOE and Dementia with Lewy Bodies (DLB)
DLB shares clinical and pathological characteristics with both PD and AD.146 Mounting evidence suggests that APOE ε4 is associated with increased risk in DLB, while possession of APOE ε2 has no effect.147–149 This association may be due to APOE ε4 increasing the likelihood of Aβ deposition, which is present in many cases of DLB along with synuclein pathology.
Others have reported that APOE ε4 does not influence the onset or progression of DLB,150–151 leaving the association between APOE and DLB progression uncertain, given the lack of evidence (Table 1). Interestingly, a recent study in mice overexpressing α-synuclein suggests that apoE might influence the solubility of α-synuclein to influence its aggregation.152
APOE and neurological diseases for which association is unclear
Based on associations reported in other neurological diseases, APOE has been suggested to play a role in Cerebral Palsy (CP),153–154 Huntington’s disease (HD),155–156 temporal lobe epilepsy (TL-E), 157–158 and frontotemporal dementia (FTD).159 However, while some studies have attempted to address a putative association between APOE and these diseases, a limited number of studies exist to draw conclusions (Table 1).
There are several possible mechanisms by which APOE may influence the onset rate and/or progression of neurological disorders other than AD, CAA, and TBI in which Aβ is not likely to be involved in pathogenesis; however, these mechanisms have not been directly assessed in human studies. Some of the suggested mechanisms, inferred from in vitro and animal studies, by which APOE could generally influence neurological disorders are by modulating: (1) neuroinflammation,160–162 (2) lipid metabolism,57, 163–165 (3) synaptic plasticity,166–169 and (4) neuronal toxicity. 66, 170
Conclusions
Increasing evidence has suggested a central role for apoE in modulating processes of neurodegeneration, particularly in AD and CAA. A large body of evidence has identified APOE ε4 as a major AD susceptibility and onset factor, while APOE ε2 appears to confer protection (Table 1). A convincing body of literature suggests that APOE genotype modulates AD risk largely through its effects on Aβ, probably by influencing aggregation and/or clearance from the brain parenchyma in an isoform-dependent fashion. Epidemiological as well as in vitro and animal studies suggest an Aβ-independent role for apoE which may negatively impact synaptic plasticity, response to neuronal injury, and neuroinflammation. Outcome in neurological disorders such as TBI, hemorrhagic stroke (ICH, SAH), and DAD appear to be influenced by APOE genotype. Though associations have been reported between APOE and other neurological disorders such as VaD, MS, ALS, PD, and DLB, significant controversy exists among studies with regard to risk, age of onset, and progression. Insufficient data exist to evaluate whether APOE genotype associates with risk, age of onset, and progression in CP, HD, TL-E, and FTD. Publication bias and population admixture in meta-analyses, Type I error due to multiple hypothesis testing, and non-uniformity in clinical diagnosis all represent limitations to our current understanding of the association of APOE with other neurological diseases. Understanding the detailed mechanism(s) as to how apoE influences Aβ metabolism will be important to better understand AD and CAA pathogenesis and treatment. Further humans studies to understand whether or not apoE plays a role in other neurological diseases are warranted. Emerging evidence suggests that drugs designed for the treatment of neurological disorders have the potential to interact with APOE genotype, resulting in differential efficacy and outcome. This interaction may be an important factor to consider in trial design.
Acknowledgments
PBV and JMC are supported by an American Health Assistance postdoctoral fellowship (AHAF 3857-43287) and an NIH F31 pre-doctoral fellowship (AG034004), respectively. DMH is supported by NIH grants AG13956, AG03991, AG026276, and NS034467.
Footnotes
Contributors: PBV and JMC wrote the first draft and DMH critically evaluated and edited the manuscript.
Search Strategy and Selection Criteria: PubMed was accessed to identify original and review articles published up to December 2010, with the terms “ApoE AND neurological disorders,” “ApoE AND Alzheimer’s disease,” “ApoE AND drug AND AD,” “ApoE AND cerebral amyloid angiopathy,” “ApoE AND traumatic brain injury,” “ApoE AND stroke,” “ApoE AND and Down’s syndrome,” “ApoE AND vascular dementia,” “ApoE AND Lewy body dementia,” “ApoE AND inclusion-body myositis,” “ApoE AND Creutzfeldt-Jakob disease,” “ApoE AND multiple sclerosis,” “ApoE and amyotrophic lateral sclerosis,” “ApoE AND Parkinson’s disease,” “ApoE AND cerebral palsy,” “ApoE AND Huntington’s disease,” “ApoE AND epilepsy,” “ApoE AND Frontotemporal dementia,” “ApoE AND Dementia.” Only papers published in English were reviewed.
Conflict of interest: The authors PBV and JMC have no conflicts of interests. DMH receives payment for being on the scientific advisory boards of En Vivo and Satori. He has also received payments from C2N Diagnostics, LLC as a scientific advisor as well as royalties from a Washington University patent licensed to C2N. Washington University receives grants from Eli-Lilly and Pfizer that supports research in the laboratory of DMH.
References
- 1.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 3.Saunders AM. Apolipoprotein E and Alzheimer disease: an update on genetic and functional analyses. J Neuropathol Exp Neurol. 2000;59:751–8. doi: 10.1093/jnen/59.9.751. [DOI] [PubMed] [Google Scholar]
- 4.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 5.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 6.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 7.Zannis VI, Breslow JL, Utermann G, Mahley RW, Weisgraber KH, Havel RJ, et al. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J Lipid Res. 1982;23:911–4. [PubMed] [Google Scholar]
- 8.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–60. [PubMed] [Google Scholar]
- 11.Bandaru VV, Troncoso J, Wheeler D, Pletnikova O, Wang J, Conant K, et al. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol Aging. 2009;30:591–9. doi: 10.1016/j.neurobiolaging.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaDu MJ, Gilligan SM, Lukens JR, Cabana VG, Reardon CA, Van Eldik LJ, et al. Nascent astrocyte particles differ from lipoproteins in CSF. J Neurochem. 1998;70:2070–81. doi: 10.1046/j.1471-4159.1998.70052070.x. [DOI] [PubMed] [Google Scholar]
- 13.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 14.Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G, et al. Apolipoprotein Eepsilon4 modifies Alzheimer’s disease onset in an E280A PS1 kindred. Ann Neurol. 2003;54:163–9. doi: 10.1002/ana.10636. [DOI] [PubMed] [Google Scholar]
- 15.West HL, Rebeck GW, Hyman BT. Frequency of the apolipoprotein E epsilon 2 allele is diminished in sporadic Alzheimer disease. Neurosci Lett. 1994;175:46–8. doi: 10.1016/0304-3940(94)91074-x. [DOI] [PubMed] [Google Scholar]
- 16.Meyer MR, Tschanz JT, Norton MC, Welsh-Bohmer KA, Steffens DC, Wyse BW, et al. APOE genotype predicts when--not whether--one is predisposed to develop Alzheimer disease. Nat Genet. 1998;19:321–2. doi: 10.1038/1206. [DOI] [PubMed] [Google Scholar]
- 17.Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology. 2004;62:1977–83. doi: 10.1212/01.wnl.0000128091.92139.0f. [DOI] [PubMed] [Google Scholar]
- 18.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, et al. Apolipoprotein E, survival in Alzheimer’s disease patients, and the competing risks of death and Alzheimer’s disease. Neurology. 1995;45:1323–8. doi: 10.1212/wnl.45.7.1323. [DOI] [PubMed] [Google Scholar]
- 19.Craft S, Teri L, Edland SD, Kukull WA, Schellenberg G, McCormick WC, et al. Accelerated decline in apolipoprotein E-epsilon4 homozygotes with Alzheimer’s disease. Neurology. 1998;51:149–53. doi: 10.1212/wnl.51.1.149. [DOI] [PubMed] [Google Scholar]
- 20.Hoyt BD, Massman PJ, Schatschneider C, Cooke N, Doody RS. Individual growth curve analysis of APOE epsilon 4-associated cognitive decline in Alzheimer disease. Arch Neurol. 2005;62:454–9. doi: 10.1001/archneur.62.3.454. [DOI] [PubMed] [Google Scholar]
- 21.Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, et al. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007;6:494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- 22.Bookheimer S, Burggren A. APOE-4 genotype and neurophysiological vulnerability to Alzheimer’s and cognitive aging. Annu Rev Clin Psychol. 2009;5:343–62. doi: 10.1146/annurev.clinpsy.032408.153625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang GC, Insel PS, Tosun D, Schuff N, Truran-Sacrey D, Raptentsetsang ST, et al. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 2010 doi: 10.1212/WNL.0b013e3181ffe4d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caselli RJ, Dueck AC, Osborne D, Sabbagh MN, Connor DJ, Ahern GL, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med. 2009;361:255–63. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Love S, Miners S, Palmer J, Chalmers K, Kehoe P. Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front Biosci. 2009;14:4778–92. doi: 10.2741/3567. [DOI] [PubMed] [Google Scholar]
- 26.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–24. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 27.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998;57:353–9. doi: 10.1097/00005072-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Greenberg SM, Vonsattel JP, Segal AZ, Chiu RI, Clatworthy AE, Liao A, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology. 1998;50:961–5. doi: 10.1212/wnl.50.4.961. [DOI] [PubMed] [Google Scholar]
- 29.Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol. 1996;148:2083–95. [PMC free article] [PubMed] [Google Scholar]
- 30.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–9. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 31.Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, et al. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology. 1996;47:190–6. doi: 10.1212/wnl.47.1.190. [DOI] [PubMed] [Google Scholar]
- 32.Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW, et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol. 1997;41:716–21. doi: 10.1002/ana.410410607. [DOI] [PubMed] [Google Scholar]
- 33.Love S, Nicoll JA, Hughes A, Wilcock GK. APOE and cerebral amyloid angiopathy in the elderly. Neuroreport. 2003;14:1535–6. doi: 10.1097/00001756-200308060-00027. [DOI] [PubMed] [Google Scholar]
- 34.Yamada M, Itoh Y, Suematsu N, Matsushita M, Otomo E. Lack of an association between apolipoprotein E epsilon 4 and cerebral amyloid angiopathy in elderly Japanese. Ann Neurol. 1996;39:683–4. doi: 10.1002/ana.410390523. [DOI] [PubMed] [Google Scholar]
- 35.Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, et al. Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306 ( Pt 2):599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–4. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 37.Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–5. [PMC free article] [PubMed] [Google Scholar]
- 38.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–4. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 39.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96:15233–8. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, et al. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol. 2000;47:739–47. [PubMed] [Google Scholar]
- 41.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–18. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 43.Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–10. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, et al. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–9. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trommer BL, Shah C, Yun SH, Gamkrelidze G, Pasternak ES, Stine WB, et al. ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-beta1–42. Neurobiol Dis. 2005;18:75–82. doi: 10.1016/j.nbd.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 46.Thal DR, Larionov S, Abramowski D, Wiederhold KH, Van Dooren T, Yamaguchi H, et al. Occurrence and co-localization of amyloid beta-protein and apolipoprotein E in perivascular drainage channels of wild-type and APP-transgenic mice. Neurobiol Aging. 2007;28:1221–30. doi: 10.1016/j.neurobiolaging.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 47.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77. doi: 10.1146/annurev.ne.19.030196.000413. [DOI] [PubMed] [Google Scholar]
- 48.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333:1242–7. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 51.Hyman BT, West HL, Rebeck GW, Buldyrev SV, Mantegna RN, Ukleja M, et al. Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome) Proc Natl Acad Sci U S A. 1995;92:3586–90. doi: 10.1073/pnas.92.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rebeck GW, Perls TT, West HL, Sodhi P, Lipsitz LA, Hyman BT. Reduced apolipoprotein epsilon 4 allele frequency in the oldest old Alzheimer’s patients and cognitively normal individuals. Neurology. 1994;44:1513–6. doi: 10.1212/wnl.44.8.1513. [DOI] [PubMed] [Google Scholar]
- 53.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009;461:916–22. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:6820–5. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sunderland T, Mirza N, Putnam KT, Linker G, Bhupali D, Durham R, et al. Cerebrospinal fluid beta-amyloid1–42 and tau in control subjects at risk for Alzheimer’s disease: the effect of APOE epsilon4 allele. Biol Psychiatry. 2004;56:670–6. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 56.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res. 2005;46:949–68. doi: 10.1194/jlr.M400486-JLR200. [DOI] [PubMed] [Google Scholar]
- 58.Drzezga A, Riemenschneider M, Strassner B, Grimmer T, Peller M, Knoll A, et al. Cerebral glucose metabolism in patients with AD and different APOE genotypes. Neurology. 2005;64:102–7. doi: 10.1212/01.WNL.0000148478.39691.D3. [DOI] [PubMed] [Google Scholar]
- 59.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am J Psychiatry. 2002;159:738–45. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 60.Reiman EM, Chen K, Langbaum JB, Lee W, Reschke C, Bandy D, et al. Higher serum total cholesterol levels in late middle age are associated with glucose hypometabolism in brain regions affected by Alzheimer’s disease and normal aging. Neuroimage. 2010;49:169–76. doi: 10.1016/j.neuroimage.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–76. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- 62.Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009;106:7209–14. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fleisher AS, Sherzai A, Taylor C, Langbaum JB, Chen K, Buxton RB. Resting-state BOLD networks versus task-associated functional MRI for distinguishing Alzheimer’s disease risk groups. Neuroimage. 2009;47:1678–90. doi: 10.1016/j.neuroimage.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D’Angelo G, et al. APOE4 Allele Disrupts Resting State fMRI Connectivity in the Absence of Amyloid Plaques or Decreased CSF A{beta}42. J Neurosci. 2010;30:17035–40. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31:445–54. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 66.Huang Y. Abeta-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2010;16:287–94. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 67.Santoro A, Siviero P, Minicuci N, Bellavista E, Mishto M, Olivieri F, et al. Effects of donepezil, galantamine and rivastigmine in 938 Italian patients with Alzheimer’s disease: a prospective, observational study. CNS Drugs. 24:163–76. doi: 10.2165/11310960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 68.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–88. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 69.Farlow MR, Lahiri DK, Poirier J, Davignon J, Hui S. Apolipoprotein E genotype and gender influence response to tacrine therapy. Ann N Y Acad Sci. 1996;802:101–10. doi: 10.1111/j.1749-6632.1996.tb32603.x. [DOI] [PubMed] [Google Scholar]
- 70.Farlow MR, Lahiri DK, Poirier J, Davignon J, Schneider L, Hui SL. Treatment outcome of tacrine therapy depends on apolipoprotein genotype and gender of the subjects with Alzheimer’s disease. Neurology. 1998;50:669–77. doi: 10.1212/wnl.50.3.669. [DOI] [PubMed] [Google Scholar]
- 71.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–54. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 72.Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, et al. Rosiglitazone monotherapy in mild-to-moderate alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 30:131–46. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 74.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, et al. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9096–101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 77.Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA. Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 2006;20:426–33. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- 78.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laskowitz DT, Kolls BJ. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2010;74:2026. doi: 10.1212/WNL.0b013e3181e03844. author reply -7. [DOI] [PubMed] [Google Scholar]
- 80.Kaufer D, Gandy S. APOE {epsilon}4 and bapineuzumab: Infusing pharmacogenomics into Alzheimer disease therapeutics. Neurology. 2009;73:2052–3. doi: 10.1212/WNL.0b013e3181c6784a. [DOI] [PubMed] [Google Scholar]
- 81.Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, Garcia-Alloza M, Finn MB, Hyman BT, et al. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–64. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mayeux R, Ottman R, Tang MX, Noboa-Bauza L, Marder K, Gurland B, et al. Genetic susceptibility and head injury as risk factors for Alzheimer’s disease among community-dwelling elderly persons and their first-degree relatives. Ann Neurol. 1993;33:494–501. doi: 10.1002/ana.410330513. [DOI] [PubMed] [Google Scholar]
- 83.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20 (Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 84.Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–3. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 85.Mayeux R, Ottman R, Maestre G, Ngai C, Tang MX, Ginsberg H, et al. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer’s disease. Neurology. 1995;45:555–7. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 86.Katzman R, Galasko DR, Saitoh T, Chen X, Pay MM, Booth A, et al. Apolipoprotein-epsilon4 and head trauma: Synergistic or additive risks? Neurology. 1996;46:889–91. [PubMed] [Google Scholar]
- 87.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat Med. 1995;1:135–7. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 88.Leclercq PD, Murray LS, Smith C, Graham DI, Nicoll JA, Gentleman SM. Cerebral amyloid angiopathy in traumatic brain injury: association with apolipoprotein E genotype. J Neurol Neurosurg Psychiatry. 2005;76:229–33. doi: 10.1136/jnnp.2003.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–71. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–92. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 91.Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–8. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 92.Sorbi S, Nacmias B, Piacentini S, Repice A, Latorraca S, Forleo P, et al. ApoE as a prognostic factor for post-traumatic coma. Nat Med. 1995;1:852. doi: 10.1038/nm0995-852. [DOI] [PubMed] [Google Scholar]
- 93.Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–71. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 94.Liaquat I, Dunn LT, Nicoll JA, Teasdale GM, Norrie JD. Effect of apolipoprotein E genotype on hematoma volume after trauma. J Neurosurg. 2002;96:90–6. doi: 10.3171/jns.2002.96.1.0090. [DOI] [PubMed] [Google Scholar]
- 95.Diaz-Arrastia R, Gong Y, Fair S, Scott KD, Garcia MC, Carlile MC, et al. Increased risk of late posttraumatic seizures associated with inheritance of APOE epsilon4 allele. Arch Neurol. 2003;60:818–22. doi: 10.1001/archneur.60.6.818. [DOI] [PubMed] [Google Scholar]
- 96.Lichtman SW, Seliger G, Tycko B, Marder K. Apolipoprotein E and functional recovery from brain injury following postacute rehabilitation. Neurology. 2000;55:1536–9. doi: 10.1212/wnl.55.10.1536. [DOI] [PubMed] [Google Scholar]
- 97.Teasdale GM, Murray GD, Nicoll JA. The association between APOE epsilon4, age and outcome after head injury: a prospective cohort study. Brain. 2005;128:2556–61. doi: 10.1093/brain/awh595. [DOI] [PubMed] [Google Scholar]
- 98.Zhou W, Xu D, Peng X, Zhang Q, Jia J, Crutcher KA. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279–90. doi: 10.1089/neu.2007.0489. [DOI] [PubMed] [Google Scholar]
- 99.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–35. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136–40. [PubMed] [Google Scholar]
- 101.Kutner KC, Erlanger DM, Tsai J, Jordan B, Relkin NR. Lower cognitive performance of older football players possessing apolipoprotein E epsilon4. Neurosurgery. 2000;47:651–7. doi: 10.1097/00006123-200009000-00026. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 102.Mann DM, Esiri MM. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci. 1989;89:169–79. doi: 10.1016/0022-510x(89)90019-1. [DOI] [PubMed] [Google Scholar]
- 103.Khachaturian AS, Corcoran CD, Mayer LS, Zandi PP, Breitner JC. Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: The Cache County Study. Arch Gen Psychiatry. 2004;61:518–24. doi: 10.1001/archpsyc.61.5.518. [DOI] [PubMed] [Google Scholar]
- 104.van Gool WA, Evenhuis HM, van Duijn CM. A case-control study of apolipoprotein E genotypes in Alzheimer’s disease associated with Down’s syndrome. Dutch Study Group on Down’s Syndrome and Ageing. Ann Neurol. 1995;38:225–30. doi: 10.1002/ana.410380215. [DOI] [PubMed] [Google Scholar]
- 105.Prasher VP, Sajith SG, Rees SD, Patel A, Tewari S, Schupf N, et al. Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer’s disease and mortality in persons with Down syndrome. Int J Geriatr Psychiatry. 2008;23:1134–40. doi: 10.1002/gps.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martinez-Gonzalez NA, Sudlow CL. Effects of apolipoprotein E genotype on outcome after ischaemic stroke, intracerebral haemorrhage and subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2006;77:1329–35. doi: 10.1136/jnnp.2006.097543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McCarron MO, Muir KW, Nicoll JA, Stewart J, Currie Y, Brown K, et al. Prospective study of apolipoprotein E genotype and functional outcome following ischemic stroke. Arch Neurol. 2000;57:1480–4. doi: 10.1001/archneur.57.10.1480. [DOI] [PubMed] [Google Scholar]
- 108.Waters RJ, Nicoll JA. Genetic influences on outcome following acute neurological insults. Curr Opin Crit Care. 2005;11:105–10. doi: 10.1097/01.ccx.0000155354.78617.91. [DOI] [PubMed] [Google Scholar]
- 109.Morris PG, Wilson JT, Dunn LT, Nicoll JA. Apolipoprotein E polymorphism and neuropsychological outcome following subarachnoid haemorrhage. Acta Neurol Scand. 2004;109:205–9. doi: 10.1034/j.1600-0404.2003.00206.x. [DOI] [PubMed] [Google Scholar]
- 110.Lanterna LA, Rigoldi M, Tredici G, Biroli F, Cesana C, Gaini SM, et al. APOE influences vasospasm and cognition of noncomatose patients with subarachnoid hemorrhage. Neurology. 2005;64:1238–44. doi: 10.1212/01.WNL.0000156523.77347.B4. [DOI] [PubMed] [Google Scholar]
- 111.McCarron MO, Weir CJ, Muir KW, Hoffmann KL, Graffagnino C, Nicoll JA, et al. Effect of apolipoprotein E genotype on in-hospital mortality following intracerebral haemorrhage. Acta Neurol Scand. 2003;107:106–9. doi: 10.1034/j.1600-0404.2003.01365.x. [DOI] [PubMed] [Google Scholar]
- 112.Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR, Ofstedal MB, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology. 2007;29:125–32. doi: 10.1159/000109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chuang YF, Hayden KM, Norton MC, Tschanz J, Breitner JC, Welsh-Bohmer KA, et al. Association between APOE epsilon4 allele and vascular dementia: The Cache County study. Dement Geriatr Cogn Disord. 29:248–53. doi: 10.1159/000285166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Davidson Y, Gibbons L, Purandare N, Byrne J, Hardicre J, Wren J, et al. Apolipoprotein E epsilon4 allele frequency in vascular dementia. Dement Geriatr Cogn Disord. 2006;22:15–9. doi: 10.1159/000092960. [DOI] [PubMed] [Google Scholar]
- 115.Baum L, Lam LC, Kwok T, Lee J, Chiu HF, Mok VC, et al. Apolipoprotein E epsilon4 allele is associated with vascular dementia. Dement Geriatr Cogn Disord. 2006;22:301–5. doi: 10.1159/000095246. [DOI] [PubMed] [Google Scholar]
- 116.Luthra K, Tripathi M, Grover R, Dwivedi M, Kumar A, Dey AB. Apolipoprotein E gene polymorphism in Indian patients with Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord. 2004;17:132–5. doi: 10.1159/000076345. [DOI] [PubMed] [Google Scholar]
- 117.Kim KW, Youn JC, Han MK, Paik NJ, Lee TJ, Park JH, et al. Lack of association between apolipoprotein E polymorphism and vascular dementia in Koreans. J Geriatr Psychiatry Neurol. 2008;21:12–7. doi: 10.1177/0891988707311028. [DOI] [PubMed] [Google Scholar]
- 118.Sulkava R, Kainulainen K, Verkkoniemi A, Niinisto L, Sobel E, Davanipour Z, et al. APOE alleles in Alzheimer’s disease and vascular dementia in a population aged 85+ Neurobiol Aging. 1996;17:373–6. doi: 10.1016/0197-4580(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 119.Luchsinger JA, Mayeux R. Cardiovascular risk factors and Alzheimer’s disease. Curr Atheroscler Rep. 2004;6:261–6. doi: 10.1007/s11883-004-0056-z. [DOI] [PubMed] [Google Scholar]
- 120.Prince M, Lovestone S, Cervilla J, Joels S, Powell J, Russ C, et al. The association between APOE and dementia does not seem to be mediated by vascular factors. Neurology. 2000;54:397–402. doi: 10.1212/wnl.54.2.397. [DOI] [PubMed] [Google Scholar]
- 121.Amouyel P, Vidal O, Launay JM, Laplanche JL. The apolipoprotein E alleles as major susceptibility factors for Creutzfeldt-Jakob disease. The French Research Group on Epidemiology of Human Spongiform Encephalopathies. Lancet. 1994;344:1315–8. doi: 10.1016/s0140-6736(94)90691-2. [DOI] [PubMed] [Google Scholar]
- 122.Nakagawa Y, Kitamoto T, Furukawa H, Ogomori K, Tateishi J. Apolipoprotein E in Creutzfeldt-Jakob disease. Lancet. 1995;345:68. [PubMed] [Google Scholar]
- 123.Salvatore M, Seeber AC, Nacmias B, Petraroli R, D’Alessandro M, Sorbi S, et al. Apolipoprotein E in sporadic and familial Creutzfeldt-Jakob disease. Neurosci Lett. 1995;199:95–8. doi: 10.1016/0304-3940(95)12030-8. [DOI] [PubMed] [Google Scholar]
- 124.Zerr I, Helmhold M, Poser S, Armstrong VW, Weber T. Apolipoprotein E phenotype frequency and cerebrospinal fluid concentration are not associated with Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:1233–8. doi: 10.1001/archneur.1996.00550120041014. [DOI] [PubMed] [Google Scholar]
- 125.Hogh P, Oturai A, Schreiber K, Blinkenberg M, Jorgensen OS, Ryder L, et al. Apoliprotein E and multiple sclerosis: impact of the epsilon-4 allele on susceptibility, clinical type and progression rate. Mult Scler. 2000;6:226–30. doi: 10.1177/135245850000600403. [DOI] [PubMed] [Google Scholar]
- 126.Burwick RM, Ramsay PP, Haines JL, Hauser SL, Oksenberg JR, Pericak-Vance MA, et al. APOE epsilon variation in multiple sclerosis susceptibility and disease severity: some answers. Neurology. 2006;66:1373–83. doi: 10.1212/01.wnl.0000210531.19498.3f. [DOI] [PubMed] [Google Scholar]
- 127.Pinholt M, Frederiksen JL, Christiansen M. The association between apolipoprotein E and multiple sclerosis. Eur J Neurol. 2006;13:573–80. doi: 10.1111/j.1468-1331.2006.01360.x. [DOI] [PubMed] [Google Scholar]
- 128.Ghaffar O, Reis M, Pennell N, O’Connor P, Feinstein A. APOE epsilon4 and the cognitive genetics of multiple sclerosis. Neurology. 2010;74:1611–8. doi: 10.1212/WNL.0b013e3181e074a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mui S, Rebeck GW, McKenna-Yasek D, Hyman BT, Brown RH., Jr Apolipoprotein E epsilon 4 allele is not associated with earlier age at onset in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:460–3. doi: 10.1002/ana.410380318. [DOI] [PubMed] [Google Scholar]
- 130.Drory VE, Birnbaum M, Korczyn AD, Chapman J. Association of APOE epsilon4 allele with survival in amyotrophic lateral sclerosis. J Neurol Sci. 2001;190:17–20. doi: 10.1016/s0022-510x(01)00569-x. [DOI] [PubMed] [Google Scholar]
- 131.Li YJ, Pericak-Vance MA, Haines JL, Siddique N, McKenna-Yasek D, Hung WY, et al. Apolipoprotein E is associated with age at onset of amyotrophic lateral sclerosis. Neurogenetics. 2004;5:209–13. doi: 10.1007/s10048-004-0193-0. [DOI] [PubMed] [Google Scholar]
- 132.Zetterberg H, Jacobsson J, Rosengren L, Blennow K, Andersen PM. Association of APOE with age at onset of sporadic amyotrophic lateral sclerosis. J Neurol Sci. 2008;273:67–9. doi: 10.1016/j.jns.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 133.Askanas V, Engel WK. New advances in the understanding of sporadic inclusion-body myositis and hereditary inclusion-body myopathies. Curr Opin Rheumatol. 1995;7:486–96. doi: 10.1097/00002281-199511000-00005. [DOI] [PubMed] [Google Scholar]
- 134.Askanas V, Mirabella M, Engel WK, Alvarez RB, Weisgraber KH. Apolipoprotein E immunoreactive deposits in inclusion-body muscle diseases. Lancet. 1994;343:364–5. doi: 10.1016/s0140-6736(94)91208-4. [DOI] [PubMed] [Google Scholar]
- 135.Mirabella M, Alvarez RB, Engel WK, Weisgraber KH, Askanas V. Apolipoprotein E and apolipoprotein E messenger RNA in muscle of inclusion body myositis and myopathies. Ann Neurol. 1996;40:864–72. doi: 10.1002/ana.410400608. [DOI] [PubMed] [Google Scholar]
- 136.Garlepp MJ, Tabarias H, van Bockxmeer FM, Zilko PJ, Laing B, Mastaglia FL. Apolipoprotein E epsilon 4 in inclusion body myositis. Ann Neurol. 1995;38:957–9. doi: 10.1002/ana.410380619. [DOI] [PubMed] [Google Scholar]
- 137.Harrington CR, Anderson JR, Chan KK. Apolipoprotein E type epsilon 4 allele frequency is not increased in patients with sporadic inclusion-body myositis. Neurosci Lett. 1995;183:35–8. doi: 10.1016/0304-3940(94)11108-u. [DOI] [PubMed] [Google Scholar]
- 138.Love S, Nicoll JA, Lowe J, Sherriff F. Apolipoprotein E allele frequencies in sporadic inclusion body myositis. Muscle Nerve. 1996;19:1605–7. doi: 10.1002/(SICI)1097-4598(199612)19:12<1605::AID-MUS11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 139.Needham M, Hooper A, James I, van Bockxmeer F, Corbett A, Day T, et al. Apolipoprotein epsilon alleles in sporadic inclusion body myositis: a reappraisal. Neuromuscul Disord. 2008;18:150–2. doi: 10.1016/j.nmd.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 140.Ezquerra M, Campdelacreu J, Gaig C, Compta Y, Munoz E, Marti MJ, et al. Lack of association of APOE and tau polymorphisms with dementia in Parkinson’s disease. Neurosci Lett. 2008;448:20–3. doi: 10.1016/j.neulet.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 141.Jasinska-Myga B, Opala G, Goetz CG, Tustanowski J, Ochudlo S, Gorzkowska A, et al. Apolipoprotein E gene polymorphism, total plasma cholesterol level, and Parkinson disease dementia. Arch Neurol. 2007;64:261–5. doi: 10.1001/archneur.64.2.261. [DOI] [PubMed] [Google Scholar]
- 142.Pankratz N, Byder L, Halter C, Rudolph A, Shults CW, Conneally PM, et al. Presence of an APOE4 allele results in significantly earlier onset of Parkinson’s disease and a higher risk with dementia. Mov Disord. 2006;21:45–9. doi: 10.1002/mds.20663. [DOI] [PubMed] [Google Scholar]
- 143.Papapetropoulos S, Farrer MJ, Stone JT, Milkovic NM, Ross OA, Calvo L, et al. Phenotypic associations of tau and ApoE in Parkinson’s disease. Neurosci Lett. 2007;414:141–4. doi: 10.1016/j.neulet.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 144.Huang X, Chen PC, Poole C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology. 2004;62:2198–202. doi: 10.1212/01.wnl.0000130159.28215.6a. [DOI] [PubMed] [Google Scholar]
- 145.Jasinska-Myga B, Opala G, Ochudlo S, Tustanowski J. Assessment of apolipoprotein E genotype in Parkinson disease patients with and without dementia. Wiad Lek. 2004;57:20–4. [PubMed] [Google Scholar]
- 146.McKeith IG, Burn DJ, Ballard CG, Collerton D, Jaros E, Morris CM, et al. Dementia with Lewy bodies. Semin Clin Neuropsychiatry. 2003;8:46–57. doi: 10.1053/scnp.2003.50006. [DOI] [PubMed] [Google Scholar]
- 147.Morris CM, Massey HM, Benjamin R, Leake A, Broadbent C, Griffiths M, et al. Molecular biology of APO E alleles in Alzheimer’s and non-Alzheimer’s dementias. J Neural Transm Suppl. 1996;47:205–18. doi: 10.1007/978-3-7091-6892-9_14. [DOI] [PubMed] [Google Scholar]
- 148.Harrington CR, Louwagie J, Rossau R, Vanmechelen E, Perry RH, Perry EK, et al. Influence of apolipoprotein E genotype on senile dementia of the Alzheimer and Lewy body types. Significance for etiological theories of Alzheimer’s disease. Am J Pathol. 1994;145:1472–84. [PMC free article] [PubMed] [Google Scholar]
- 149.Hardy J, Crook R, Prihar G, Roberts G, Raghavan R, Perry R. Senile dementia of the Lewy body type has an apolipoprotein E epsilon 4 allele frequency intermediate between controls and Alzheimer’s disease. Neurosci Lett. 1994;182:1–2. doi: 10.1016/0304-3940(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 150.Nielsen AS, Ravid R, Kamphorst W, Jorgensen OS. Apolipoprotein E epsilon 4 in an autopsy series of various dementing disorders. J Alzheimers Dis. 2003;5:119–25. doi: 10.3233/jad-2003-5206. [DOI] [PubMed] [Google Scholar]
- 151.Carrillo Garcia F, Gil Neciga E, Alberca R, Garcia-Solis D, Millan J, Rodriguez Uranga JJ, et al. Apolipoprotein E4 in dementia with Lewy bodies. Neurologia. 2008;23:152–6. [PubMed] [Google Scholar]
- 152.Gallardo G, Schluter OM, Sudhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–8. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- 153.Kuroda MM, Weck ME, Sarwark JF, Hamidullah A, Wainwright MS. Association of apolipoprotein E genotype and cerebral palsy in children. Pediatrics. 2007;119:306–13. doi: 10.1542/peds.2006-1083. [DOI] [PubMed] [Google Scholar]
- 154.McMichael GL, Gibson CS, Goldwater PN, Haan EA, Priest K, Dekker GA, et al. Association between Apolipoprotein E genotype and cerebral palsy is not confirmed in a Caucasian population. Hum Genet. 2008;124:411–6. doi: 10.1007/s00439-008-0564-y. [DOI] [PubMed] [Google Scholar]
- 155.Panegyres PK, Beilby J, Bulsara M, Toufexis K, Wong C. A study of potential interactive genetic factors in Huntington’s disease. Eur Neurol. 2006;55:189–92. doi: 10.1159/000093867. [DOI] [PubMed] [Google Scholar]
- 156.Kehoe P, Krawczak M, Harper PS, Owen MJ, Jones AL. Age of onset in Huntington disease: sex specific influence of apolipoprotein E genotype and normal CAG repeat length. J Med Genet. 1999;36:108–11. [PMC free article] [PubMed] [Google Scholar]
- 157.Busch RM, Lineweaver TT, Naugle RI, Kim KH, Gong Y, Tilelli CQ, et al. ApoE-epsilon4 is associated with reduced memory in long-standing intractable temporal lobe epilepsy. Neurology. 2007;68:409–14. doi: 10.1212/01.wnl.0000253021.60887.db. [DOI] [PubMed] [Google Scholar]
- 158.Yeni SN, Ozkara C, Buyru N, Baykara O, Hanoglu L, Karaagac N, et al. Association between APOE polymorphisms and mesial temporal lobe epilepsy with hippocampal sclerosis. Eur J Neurol. 2005;12:103–7. doi: 10.1111/j.1468-1331.2004.00956.x. [DOI] [PubMed] [Google Scholar]
- 159.Agosta F, Vossel KA, Miller BL, Migliaccio R, Bonasera SJ, Filippi M, et al. Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc Natl Acad Sci U S A. 2009;106:2018–22. doi: 10.1073/pnas.0812697106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guo L, LaDu MJ, Van Eldik LJ. A dual role for apolipoprotein e in neuroinflammation: anti- and pro-inflammatory activity. J Mol Neurosci. 2004;23:205–12. doi: 10.1385/JMN:23:3:205. [DOI] [PubMed] [Google Scholar]
- 161.LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J, et al. Apolipoprotein E and apolipoprotein E receptors modulate A beta-induced glial neuroinflammatory responses. Neurochem Int. 2001;39:427–34. doi: 10.1016/s0197-0186(01)00050-x. [DOI] [PubMed] [Google Scholar]
- 162.Colton CA, Needham LK, Brown C, Cook D, Rasheed K, Burke JR, et al. APOE genotype-specific differences in human and mouse macrophage nitric oxide production. J Neuroimmunol. 2004;147:62–7. doi: 10.1016/j.jneuroim.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 163.Pedro-Botet J, Senti M, Nogues X, Rubies-Prat J, Roquer J, D’Olhaberriague L, et al. Lipoprotein and apolipoprotein profile in men with ischemic stroke. Role of lipoprotein(a), triglyceride-rich lipoproteins, and apolipoprotein E polymorphism. Stroke. 1992;23:1556–62. doi: 10.1161/01.str.23.11.1556. [DOI] [PubMed] [Google Scholar]
- 164.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–21. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 165.Miyata M, Smith JD. Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 166.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–2. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 167.Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, et al. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci U S A. 1998;95:10914–9. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Raber J, Wong D, Yu GQ, Buttini M, Mahley RW, Pitas RE, et al. Apolipoprotein E and cognitive performance. Nature. 2000;404:352–4. doi: 10.1038/35006165. [DOI] [PubMed] [Google Scholar]
- 169.Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18:390–8. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 170.Tolar M, Keller JN, Chan S, Mattson MP, Marques MA, Crutcher KA. Truncated apolipoprotein E (ApoE) causes increased intracellular calcium and may mediate ApoE neurotoxicity. J Neurosci. 1999;19:7100–10. doi: 10.1523/JNEUROSCI.19-16-07100.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Holtzman DM. Alzheimer’s disease: Moving towards a vaccine. Nature. 2008;454:418–20. doi: 10.1038/454418a. [DOI] [PubMed] [Google Scholar]
- 172.Tarawneh R, Holtzman DM. Biomarkers in translational research of Alzheimer’s disease. Neuropharmacology. 2010;59:310–22. doi: 10.1016/j.neuropharm.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]