

Abstract
Spiral ganglion neurons (SGNs) are postsynaptic to hair cells and project to the brainstem. The inner hair cell (IHC) to SGN synapse is susceptible to glutamate excitotoxicity and to acoustic trauma, with potentially adverse consequences to long-term SGN survival. We used a cochlear explant culture from P6 rat pups consisting of a portion of organ of Corti maintained intact with the corresponding portion of spiral ganglion to investigate excitotoxic damage to IHC–SGN synapses in vitro. The normal innervation pattern is preserved in vitro. Brief treatment with NMDA and kainate results in loss of IHC–SGN synapses and degeneration of the distal type 1 SGN peripheral axons, mimicking damage to SGN peripheral axons caused by excitotoxicity or noise in vivo. The number of IHC presynaptic ribbons is not significantly altered. Reinnervation of IHCs occurs and regenerating axons remain restricted to the IHC row. However, the number of postsynaptic densities (PSDs) does not fully recover and not all axons regrow to the IHCs. Addition of either neurotrophin-3 (NT-3) or BDNF increases axon growth and synaptogenesis. Selective blockade of endogenous NT-3 signaling with TrkC–IgG reduced regeneration of axons and PSDs, but TrkB–IgG, which blocks BDNF, has no such effect, indicating that endogenous NT-3 is necessary for SGN axon growth and synaptogenesis. Remarkably, TrkC–IgG reduced axon growth and synaptogenesis even in the presence of BDNF, indicating that endogenous NT-3 has a distinctive role, not mimicked by BDNF, in promoting SGN axon growth in the organ of Corti and synaptogenesis on IHCs.
Introduction
The peripheral axons (sometimes termed “dendrites”) of spiral ganglion neurons (SGNs) contact the hair cells of the organ of Corti (Slepecky, 1996). Approximately 95% of SGNs are type 1 and innervate the inner hair cells (IHCs). In the mature cochlea, each type 1 SGN contacts just a single IHC, but each IHC is contacted by 5–20 SGNs. Type 2 SGN peripheral axons project past the IHC row, and each innervates many outer hair cells (OHCs).
Innervation of the organ of Corti (OC) depends on neurotrophins BDNF and neurotrophin-3 (NT-3), which are expressed in the developing OC (Fritzsch et al., 2004). BDNF expression declines postnatally (Wheeler et al., 1994), but NT-3 remains expressed in mature IHCs and adjacent supporting cells (Ylikoski et al., 1993; Wheeler et al., 1994; Stankovic et al., 2004; Sugawara et al., 2007). TrkB and TrkC, the cognate receptors for BDNF and NT-3, respectively, are expressed in SGNs (Ylikoski et al., 1993; Pirvola et al., 1994, 1997; Mou et al., 1997). BDNF and NT-3 promote SGN neurite growth in vitro (Malgrange et al., 1996; Hegarty et al., 1997) and in vivo (Wise et al., 2005). However, the roles of the neurotrophins in cochlear synapse formation and maintenance have not been defined. Here we ask whether BDNF or NT-3 is necessary for SGN synaptogenesis in the cochlea. To do so, we take advantage of a reinnervation process in cultured postnatal rat cochlea explants, exploiting the accessibility of the culture system to experimentally manipulate neurotrophin signaling.
IHC–SGN synaptic transmission is excitatory and glutamatergic, with both NMDA and kainate/AMPA-type glutamate receptors in SGNs (Eybalin, 1993). Like other glutamatergic synapses, the IHC–SGN synapse is susceptible to excitotoxicity: intracochlear perfusion of glutamate agonists in vivo results in degeneration of SGN synaptic terminals on IHCs (Pujol et al., 1985, 1996; Gil-Loyzaga and Pujol, 1990; Puel et al., 1994; Ruel et al., 2007). Acoustic trauma results in similar damage to IHC–SGN synapses (Puel et al., 1998), presumably as a result of excessive glutamate release from IHCs because glutamate antagonists mitigate noise damage to the synapses (Puel et al., 1998), whereas inhibiting glutamate reuptake exacerbates noise damage (Hakuba et al., 2000).
We show here that excitotoxic damage to type 1 SGNs, loss of IHC–SGN synapses, and subsequent reinnervation of IHCs occur in vitro after exposure to glutamate. However, synaptic density after reinnervation is much lower than the original density. Exogenous BDNF or NT-3 enhances neurite growth and synaptogenesis. Nevertheless, by using Trk–IgG neurotrophin scavengers, we show, remarkably, that NT-3, the principal endogenous neurotrophin in the postnatal OC, distinctively promotes IHC–SGN synaptogenesis and cannot be replaced by BDNF.
Acoustic trauma results in delayed loss of SGNs (Kujawa and Liberman, 2006), possibly as a result of excitotoxic damage to SGN contacts with IHCs (Kujawa and Liberman, 2009), their principal source of neurotrophic support. Here, we provide a culture model for investigation of excitotoxic trauma to SGN postsynaptic terminals. The results show a distinctive role for NT-3 in mitigating damage, and the novel system may facilitate identification of therapeutic interventions for acoustic trauma.
Materials and Methods
Culture methods.
Protocols for all experiments involving animals were approved by the University Animal Care and Use Committee. Postnatal day 4 (P4) to P6 rat cochleae of both sexes were dissected in ice-cold PBS. The membranous labyrinth was separated from the modiolus and the stria vascularis and spiral ligament were carefully removed. With the organ of Corti and spiral ganglion kept intact, Reissner's membrane and tectorial membrane were removed with fine forceps. Only the middle part of the middle turn was used for each culture in our studies to avoid differing region-specific effects of neurotrophins. The explants were placed in a well on a cover glass (Lab-Tek catalog #155411 multichambered cover-glass system) coated with polyornithine (100 μg/ml) and laminin (20 μg/ml), to which they adhere. The cultures were evaluated the following day using differential interference contrast optics for adhesion to the substrate. Explants that failed to completely adhere to the coverslip were discarded; all others were used for these experiments.
Unless otherwise indicated, the explants were cultured in N2-supplemented DMEM with 10% fetal bovine serum at 37°, 6.5% CO2. The explants can be cultured for at least 2 weeks (data not shown); however, in this paper, we used cultures of, at most, 4 d in vitro (DIV). All procedures, including imaging, were performed in the culture well with the explants remaining attached to the coverslip. This facilitates preservation of the morphology.
To cause excitotoxic trauma, we exposed the explants, after 24 h in culture (1 DIV), to glutamate receptor agonists NMDA (0.5 mm; catalog #0114; Tocris Bioscience) and kainic acid (0.5 mm; #0222; Tocris Bioscience; or catalog #K2389; Sigma) for 2 h (“NK” treatment). After NK treatment, the explants were maintained up to 72 h (4 DIV in total) in control culture medium or in culture medium containing BDNF (3.6 nm; catalog #10908010; Invitrogen) or NT-3 (3.6 nm; catalog #N1905; Sigma) or TrkC–IgG protein (2 μg/ml) or TrkB–IgG protein (2 μg/ml). The cultures were then fixed and prepared for immunofluorescence.
Synthesis of TrkC–IgG and TrkB–IgG.
TrkC–IgG and TrkB–IgG (also known as TrkC–Fc and TrkB–Fc) are chimeric proteins consisting of the ectodomain of the Trk family receptors containing the ligand-binding domain fused to the Fc domain of human IgG. We have previously established efficacy and specificity of these reagents for cultured SGNs (Hansen et al., 2001). In particular, we have demonstrated that TrkC–IgG effectively blocks NT-3 signaling but not BDNF signaling (Hansen et al., 2001). A cDNA encoding a TrkC–IgG fusion protein (“receptor body” or “immunoadhesin”) was constructed similarly to that described by Shelton et al. (1995): the rat TrkC extracellular domain up to Thr429 was fused to the C-terminal portion of the human IgG Fc domain, starting at Asp216 (Ashkenazi et al., 1993). TrkB–IgG was constructed identically except that the rat TrkB extracellular domain up to His429 was fused to the C-terminal portion of the human IgG Fc domain. The genes encoding the fusion proteins were placed downstream of a human CMV early promoter. The constructs were transfected into HEK293 cells. After 24 h, the culture medium was changed to one lacking serum. After an additional 24 h, the supernatant culture medium was removed and passed through a protein A-Sepharose column to isolate the secreted TrkC–IgG or TrkB–IgG proteins. Protein concentration was adjusted to 0.5 mg/ml in culture medium by vacuum dialysis for storage at −86°. The stock TrkB–IgG or TrkC–IgG were added to cultures to a final concentration of 2 μg/ml (1:250 dilution). Specificity and efficacy were assessed by an in vitro SGN survival bioassay as in the study by Hansen et al. (2001).
Fluorescent labeling of cells.
All procedures were performed in the culture well with the explants remaining attached to the coverslip. After fixation with 4% paraformaldehyde for 15 min, the cultures were permeabilized with 1% Triton X-100 in PBS for 45 min, incubated 30 min in 50 mm NH4Cl, and 30 min in 20% horse serum in PBS. For immunofluorescence imaging, the cultures were incubated sequentially in primary and secondary antibodies diluted in 1% horse serum in PBS, primary antibodies for 24 h at 4° and secondary antibodies for 1 h at room temperature, with three intervening PBS washes. Primary antibodies used were as follows: anti-myosin VI (1:2000; catalog #KA-15; Sigma) to label hair cells, anti-NF200 (1:400; catalog #N0142; Sigma) to label the SGNs and their peripheral axons, anti-peripherin (1:2000; catalog #AB1530; Millipore Bioscience Research Reagents) to selectively label type 2 SGNs and their peripheral axons (Hafidi et al., 1993), anti-PSD-95 (mouse monoclonal IgG2a, 1:2000; catalog #MABN68 and #MAB1569; Millipore) to label postsynaptic densities (PSDs) in SGNs (Bao et al., 2004), and anti-CTBP2 (mouse monoclonal IgG1, 1:100; catalog #612044; BD Biosciences) to label ribbon synapses in hair cells (Khimich et al., 2005). Secondary antibodies were Alexa Fluor conjugates (1:1000; Invitrogen). For CTBP2/PSD-95 double labeling, isoform-specific secondary antibodies were used sequentially to distinguish the two mouse primary antibodies.
Hair cell apical structures were labeled with phalloidin-647 (1:40; catalog #A22287; Invitrogen). Viable hair cells were labeled by uptake of the fixable fluorescent styryl dye AM1-43 (catalog #70024; Biotium) (Meyers et al., 2003): explants were cultured, before fixation, in medium containing 30 mm KCl and 40 nm AM1-43 at 37°/5% CO2 for 1 min. The explants were then washed twice (5 min each) with DMEM and fixed.
Quantitative analysis of SGN neurites and synapses in digital images.
After each explant was fluorescently labeled to detect SGN somata and axons, IHCs, synapses, or other structures, the explants were viewed in the culture well using a Leica TCS P2 Spectra confocal system with Leica DMIRE2 inverted microscope. Optical sections in the x–y plane (z-sections) were generally imaged and recorded at 0.6 μm intervals in the z-axis. The resulting confocal image series (z-stack) contains a three-dimensional record of the imaged information in the entire volume of the explant. The z-stack was viewed, rotated, and “resliced” (to view a plane perpendicular to the x–y plane) as necessary using Leica CS software or NIH ImageJ software (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Thus, a sequence of planes or optical “slices” perpendicular to any of the axes was generated, allowing us to follow individual axons along their length, identify IHCs contacted by each axon, and identify synapses formed by each IHC on each axon. We counted all SGN–IHC contacts and synapses in the volume. Two structures were considered to be in contact if they touched within the same z-section (i.e., within 0.6 μm). Every IHC, NF200-labeled fiber, fiber–IHC contact, and PSD-95 punctum (>1 pixel in size) was counted within randomly chosen volumes of organ of Corti, each volume having a length of 10–15 IHCs.
PSDs were visualized as PSD-95 immunoreactive puncta. Innervation was quantified by counting the number of PSDs on each IHC in z-stacks containing the bases of the IHCs. We can resolve individual PSDs in the stacks and count them using an unbiased stereological method, in this case an optical disector (Mouton, 2002). In NIH ImageJ, each PSD is counted in the first optical section in which it appears in focus, and the location is marked to prevent its being counted in subsequent sections. Supplemental Figure 1B–D (available at www.jneurosci.org as supplemental material) illustrates the counting method.
Axonal growth is quantified as the average number of NF200-labeled fibers in contact with an IHC and is obtained by counting contacts in multiple confocal series, each containing 10–15 hair cells. This number is graphed (see Figs. 2E, 5E, 8) as fibers/IHC. The number of synapses made by each IHC was quantified by counting PSD-95 puncta in contact (≤0.6 μm distance) with the IHC. This number is graphed (see Fig. 7) as PSD-95 puncta/IHC. The number of IHCs contacted by each fiber was determined by following a fiber in the 3D reconstructions and counting the IHCs with which it makes contact. This is given in supplemental Figure 3 (available at www.jneurosci.org as supplemental material) as IHCs/fiber. In summary, for each post-NK experimental condition, we quantified PSDs/IHC, fibers/IHC, and IHCs/fiber for two to three separate confocal series in each explant. The number of confocal series used for each experimental condition is in the corresponding column in each figure. One explant was prepared from the middle turn of each cochlea. In the event that both cochleae were used from a single rat, they were placed in different experimental or control groups.
Figure 2.

Type 1, but not type 2, SGN peripheral processes degenerate after excitotoxic trauma. A–D, Explants shown were fixed at 1 DIV, immediately after NK treatment. As in Figure 1B–D, hair cells are labeled with anti-myosin VI (blue), type 1 and type 2 SGN peripheral processes with anti-NF200 (A, B, D, red), and type 2 SGN peripheral axons with anti-peripherin (C, D, green). Merged NF200 and peripherin images are shown in D. A, Low-magnification z-projection image (scale bar, 20 μm) showing damaged SGN peripheral processes and disrupted contacts with inner hair cells. A dotted line is drawn through the bases of the IHCs to indicate the gap between the IHCs and the axon ends. B–D, Higher-magnification images (scale bar, 10 μm) showing that damaged fibers are peripherin negative (type 1), whereas the intact fibers in the radial bundles are peripherin positive (type 2). E, Counts of contacts between SGN axons and IHCs in 1 DIV cultures immediately after 2 h NMDA/kainate (NK) or kainate (KA) treatment or no-NK controls. Shown are means ± SEM.
Figure 5.

BDNF and NT-3 increase the number of initial SGN–IHC contacts 18 h after excitotoxic trauma. A–D, Representative images of explants 18 h after NK. Scale bar, 20 μm. Hair cells, SGN peripheral axons, and PSDs were visualized by immunofluorescence as in Figure 6. A, A non-NK-treated control 2 DIV explant. B, An explant maintained 18 h after NK in medium without added neurotrophins. C, An explant maintained 18 h after NK in medium containing 3.6 nm NT-3. D, An explant maintained 18 h after NK in medium containing 3.6 nm BDNF. SGN–IHC contacts were counted in 3D reconstructions of confocal stacks. The average number of axons per IHC is shown in E for 2 DIV non-NK-treated control explants (2 DIV no NK), explants maintained 18 h after NK in medium without added neurotrophins (NK/no NT), explants maintained 18 h after NK in medium containing 3.6 nm NT-3 (NK/NT-3), and explants maintained 18 h after NK in medium containing 3.6 nm BDNF (NK/BDNF). NK/NT-3 and NK/BDNF are significantly different from NK/no NT and from no NK (p < 0.01, ANOVA) but are not different from each other.
Figure 8.

Quantitation of SGN neurite regrowth in the organ of Corti. Neurite regrowth is quantified by counting contacts between IHCs and NF200-immunofluorescent fibers, comparing cultures maintained for 72 h after NK without neurotrophins (no NT), with BDNF or NT-3, with TrkB–IgG or TrkC–IgG fusion proteins, or maintained similarly 4 DIV but with no excitotoxic treatment. Statistical analysis was done by Kruskal–Wallis (nonparametric ANOVA) test and Dunn's post hoc comparison using GraphPad InStat software. The number of independent confocal series, each 10–15 hair cells in width, is given as n. Columns under the same horizontal bar are not significantly different from each other. ap < 0.005 indicates a significant difference between these two groups, except the significance of the difference between NK/BDNF and NK/BDNF/TrkC–IgG is p = 0.01. bp < 0.0001 indicates a significant difference between NK/TrkC–IgG and all other conditions.
Figure 7.

Quantitation of synapse regeneration in the organ of Corti. PSD-95 puncta were counted on each inner hair cell, comparing cultures maintained for 72 h after NK without neurotrophins (no NT), with BDNF or NT-3, with TrkB–IgG or TrkC–IgG fusion proteins, or maintained similarly 4 DIV but with no excitotoxic treatment. Statistical analysis was done by Kruskal–Wallis (nonparametric ANOVA) test and Dunn's post hoc comparison using GraphPad InStat software. The number of independent confocal series, each 10–15 hair cells in width, is given as n. Columns under the same horizontal bar are not significantly different from each other. ap < 0.0001 indicates a significant difference between 4 DIV control and all other groups for all comparisons, except between 4 DIV control and NK/NT-3 for which p = 0.0005. bp < 0.001 indicates significant differences between these two groups for differences between NK/NT-3 and all members of the other group; p < 0.01 for differences between NK/BDNF and all members of the other group, except between NK/BDNF and NK/TrkC–IgG/BDNF for which p = 0.03. cp < 0.03 indicates a significant difference between NK/TrkC–IgG and the three members of the adjacent group; the difference between NK/TrkC–IgG and all other conditions is significant with p < 0.0001.
Assessment of SGN survival.
Dying cells were labeled using the vital fluorescent dye Sytox green (catalog #S-7020; Invitrogen). Explants were incubated with Sytox green (diluted 1:40,000 in culture medium from a 5 mm stock) at 37°C for 20 min, washed (5 min) with culture medium, and fixed. Ouabain was purchased from Sigma (catalog #O3125). TUNEL, to detect apoptotic cells, was performed essentially as we have described previously (Alam et al., 2007) after fixation and permeabilization. Immunohistochemistry was done after the TUNEL procedure.
SGNs were counted using an optical disector method (Mouton, 2002, their Ch. 6) in explants that had been fixed, permeabilized, and labeled with anti-NF200. A z-stack was made of the spiral ganglion region of the explant at 0.8 μm intervals on the z-axis; the typical thickness was 35 μm, yielding ≈45 optical sections in each z-stack. Square or rectangular areas (typically ≈150 × 150 μm) were randomly selected in the z-stack. Within each selected area, we viewed the z-sections in sequence, counting each nucleus in the first section in which it appeared in focus. This number was divided by the total volume to obtain the SGN density. The densities in two to four volumes were averaged for each explant, with three to four explants per group.
Results
An organotypic cochlea culture preserves synaptic interactions between SGNs and hair cells
The number of afferent synapses on each IHC varies according to location, with a greater number in the middle than in the apex or base (Meyer et al., 2009). Moreover, SGNs are known to vary in their physiology depending on their location in the cochlea, in a neurotrophin-dependent manner (Adamson et al., 2002; Zhou et al., 2005). We therefore restricted our study to the middle of the cochlea to minimize any location-dependent effects.
In a typical organotypic explant, a segment of the organ of Corti with one intact row of IHCs and three intact rows of OHCs, with the corresponding SGN somata and axons, were visualized. Similar to the arrangement in vivo, each IHC is contacted by multiple type 1 SGN axons, labeled by anti-NF200, each of which follows a radial trajectory terminating at the IHCs (Fig. 1A,B). OHCs are innervated by type 2 SGNs, which are selectively labeled by anti-peripherin antibodies (Hafidi et al., 1993) (Fig. 1C). As in vivo, each type 2 SGN axon passes the IHC row and then takes a spiral trajectory in apparent contact with multiple outer hair cells in the same row. Peripherin and NF200 immunofluorescence are overlaid in Figure 1B–D to show the trajectories of the two types of fibers in the explant, confirming that they have not been disrupted by culture. We did no additional quantitation of synapses between OHCs and type 2 SGNs because they were apparently unaffected by the excitotoxic agents used in this study, by these excitotoxic agents in vivo (Pujol et al., 1985; Puel et al., 1995; Pujol and Puel, 1999), and by noise (Kujawa and Liberman, 2009).
Figure 1.
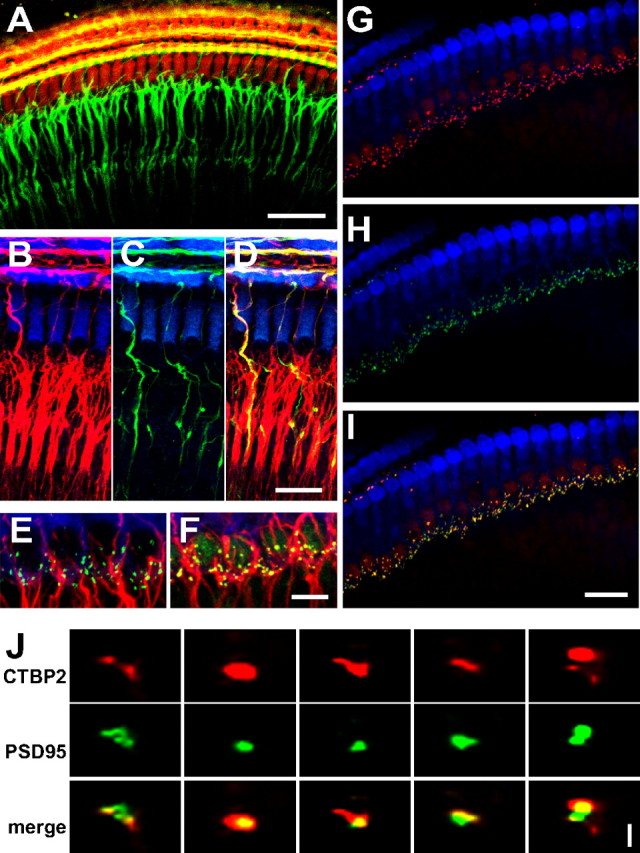
Synaptic interactions between hair cells and SGNs are preserved in organotypic culture. Organotypic cochlear explants, composed of portions of the organ of Corti and associated spiral ganglion, prepared from the middle turn of P4–P6 rat pups and cultured up to 4 d. Hair cells are labeled with anti-myosin VI (red in A, blue in B–I) and SGN peripheral axons with anti-NF200 (green in A, red in B–F). A, Low-magnification projection image (scale bar, 50 μm) of a confocal series through an explant, including the IHC row and the three OHC rows, showing the characteristic innervation pattern. Type 1 SGN peripheral processes are the radial fibers that innervate the IHC row; type 2 SGN peripheral processes continue past the IHC row and enter one of the three spiral bundles to contact OHCs in one of the rows. B–D, High-magnification projection images (scale bar, 20 μm) of an explant labeled with anti-NF200 (B, red) and anti-peripherin (C, green)—merged in D (yellow)—to confirm that fibers bypassing the IHCs and entering the spiral bundles are type 2 (peripherin-positive). E, F, High-magnification projection images (scale bar, 10 μm) of an explant with hair cells and SGN peripheral axons labeled as in B. E, PSDs labeled by anti-PSD-95 (green) are on the SGN peripheral processes closely apposed to the basolateral region of the IHCs. F, Presynaptic ribbons in the IHCs, labeled by anti-CTBP2 (green), are on the basolateral region of the IHCs closely apposed to the SGN peripheral axons. G–I, Colocalization of presynaptic ribbons (CTBP2-immunoreactive puncta, red, shown in G) and postsynaptic densities (PSD-95-immunoreactive puncta, green, shown in H) superimposed together in I on myosin VI immunofluorescence (blue). Scale bar, 20 μm. J, Five examples of colocalized presynaptic ribbons and PSDs shown at higher magnification (scale bar, 2 μm). These are in the y–z plane, resliced from confocal z-stacks.
Because this study focuses on the postsynaptic response to excitotoxic trauma, we quantified PSDs for all experimental conditions. These were visualized by anti-PSD-95 immunoreactivity (Bao et al., 2004), shown overlaid on myosin 6 and NF200 immunofluorescence in Figure 1E. In some cases, we labeled presynaptic ribbons with anti-CTBP2 (Khimich et al., 2005) (which also labels hair cell nuclei.) CTBP2 immunoreactivity is shown overlaid on myosin 6 and NF200 immunofluorescence in Figure 1F. We counted CTBP2 and PSD-95 puncta, as described in Materials and Methods (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), in 64 IHCs in nine confocal series each taken from the midpoint of a separate explant after 1 DIV. We obtained counts of 20.67 ± 2.68 CTBP2 puncta/IHC and 19.61 ± 4.89 PSD-95 puncta/IHC. Colocalization of PSDs (PSD-95 puncta) and presynaptic ribbons (CTBP2 puncta) in control explants is shown in Figure 1G–I, which demonstrates that synapses remain intact after transfer of the organ of Corti to culture. Figure 1J shows five examples of ribbon-PSD pairs at higher magnification, illustrating typical variation in size, shape, and relative position in the reconstructions from confocal microscope optical slices.
We found in control explants that every PSD colocalized with a ribbon, but ≈1 ribbon/IHC does not colocalize with a PSD. However, as discussed below, after excitotoxic trauma, the number of ribbons not colocalizing with PSDs increases. The numbers of PSDs and ribbons that we obtained in control 1 DIV cultures are similar to counts of nerve terminals or of synaptic bodies in mouse cochleae obtained using disector pairs, another unbiased method, by Francis et al. (2006), to counts of nerve terminals obtained in 3D reconstructions of mouse cochleae (Francis et al., 2004), and to counts of ribbons (Kujawa and Liberman, 2009; Meyer et al., 2009) in mouse cochleae.
The number of PSDs is much greater than the average number of fibers we counted in contact with each IHC. In explants after 1 DIV, 3.5 ± 0.5 NF200-labeled fibers contact each IHC, based on counting contacts on 319 IHCs in 19 different confocal series from seven cochleae (Fig. 2E). There may be several reasons for this discrepancy. Possibly, some SGN axons make more than one synapse with an IHC. Also, we are not always able to resolve individual axons in tight bundles, so that some NF200-labeled fibers, particularly in control cultures, may be small bundles of axons rather than individual axons. Because of the uncertainty in the exact number of axons, we rely, in this study, on the number of PSDs for quantitative conclusions about synapse number. The number of fibers is always correlated with the number of PSDs and supports the quantitative results but is, for this purpose, a qualitative measure.
The organ of Corti is also innervated by olivocochlear efferent fibers; however, these degenerate after dissection of the cochlea. We verified absence of efferents by using an anti-choline acetyltransferase antibody that detected efferents in vivo (data not shown).
A brief application of glutamate agonists causes rapid degeneration of type 1 SGN synapses on IHCs, mimicking in vivo excitotoxicity
To cause excitotoxic trauma, we used NK treatment of 1 DIV cultures with NMDA and kainate, as described in Materials and Methods. This caused rapid degeneration of type 1 SGN peripheral axons. By the end of the 2 h NK treatment, >99% of type 1 (radial, peripherin-negative) axons no longer contact IHCs (Fig. 2A) (see also Fig. 4A). This is illustrated in Figure 2A by a dotted line drawn through the bases of the IHCs in the row, showing a gap of 10–20 μm between the dotted line and the ends of the type 1 SGN fibers. In contrast, type 2 (spiral, peripherin-positive) SGN fibers remain intact after NK treatment (Fig. 2A–D), as had been observed in vivo (Pujol et al., 1985). In quantitative terms, of 606 type 1 SGN fibers scored in nine confocal stacks from three separate NK-treated cochleae, only five remained in contact with IHCs (Fig. 2E). Also, as observed in vivo, transient excitotoxicity from exposure to glutamate agonists did not cause acute death of SGNs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 4.
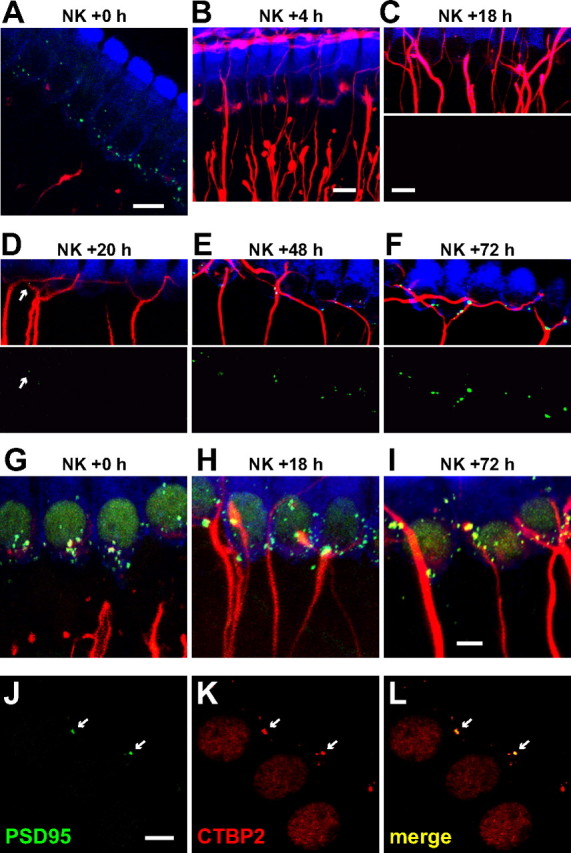
Neurite and synapse recovery after NK treatment. Explants were maintained in culture up to 72 h after NK before fixation. In A–F, hair cells are labeled with anti-myosin VI (blue), SGN peripheral processes with anti-NF200 (red), and postsynaptic densities with anti-PSD-95 (green). A, A single confocal image (optical slice) of an explant fixed immediately after NK exposure. B, z-Projection of a confocal image stack of IHCs in an explant fixed 4 h after the end of NK exposure. C–F, z-Projections of confocal image stacks of IHCs in explants fixed at the indicated times after the end of NK exposure: 18 h (C), 20 h (D), 48 h (E), and 72 h (F). The PSD-95 puncta are shown superimposed on the hair cells and SGNs in the top and separately below. The arrow in D indicates a PSD-95 punctum; puncta are first observed at 20 h after NK. G–I, z-Projections of confocal image stacks of IHCs in explants fixed immediately (G), 18 h (H), or 72 h (I) after NK exposure. Hair cells and SGN axons labeled as in A–F; presynaptic ribbons labeled with anti-CTBP2 (green). Not all CTBP2 puncta are associated with SGN axons. J–L, Confocal images of inner hair cells 72 h after NK showing juxtaposition of presynaptic and postsynaptic densities. J, PSDs only. K, IHC nuclei and synaptic ribbons labeled with anti-CTBP2. L, Merged presynaptic and postsynaptic labeling. Arrows mark location of two labeled PSDs. All PSDs colocalize with ribbons but not all ribbons colocalize with PSDs. Scale bars: A–F, 10 μm; G–L, 5 μm.
Puel et al. (1994) showed that excitotoxic damage to the postsynaptic terminals in vivo is attributable primarily to non-NMDA receptors. We therefore tested the effects of kainate alone. Treatment with 0.5 mm kainate for 2 h caused loss of terminals on IHCs comparable with the loss caused by NK treatment (Fig. 2E). These data suggest that NMDA is not necessary for excitotoxic damage. Nevertheless, experiments here were performed with both NMDA and kainate to more precisely mimic the response of SGNs to glutamate.
Presynaptic changes after transient excitotoxic trauma
Transient excitotoxic trauma in vivo does not result in loss of hair cell or SGN viability so we asked whether this was also the case for NK treatment of organotypic cultures. Although higher concentrations of NMDA and kainate (e.g., 100 mm) and long exposures resulted in death of IHCs (data not shown), the NK treatment used here did not cause hair cell death. Because of the precise arrangement of IHCs and OHCs in aligned rows, loss of individual hair cells is easily detected as a gap in the row. Such gaps were not observed, indicating that hair cells do not die during the course of these experiments (up to 4 DIV), even after NK treatment (Fig. 3).
Figure 3.

Presynaptic changes in IHCs after NK treatment in vitro. Viability of hair cells 72 h after NK (4 DIV in total) is shown by uptake of AM1-43 dye by IHCs (A) and OHCs (B). Hair cell stereocilia bundles are visualized by phalloidin labeling (green) in C: the top only the stereocilia, and the bottom shows the stereocilia superimposed on myosin VI immunoreactivity (red), allowing the hair cell to be visualized. Explants were maintained in culture up to 72 h after NK before fixation. D–F are 2D z-projections of confocal image stacks showing inner hair cells, labeled with anti-myosin VI (red), fixed immediately after NK treatment (D), 18 h after NK (E), or 72 h after NK (F). Presynaptic ribbons are labeled with anti-CTBP2 (green). Quantitation of synaptic ribbons (CTBP2-immunoreactive puncta) in confocal image stacks is plotted (mean ± SD) in G for the indicated number of control (no NK) explants and of explants immediately after NK (NK0), 18 h after NK (NK18), and 72 h after NK (NK72). Typical and apical ribbons are defined in Results. Statistical comparison was done by one-way ANOVA with Tukey–Kramer multiple comparisons test. Significant differences are indicated as *p < 0.05 or ***p < 0.001, except that the difference between NK0 and NK72 apical ribbons is p < 0.05 and not p < 0.001. Note that there is no significant difference in the total number of ribbons among the time points, but there is a statistically significant shift from typical to apical location by 72 h after NK. Scale bars: A, B, 15 μm; C–F, 10 μm.
We further asked whether the hair cells remained functionally and structurally sound. The styryl dye FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] and its fixable analog AM1-43 are selectively taken up by hair cells via the nonselective cation mechanotransduction channels and can be used to verify the presence of functional channels (Géléoc and Holt, 2003; Meyers et al., 2003). We incubated organotypic cultures 72 h after NK in AM1-43, as described in Materials and Methods, and observed selective uptake by hair cells, both IHCs (Fig. 3A) and OHCs (Fig. 3B). This indicates that the hair cells retain functional mechanotransduction channels. Structural integrity at 72 h after NK was verified by labeling with fluorescent phalloidin to show intact stereocilia on both IHCs and OHCs (Fig. 3C).
Presynaptic ribbons remained present in the hair cells immediately after NK treatment (Fig. 3D), 18 h after NK (Fig. 3E), and 72 h after NK (Fig. 3F), although PSDs are absent 18 h after NK (Fig. 4C). That is, presynaptic structures persist in the hair cells even in the absence of a neuronal PSD. We quantified presynaptic effects of excitotoxic trauma, with regard to number and location of synaptic ribbons, visualized as CTBP2-immunoreactive puncta in IHCs. We counted ribbons in six to eight IHCs at the middle point of each explant in confocal z-stacks using the same optical disector method described for counting PSDs (supplemental Fig. 1B–D, available at www.jneurosci.org as supplemental material). Ribbons were counted in the indicated number of control (no NK) explants and of explants immediately after NK, 18 h after NK, and 72 h after NK. The mean numbers of ribbons/IHC are shown in Figure 3G. With regard to the total number of synaptic ribbons/IHC, there is no significant difference between the no-NK control group and any post-NK group. However, similar to what was observed after acoustic trauma in vivo (Kujawa and Liberman, 2009), we did observe a change in the spatial distribution of ribbons within the IHC (Fig. 3D–F). In IHCs in control explants, ≈95% of the CTBP2-immunoreactive puncta are located near the basal surface of the cell, mainly below or surrounding the cell nucleus (20.0 ± 2.9 of 21.0 ± 2.7 ribbons/IHC from 70 IHCs in 10 explants). We term ribbons in this location “typical” ribbons. Only 5% of the CTBP2-immunoreactive puncta are located above the cell nucleus in the base to apex axis of the cell, a location we term “apical.” Although there was no significant decrease in the percentage of ribbons that are typical or apical immediately after (NK0) or 18 h after (NK18) excitotoxic trauma, at 72 h after NK, a significant shift from typical to apical was apparent (Fig. 3G): the number of apical ribbons increased from 1.00 ± 0.70 to 5.6 ± 2.5 ribbons/IHC.
In contrast to these observations, Kujawa and Liberman (2009) reported that the number of CTBP2 puncta declines in IHCs after noise exposure. This might be a consequence of the difference between noise and in vitro exposure to glutamate agonists with respect to their effects on hair cells. Hair cells are directly responsive to sound, and the loss of ribbons after noise may be a result of damage to the IHCs caused directly by the noise. Noise damage to SGN synaptic terminals is attributable to excess glutamate released by the IHCs (Puel et al., 1998) so is an indirect consequence of noise. NK exposure in vitro mimics noise-induced excitotoxic damage to the synaptic terminals but cannot mimic direct effects of noise on hair cells. This could account for the persistence of ribbons after excitotoxic trauma but not after noise.
Recovery from excitotoxic trauma
Although the distal ends of the type 1 SGN peripheral axons degenerate, some NF200-immunoreactive material (Fig. 4B) as well as many PSD-95 immunoreactive puncta (Fig. 4A) remain at the end of NK treatment, apparently as debris associated with the base of the IHC. This resembles in vivo excitotoxic trauma in which debris, including PSDs and postsynaptic membrane, remains temporarily at the base of the IHCs, evident by electron microscopy (Puel et al., 1994, their Fig. 1F). Most PSD debris is cleared by 4 h after NK (Fig. 4B), and, by 18 h after NK, PSD debris is not evident (Fig. 4C). Because regenerating peripheral processes are just beginning to contact the IHC row at 18 h after NK (Fig. 4C), PSDs seen at later times (Fig. 4D–F) must be new PSDs because of reinnervation. Presynaptic densities within the IHCs—visualized as CTBP2-immunoreactive puncta—are retained after NK (Fig. 3D–G), although postsynaptic partners are absent. However, in the absence of PSDs, ribbons may become mislocalized within the IHC (Fig. 3D–G).
Synapse regeneration after glutamate agonist- or noise-induced loss of synapses on IHCs has been reported to occur in vivo (Pujol et al., 1996; Puel et al., 1998; Pujol and Puel, 1999). We asked whether reinnervation of IHCs occurs in the in vitro model. After NK treatment, the explants were maintained in culture for various times up to 96 h. SGN–IHC contacts and PSD-95 puncta were counted in 3D reconstructions of confocal z-stacks.
At 4 h after NK, few contacts are observed between SGN peripheral axons and IHCs (Fig. 4B). Axonal regrowth is occurring because, by 18 h after NK, SGN peripheral axons are in contact with IHCs (Fig. 4C). However, at 18 h after NK, PSD-95 puncta are not detectable, indicating that debris from preexisting PSDs has been cleared by this time and that new synapses have not yet formed. Some new PSDs can be observed by 20 h after NK, although they are rare at this time (Fig. 4D). PSD number is greater at 48 and 72 h after NK (Fig. 4E,F). By 72 h after NK, most IHCs have established synaptic contact with at least one SGN fiber and have closely apposed PSDs (Fig. 4F).
The reinnervated pattern exhibits at least two significant differences from the original pattern, both evident in Figure 4F. First, innervation of the IHCs is much sparser than in control non-NK-treated explants, with respect to the number of PSDs and to the number of fibers that contact IHCs. Because the total number of synaptic ribbons does not decline significantly after trauma (Fig. 3G), this implies that there must be ribbons not apposed to PSDs. Indeed, Figure 4G–I shows that not all CTBP2 puncta are in contact with SGN fibers. Figure 4J–L shows colabeling of CTBP2 and PSD-95: there are fewer PSDs than ribbons puncta, and not all of the latter are colocalized with PSDs. All of the PSDs are colocalized with CTBP2 puncta. Thus, although fewer in number than the original PSDs, the regenerated PSDs directly appose ribbons and, presumably, represent structurally intact synapses.
A second difference from the original pattern is that the SGN fibers appear to contact multiple IHCs rather than just one (quantified in supplemental Fig. 3, available at www.jneurosci.org as supplemental material). At 72 h after NK, we observed SGN fibers running in a spiral direction along the bases of IHCs and contacting multiple IHCs at the base of each. Although it is difficult to follow individual axons and verify that a given axon synapses on more than one IHC, SGN fibers with such trajectories orthogonal to the normal radial trajectory were never observed in control non-NK-exposed explants.
Exogenous neurotrophins BDNF and NT-3 promote reinnervation of IHCs
As noted above, by 18 h after NK, regrowing type 1 SGN peripheral axons have reached the IHC row and are in contact (i.e., within 0.6 μm, the limit of our resolution in the z-axis) with IHCs, although no PSDs are evident at this time. Representative images of contacts between IHCs and NF200-immunoreactive fibers at 18 h after NK are shown in Figure 5A–D and quantified in Figure 5E. The number of fiber–IHC contacts at 18 h after NK is 0.88 ± 0.68, significantly less than the number of fiber–IHC contacts in non-NK-treated explants cultured for a similar length of time, 2 DIV, which is 3.33 ± 0.41.
As is evident from the images, addition of neurotrophins BDNF or NT-3 to the culture medium during the post-NK recovery increases axonal regrowth after NK. That is, the number of fiber–IHC contacts is significantly (p < 0.01, ANOVA) greater in the presence than in the absence of neurotrophins (Fig. 5E). Nevertheless, even with the addition of neurotrophins, the number of fiber–IHC contacts is not restored to levels seen in control explants not subjected to excitotoxic trauma (no-NK).
Exogenous neurotrophins BDNF and NT-3 promote IHC–SGN synaptogenesis
We next quantified reinnervation of IHCs, allowing 72 h for reinnervation after NK treatment. Because the excitotoxic trauma is applied at 24 h in vitro, these cultures were 4 DIV total. The number of PSDs in control non-NK-treated explants at 4 DIV is lower than at 1 DIV, suggesting that there may be some PSD loss in older cultures despite the lack of SGN death. By 72 h after NK, new synapses have formed, but the number is significantly less than in control non-NK-treated explants (Figs. 6, 7). In control explants, there are 12.3 ± 4.6 PSDs/IHC, and 91% of the IHCs have more than five PSD-95 puncta; in NK-treated explants, there are 1.5 ± 1.7 PSDs/IHC, <50% of the IHCs have more than one PSD punctum, and 36% of the IHCs had no PSD-95 puncta at all (Fig. 7). We compared the number of PSD-95 puncta among NK-treated cultures maintained in BDNF, NT-3, or no neurotrophin 72 h after NK and explants not exposed to NK (no-NK) but also maintained 4 DIV. The number of PSDs is significantly increased by either BDNF (4.0 ± 3.2 PSDs/IHC) or NT-3 (5.3 ± 3.3 PSDs/IHC), with the two neurotrophins exhibiting approximately equal effectiveness (Figs. 6, 7). Even so, the number of PSD-95 puncta in neurotrophin-treated explants 72 h after NK remains significantly less than in control non-NK-treated explants. Thus, after excitotoxic trauma, the original innervation is not quantitatively regenerated, even with the addition of neurotrophins.
Figure 6.

Endogenous NT-3 drives synapse regeneration after excitotoxic trauma. Shown are representative images of 4 DIV explants. Hair cells are labeled with anti-myosin VI (blue), SGN peripheral axons with anti-NF200 (red), and postsynaptic densities with anti-PSD-95 (green). A, Low-magnification image (scale bar, 50 μm) of a non-NK-treated control explant. The area enclosed by the dotted outline is shown at higher magnification in B to illustrate the IHC–SGN synapses at the bases of the inner hair cells. In B–H, superimposed myosin VI, NF200, and PSD-95 immunofluorescence is shown in the top and PSD-95 puncta only in the bottom. C–H, Explants cultured for 72 h after NK under the indicated conditions: C, control medium with no neurotrophins added; D, 3.6 nm NT-3; E, 3.6 nm BDNF; F, 2 μg/ml TrkB–IgG; G, 2 μg/ml TrkC–IgG; H, 2 μg/ml TrkC–IgG; and 3.6 nm BDNF. Scale bar: B–H, 20 μm.
The number of fiber–IHC contacts increases only slightly between 18 and 72 h after NK (compare Figs. 5, 8) and remains much sparser than in control (no-NK) explants. For example, in the 72 h after NK explants, on average, each IHC is contacted by 1.3 ± 0.7 fibers and 18% of IHCs have no contacts, but in control 4 DIV explants, each IHC is contacted by 2.8 ± 0.8 fibers and no IHCs completely lack contacts (Figs. 6, 8). We compared the number of fiber–IHC contacts among NK-treated cultures maintained with BDNF, with NT-3, or with no neurotrophin 72 h after NK and cultures not exposed to NK (no-NK) but maintained 4 DIV. The number of fiber–IHC contacts is significantly and approximately equally increased by the addition of either BDNF or NT-3 and is not significantly smaller than in control cultures not subjected to excitotoxic trauma (Fig. 8). Counts of PSDs/IHC provide the quantitative basis for our conclusion, but the fiber/IHC counts are qualitatively consistent with the counts of PSDs and corroborate them: specifically, reinnervation after NK does not fully restore the original innervation, but BDNF and NT-3 improve reinnervation by increasing the number of fiber–IHC contacts and the number of PSDs on IHCs.
Endogenous NT-3 is distinctively required for reinnervation and synaptogenesis
Postnatally, NT-3 is produced in inner hair cells and adjacent supporting cells (Ylikoski et al., 1993; Sugawara et al., 2007). Having established that neurotrophins promote reinnervation and synaptogenesis, we asked whether endogenous neurotrophins are necessary. To block endogenous neurotrophins, we synthesized and used chimeric proteins consisting of the extracellular ligand-binding domains of TrkB or TrkC fused to the Fc domain of human IgG (Shelton et al., 1995), which dimerizes the proteins. The TrkB–IgG and TrkC–IgG fusion proteins are soluble high-affinity receptors for, respectively, BDNF and NT-3 and sequester the neurotrophins, preventing their binding to cell-surface TrkB or TrkC. We have shown previously that these reagents specifically and effectively antagonize BDNF and NT-3 in spiral ganglion cultures even when the neurotrophins are added at high (3.6 nm) concentration and that TrkC–IgG has no effect on BDNF signaling (Hansen et al., 2001). They are also referred to as TrkB–Fc and TrkC–Fc, but we have used the terms TrkB–IgG and TrkC–IgG in previous publications and continue to do so to maintain consistency.
We exposed 1 DIV cochlear explant cultures to NK and allowed 72 h for recovery, as above, but included TrkB–IgG, or TrkC–IgG during the recovery period to sequester endogenous BDNF or NT-3. We counted PSD-95-immunoreactive puncta on IHCs to test the requirements for endogenous neurotrophins in synaptogenesis (Figs. 6F–H, 7). We counted fiber–IHC contacts to test the requirement for endogenous neurotrophins in reinnervation and axon growth (Figs. 6F–H, 8).
Comparison among Figure 6, C, F, and G, suggests that, although the numbers of PSDs and of fiber–IHC contacts are relatively small after 72 h recovery in the absence of neurotrophins (Fig. 6C), they are further reduced by the inclusion of TrkC–IgG during the recovery period (Fig. 6G). In contrast, no significant reduction was observed with inclusion of TrkB–IgG (Fig. 6F). Figure 7 shows quantitation of PSDs in multiple cochlea explant cultures and confirms that TrkC–IgG significantly (p < 0.03, Kruskal–Wallis test) reduces the number of PSDs formed during reinnervation, whereas TrkB–IgG has no significant effect. Similarly, the quantitation shown in Figure 8 confirms what is suggested by the images, that TrkC–IgG significantly (p < 0.0001, Kruskal–Wallis test) reduces SGN peripheral axon growth into the organ of Corti, although TrkB–IgG has no such effect.
These data indicate that NT-3 produced in the organ of Corti, presumably in the IHCs and adjacent supporting cells, contributes significantly to axonal regrowth and synaptogenesis. We considered two possible reasons for this finding. First, NT-3 in the organ of Corti may be contributing generally to making the environment conducive to axonal growth and synaptogenesis but not necessarily acting as a specific guidance or synapse-promoting signal between IHC and axon. Alternatively, the action of NT-3 may be specific, with NT-3 acting as a direct, localized, chemotropic signal between IHCs and SGN peripheral axons: guiding the axons to the IHCs and promoting the formation of synapses.
To distinguish between these possibilities, we asked whether addition of TrkC–IgG would inhibit axon growth and synaptogenesis in the presence of high levels of BDNF. When added to cultures, BDNF promotes PSD formation as effectively as NT-3 (Figs. 6, 7) and promotes SGN neurite growth as effectively as NT-3 (Hegarty et al., 1997) (Figs. 6, 8). So, if NT-3 serves only to promote an environment generally stimulatory to axon growth and synaptogenesis, then BDNF can substitute and no inhibition by TrkC–IgG will be observed in the presence of exogenous BDNF. However, if NT-3 has a “special” specific role in axon guidance and/or synaptogenesis, then addition of BDNF with TrkC–IgG will not compensate for loss of NT-3 and inhibition by TrkC–IgG will be observed.
In fact, the number of PSDs formed in the presence of BDNF is significantly (p = 0.03) reduced by the addition of TrkC–IgG (Fig. 6, compare E, H; Fig. 7, compare NK/BDNF, NK/TrkCIgG/BDNF). Because TrkC–IgG does not block BDNF (Shelton et al., 1995; Hansen et al., 2001), this result indicates that endogenous NT-3 is still required for growth of SGN axons to IHCs despite the presence of BDNF. Similarly, the number of SGN fiber–IHC contacts formed in the presence of BDNF is significantly (p = 0.01) reduced by the addition of TrkC–IgG (Fig. 6, compare E, H; Fig. 8, compare NK/BDNF, NK/TrkC–IgG/BDNF). These results imply that, besides contributing to an environment supportive of reinnervation, NT-3 also has a specific role in promoting SGN peripheral axon growth to IHCs and promoting synaptogenesis between IHCs and SGNs. Remarkably, this role of NT-3 is distinctive and BDNF is apparently unable to substitute for NT-3.
Regenerating type 1 SGN peripheral axons do not grow past the inner hair cell row
Having observed overgrowth of axons within the IHC row during reinnervation, we asked whether some type I SGN axons grow past the IHC row without contacting IHCs and enter the spiral bundles in the OHC rows. If this indeed occurs, it could account, in part, for the reduced number of axons contacting IHCs after recovery from excitotoxic trauma. Representative images of an explant maintained in BDNF for 72 h after NK and labeled to visualize NF200 and peripherin immunoreactivity are shown in Figure 9A–C. Peripherin-negative (type 1) fibers are not observed in the spiral bundles of the post-NK explants. Therefore, all fibers in the spiral bundles are type 2 SGN peripheral axons, implying that the regenerating type 1 SGN peripheral axons do not contribute to the spiral bundles.
Figure 9.
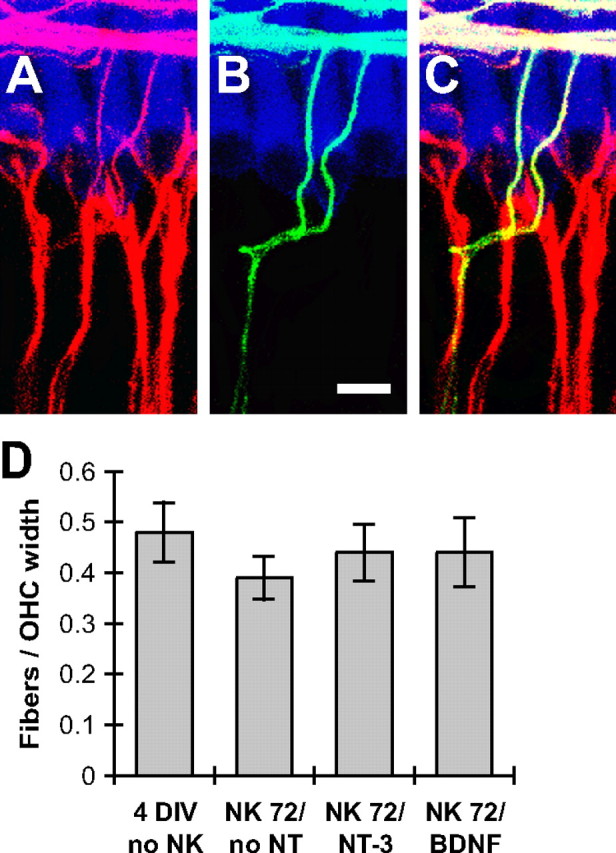
Regenerating type 1 SGN axons do not enter the spiral bundles (outer hair cell rows). A–C, Representative images of an explant maintained in medium with 3.6 nm NT-3 for 72 h after NK. Hair cells are labeled with anti-myosin VI (blue), all SGN peripheral axons with anti-NF200 (red), and type 2 SGN peripheral axons with anti-peripherin (green). NF200 immunofluorescence is shown superimposed on hair cells in A, peripherin immunofluorescence in B, and the merged images in C (superimposed green and red is yellow.) Scale bar, 10 μm. All fibers traversing the IHC row and entering the spiral bundles appear peripherin positive. These were counted in 3D reconstructions of confocal stacks from explants maintained 4 DIV without NK treatment (control), maintained 72 h after NK without neurotrophins (NK72/no NT), 72 h after NK with 3.6 nm NT-3 (NK72/NT-3), or 72 h after NK with 3.6 nm BDNF (NK72/BDNF). A graph of the number of NF200-immunoreactive fibers normalized to the average width of a single OHC (6–7 cochleae per group) is shown in D; error bars show SEM. No statistically significant difference was found among these groups (ANOVA, p > 0.05), indicating that regenerating type 1 axons make little or no contribution to innervation of OHCs.
To confirm that regenerating axons do not contribute to the spiral bundles, we counted axon–OHC contacts in confocal image stacks of control non-NK-treated 4 DIV explants or explants maintained 72 h after NK without neurotrophins, with BDNF, or with NT-3. The number of axons projecting past the IHC row in a segment of the spiral bundles corresponding to 10 OHC widths was counted in six to seven cochleae per group. Counts of axons—normalized to the width of a single OHC—are shown in Figure 9D. No statistically significant difference (ANOVA) was found among the NK-treated groups, and no statistically significant difference was found between the NK-treated groups and the 4 DIV control group. These data indicate that regenerating type 1 SGN axons do not enter the spiral bundles and contact OHCs. The reduction in the number of axons contacting IHCs must then be attributable to failure of many SGN peripheral axons to regrow to the IHC row. Also, although regenerating axons exhibit an aberrant overgrowth of their IHC targets, they do respect the IHC row as a boundary and do not cross it to enter the spiral bundles and innervate OHCs. This resembles normal cochlear development in which type I SGN peripheral processes innervate IHCs but not OHCs (Koundakjian et al., 2007), implying that there is a “stop” signal for type 1 SGN processes on the IHCs themselves or on adjacent supporting cells or extracellular matrix.
Discussion
We have shown that transient exposure to glutamatergic agonists in vitro causes degeneration of IHC–SGN synapses and distal ends of type 1 SGN peripheral axons, with subsequent synaptic regeneration. Below, we compare trauma and recovery in our in vitro excitotoxic model to excitotoxicity and noise damage in the cochlea in vivo, indicating insights gained by experimental manipulations that specifically target the postsynaptic terminal. We then discuss novel observations regarding neurotrophin function in the cochlea that were made possible by use of explant cultures: the role of neurotrophins in recovery from synaptic trauma and, in particular, the distinctive role of NT-3.
Excitotoxic trauma in vitro resembles that in vivo
The consequences of glutamatergic excitotoxic trauma in the in vitro model presented here are similar to those after intracochlear infusion of glutamate agonists in vivo (Pujol and Puel, 1999; Ruel et al., 2007): damage is restricted to distal ends of type 1 SGN peripheral processes. SGNs express both non-NMDA-type and NMDA-type glutamate receptors (Niedzielski and Wenthold, 1995), but excitotoxic damage to type 1 SGN peripheral processes in vivo was mediated by non-NMDA glutamate receptors (Pujol et al., 1985; Puel et al., 1994; Ruel et al., 2007). Although, in the present study, we combined kainate and NMDA to model the SGN response to glutamate, damage caused by kainic acid alone was identical to that caused by the combination (Fig. 2).
As is the case in vivo (Pujol et al., 1985; Puel et al., 1995; Pujol and Puel, 1999), glutamatergic agonists do not disrupt type 2 SGN–OHC synapses in vitro, although type 2 SGNs use AMPA-type glutamate receptors (Weisz et al., 2009). It is not obvious what physiological difference(s) between type 1 and type 2 SGNs could account for the insensitivity of the latter to excitotoxicity. The in vitro model described here can be used, in future studies, to determine why excitotoxic damage is caused selectively by non-NMDA agonists and why the damage is restricted to type 1 SGNs.
Excitotoxicity in vitro as a model for postsynaptic effects of acoustic trauma
After acoustic trauma in vivo, there is a rapid reduction in the number of SGN peripheral axons in contact with IHCs (Ruel et al., 2005; Kujawa and Liberman, 2009) similar to what is observed after intracochlear perfusion of AMPA agonists. Moreover, this effect of noise is blocked by a glutamate receptor antagonist (Puel et al., 1998), indicating that it is directly attributable to excitotoxicity and suggesting that excitotoxic trauma can be a model for postsynaptic effects of noise. Noise may have additional presynaptic effects in the hair cells not shared by excitotoxic trauma, so use of an excitotoxicity model allows investigation of the specifically postsynaptic effects of noise on SGNs.
As is the case in vivo, excitotoxic trauma in vitro causes rapid degeneration of IHC–SGN synapses and the culture system facilitates experimental analysis of the postsynaptic damage. In our in vitro model, there is reinnervation of completely denervated IHCs after excitotoxic trauma, and this may occur also after excitotoxicity in vivo (Pujol et al., 1985, 1996; Puel et al., 1995). In contrast, acoustic trauma reduces innervation of IHCs within 24 h, but there is no apparent subsequent reinnervation (Kujawa and Liberman, 2009). An intriguing possibility is that the SGN is intrinsically able to regenerate its bouton on IHCs provided that it has a suitable presynaptic partner, and failure of reinnervation after acoustic trauma may be because both the presynaptic and postsynaptic sides of the synapse are damaged. In the case of excitotoxicity in which damage is rather restricted to the postsynaptic terminal, reinnervation can occur. Therapeutic interventions to facilitate post-trauma reinnervation (or innervation of regenerated hair cells) may need to be directed toward both synaptic partners.
Alternatively, reinnervation potential may depend on age and occurs in our in vitro excitotoxicity model because the explants are from neonates. If so, comparison between juvenile and older cochleae may be instructive regarding requirements for reinnervation. It may also be the case that, as in vitro, some rapid reinnervation may occur after noise trauma in vivo. In this regard, it may be noted that regrowth of SGN axons to IHCs is well underway by 18 h after trauma (Figs. 4, 5). Possibly, regrowth of damaged peripheral axons to IHCs in vivo occurred within the first 24 h after acoustic trauma, the earliest time point examined (Kujawa and Liberman, 2009).
Presynaptic effects of excitotoxicity in vitro and acoustic trauma
An apparently significant difference between noise and excitotoxic damage is that, in the former, there is a significant reduction in the number of presynaptic ribbons (Kujawa and Liberman, 2009), whereas in the latter, although there is a significant increase in the number of apically mislocalized ribbons, there is no significant loss of ribbons (Fig. 3). As with other differences in outcomes, this might result from differences in species, age of the animal, or in vitro versus in vivo conditions. A likely possibility is that, as discussed above, this difference simply reflects a difference between excitotoxicity, which primarily targets the postsynaptic terminal, and noise, which primarily targets the hair cell and also causes postsynaptic damage via glutamate release from the hair cells. The data imply that ribbons may persist for several days in the absence of a postsynaptic partner and that the rapid loss of presynaptic ribbons after acoustic trauma results, at least in part, from a direct effect of noise on the IHC rather than being entirely a result of loss of SGN postsynaptic terminals on the IHC.
Role of NT-3 in reinnervation in the organ of Corti after excitotoxic trauma
Neurotrophins play important roles in neuronal survival in inner ear development (Fritzsch et al., 2004). Postnatally, NT-3 is expressed in IHCs and adjacent supporting cells (Ylikoski et al., 1993; Wheeler et al., 1994; Stankovic et al., 2004; Sugawara et al., 2007). SGNs express TrkB and TrkC (Mou et al., 1997), respectively, receptors for BDNF and NT-3, and exogenous NT-3 and BDNF both promote SGN survival in vitro (Lefebvre et al., 1994; Hegarty et al., 1997; Mou et al., 1997). In addition to supporting neuronal survival, neurotrophins promote neurite growth and synaptogenesis (Huang and Reichardt, 2001). However, these latter neurotrophin functions are often difficult to reveal because neurotrophic factor deprivation kills neurons. We exploited the accessibility of an in vitro system to investigate the roles of neurotrophins, particularly NT-3, in SGN neurite growth to IHCs and synaptogenesis on IHCs. Important, previously unanswered questions are whether neurotrophins are necessary for synaptogenesis as well as axonal growth and whether NT-3 and BDNF are functionally similar in effects on cochlear innervation.
Although exogenous NT-3 and BDNF are equally effective in promoting SGN neurite growth and synaptogenesis in vitro, we show here that growth of SGN axons to IHCs and synaptogenesis on IHCs are greatly reduced by TrkC–IgG, an NT-3 scavenger, thus establishing a role for endogenous NT-3 in reinnervation of IHCs. Because TrkB–IgG was without effect, BDNF is not necessary for reinnervation. Remarkably, TrkC–IgG inhibits SGN axon growth and synaptogenesis even when BDNF is added to the culture medium. This implies that NT-3, the principal endogenous neurotrophin in the organ of Corti, has a distinctive role in promoting SGN axon growth to IHCs and synaptogenesis on IHCs, a role for which BDNF cannot substitute even when added at high concentration. Conversely, in the vestibular system, BDNF is necessary for proper innervation of hair cells by vestibular neurons and NT-3 cannot substitute (Agerman et al., 2003). BDNF and NT-3 also differ with respect to physiological phenotypes they induce in SGNs (Adamson et al., 2002). The observations that NT-3 and BDNF differ in their effects on inner ear neurons imply that, in this context, TrkC signaling is functionally different from TrkB signaling, although they are structurally similar receptor protein-tyrosine kinases. In support of this, Postigo et al. (2002) have identified biochemical differences between TrkB and TrkC that could account for functional differences such as those discussed here. BDNF and NT-3 have been proposed as potential therapeutics to prevent SGN degeneration in deaf individuals (Green et al., 2008). These data indicating that they are in fact not equivalent must be considered when designing such therapies.
Implications for accelerated age-related hearing loss resulting from noise exposure
In humans (Gates et al., 2000) and mice (Kujawa and Liberman, 2006, 2009), exposure to noise while young, even at a level that does not compromise hair cells, is associated with accelerated age-related hearing loss in the aging individual, indicating that deleterious effects of noise continue long after the exposure has ended (Gates, 2006). In noise-exposed mice, accelerated age-related hearing loss was associated with increased SGN death (Kujawa and Liberman, 2006, 2009). A possible explanation is that the reduction in SGN postsynaptic terminals results in altered neural activity or neurotrophic support, which, in turn, compromises SGN survival. The system described here models critical aspects of noise damage to type 1 SGN postsynaptic terminals, allowing the accessibility of tissue culture to be applied to experimental investigation of its cellular mechanisms. This could identify means to prevent SGN degeneration after noise damage or to improve innervation of new hair cells should gene or stem cell therapies allow their regeneration (Edge and Chen, 2008).
Footnotes
This study was supported by NIH Grants R01 DC02961 (S.H.G.) and P30 DC010362 and a grant from the American Hearing Research Foundation (Q.W.). We thank Drs. Rémy Pujol, Bernd Fritzsch, Marlan Hansen, Pamela Roehm, Jonathan Kopelovich, and members of the Green laboratory for valuable comments. We thank Daniel Denman, Swapnil Mehta, Dylan Todd, and Catherine Kane for technical assistance.
References
- Adamson CL, Reid MA, Davis RL. Opposite actions of brain-derived neurotrophic factor and neurotrophin-3 on firing features and ion channel composition of murine spiral ganglion neurons. J Neurosci. 2002;22:1385–1396. doi: 10.1523/JNEUROSCI.22-04-01385.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agerman K, Hjerling-Leffler J, Blanchard MP, Scarfone E, Canlon B, Nosrat C, Ernfors P. BDNF gene replacement reveals multiple mechanisms for establishing neurotrophin specificity during sensory nervous system development. Development. 2003;130:1479–1491. doi: 10.1242/dev.00378. [DOI] [PubMed] [Google Scholar]
- Alam SA, Robinson BK, Huang J, Green SH. Prosurvival and proapoptotic intracellular signaling in rat spiral ganglion neurons in vivo after the loss of hair cells. J Comp Neurol. 2007;503:832–852. doi: 10.1002/cne.21430. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Capon DJ, Ward RH. Immunoadhesins. Int Rev Immunol. 1993;10:219–227. doi: 10.3109/08830189309061697. [DOI] [PubMed] [Google Scholar]
- Bao J, Lin H, Ouyang Y, Lei D, Osman A, Kim TW, Mei L, Dai P, Ohlemiller KK, Ambron RT. Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat Neurosci. 2004;7:1250–1258. doi: 10.1038/nn1342. [DOI] [PubMed] [Google Scholar]
- Edge AS, Chen ZY. Hair cell regeneration. Curr Opin Neurobiol. 2008;18:377–382. doi: 10.1016/j.conb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eybalin M. Neurotransmitters and neuromodulators of the mammalian cochlea. Physiol Rev. 1993;73:309–373. doi: 10.1152/physrev.1993.73.2.309. [DOI] [PubMed] [Google Scholar]
- Francis HW, Rivas A, Lehar M, Ryugo DK. Two types of afferent terminals innervate cochlear inner hair cells in C57BL/6J mice. Brain Res. 2004;1016:182–194. doi: 10.1016/j.brainres.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Francis HW, Rivas A, Lehar M, Saito Y, Mouton PR, Ryugo DK. Efficient quantification of afferent cochlear ultrastructure using design-based stereology. J Neurosci Methods. 2006;150:150–158. doi: 10.1016/j.jneumeth.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Tessarollo L, Coppola E, Reichardt LF. Neurotrophins in the ear: their roles in sensory neuron survival and fiber guidance. Prog Brain Res. 2004;146:265–278. doi: 10.1016/S0079-6123(03)46017-2. [DOI] [PubMed] [Google Scholar]
- Gates GA. The effect of noise on cochlear aging. Ear Hear. 2006;27:91. doi: 10.1097/01.aud.0000194512.51849.ab. author reply 92. [DOI] [PubMed] [Google Scholar]
- Gates GA, Schmid P, Kujawa SG, Nam B, D'Agostino R. Longitudinal threshold changes in older men with audiometric notches. Hear Res. 2000;141:220–228. doi: 10.1016/s0378-5955(99)00223-3. [DOI] [PubMed] [Google Scholar]
- Géléoc GS, Holt JR. Developmental acquisition of sensory transduction in hair cells of the mouse inner ear. Nat Neurosci. 2003;6:1019–1020. doi: 10.1038/nn1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Loyzaga P, Pujol R. Neurotoxicity of kainic acid in the rat cochlea during early developmental stages. Eur Arch Otorhinolaryngol. 1990;248:40–48. doi: 10.1007/BF00634780. [DOI] [PubMed] [Google Scholar]
- Green SH, Altschuler RA, Miller JM. Cell death and cochlear protection. In: Schacht J, Popper AN, Fay RR, editors. Auditory trauma, protection and repair. New York: Springer; 2008. [Google Scholar]
- Hafidi A, Després G, Romand R. Ontogenesis of type II spiral ganglion neurons during development: peripherin immunohistochemistry. Int J Dev Neurosci. 1993;11:507–512. doi: 10.1016/0736-5748(93)90024-8. [DOI] [PubMed] [Google Scholar]
- Hakuba N, Koga K, Gyo K, Usami SI, Tanaka K. Exacerbation of noise-induced hearing loss in mice lacking the glutamate transporter GLAST. J Neurosci. 2000;20:8750–8753. doi: 10.1523/JNEUROSCI.20-23-08750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MR, Zha XM, Bok J, Green SH. Multiple distinct signal pathways, including an autocrine neurotrophic mechanism, contribute to the survival-promoting effect of depolarization on spiral ganglion neurons. J Neurosci. 2001;21:2256–2267. doi: 10.1523/JNEUROSCI.21-07-02256.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty JL, Kay AR, Green SH. Trophic support of cultured spiral ganglion neurons by depolarization exceeds and is additive with that by neurotrophins or cyclic AMP, and requires elevation of [Ca2+]i within a set range. J Neurosci. 1997;17:1959–1970. doi: 10.1523/JNEUROSCI.17-06-01959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Koundakjian EJ, Appler JL, Goodrich LV. Auditory neurons make stereotyped wiring decisions before maturation of their targets. J Neurosci. 2007;27:14078–14088. doi: 10.1523/JNEUROSCI.3765-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci. 2006;26:2115–2123. doi: 10.1523/JNEUROSCI.4985-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang H, Schulte BA, Schmiedt RA. Ouabain induces apoptotic cell death in type I spiral ganglion neurons, but not type II neurons. J Assoc Res Otolaryngol. 2005;6:63–74. doi: 10.1007/s10162-004-5021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre PP, Malgrange B, Staecker H, Moghadass M, Van de Water TR, Moonen G. Neurotrophins affect survival and neuritogenesis by adult injured auditory neurons in vitro. Neuroreport. 1994;5:865–868. doi: 10.1097/00001756-199404000-00003. [DOI] [PubMed] [Google Scholar]
- Malgrange B, Lefebvre P, Van de Water TR, Staecker H, Moonen G. Effects of neurotrophins on early auditory neurones in cell culture. Neuroreport. 1996;7:913–917. doi: 10.1097/00001756-199603220-00016. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin YM, Harke B, Hell SW, Egner A, Moser T. Tuning of synapse number, structure and function in the cochlea. Nat Neurosci. 2009;12:444–453. doi: 10.1038/nn.2293. [DOI] [PubMed] [Google Scholar]
- Meyers JR, MacDonald RB, Duggan A, Lenzi D, Standaert DG, Corwin JT, Corey DP. Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J Neurosci. 2003;23:4054–4065. doi: 10.1523/JNEUROSCI.23-10-04054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou K, Hunsberger CL, Cleary JM, Davis RL. Synergistic effects of BDNF and NT-3 on postnatal spiral ganglion neurons. J Comp Neurol. 1997;386:529–539. [PubMed] [Google Scholar]
- Mouton PR. Baltimore: Johns Hopkins UP; 2002. Principles and practices of unbiased stereology: an introduction for bioscientists. [Google Scholar]
- Niedzielski AS, Wenthold RJ. Expression of AMPA, kainate and NMDA receptor subunits in cochlear and vestibular ganglia. J Neurosci. 1995;15:2338–2353. doi: 10.1523/JNEUROSCI.15-03-02338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Arumäe U, Moshnyakov M, Palgi J, Saarma M, Ylikoski J. Coordinated expression and function of neurotrophins and their receptors in the rat inner ear during target innervation. Hear Res. 1994;75:131–144. doi: 10.1016/0378-5955(94)90064-7. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Hallböök F, Xing-Qun L, Virkkala J, Saarma M, Ylikoski J. Expression of neurotrophins and Trk receptors in the developing, adult, and regenerating avian cochlea. J Neurobiol. 1997;33:1019–1033. doi: 10.1002/(sici)1097-4695(199712)33:7<1019::aid-neu11>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Postigo A, Calella AM, Fritzsch B, Knipper M, Katz D, Eilers A, Schimmang T, Lewin GR, Klein R, Minichiello L. Distinct requirements for TrkB and TrkC signaling in target innervation by sensory neurons. Genes Dev. 2002;16:633–645. doi: 10.1101/gad.217902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel JL, Pujol R, Tribillac F, Ladrech S, Eybalin M. Excitatory amino acid antagonists protect cochlear auditory neurons from excitotoxicity. J Comp Neurol. 1994;341:241–256. doi: 10.1002/cne.903410209. [DOI] [PubMed] [Google Scholar]
- Puel JL, Saffiedine S, Gervais d'Aldin C, Eybalin M, Pujol R. Synaptic regeneration and functional recovery after excitotoxic injury in the guinea pig cochlea. C R Acad Sci III. 1995;318:67–75. [PubMed] [Google Scholar]
- Puel JL, Ruel J, Gervais d'Aldin C, Pujol R. Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport. 1998;9:2109–2114. doi: 10.1097/00001756-199806220-00037. [DOI] [PubMed] [Google Scholar]
- Pujol R, Puel JL. Excitotoxicity, synaptic repair, and functional recovery in the mammalian cochlea: a review of recent findings. Ann N Y Acad Sci. 1999;884:249–254. doi: 10.1111/j.1749-6632.1999.tb08646.x. [DOI] [PubMed] [Google Scholar]
- Pujol R, Lenoir M, Robertson D, Eybalin M, Johnstone BM. Kainic acid selectively alters auditory dendrites connected with cochlear inner hair cells. Hear Res. 1985;18:145–151. doi: 10.1016/0378-5955(85)90006-1. [DOI] [PubMed] [Google Scholar]
- Pujol R, Gervais d'Aldin C, Saffiedine S, Eybalin M, Puel JL. Repair of inner hair cell-auditory nerve synapses and recovery of function after an excitotoxic injury. In: Salvi RJ, editor. Auditory plasticity and regeneration. New York: Thieme Medical Publishers; 1996. pp. 100–107. [Google Scholar]
- Ruel J, Wang J, Pujol R, Hameg A, Dib M, Puel JL. Neuroprotective effect of riluzole in acute noise-induced hearing loss. Neuroreport. 2005;16:1087–1090. doi: 10.1097/00001756-200507130-00011. [DOI] [PubMed] [Google Scholar]
- Ruel J, Wang J, Rebillard G, Eybalin M, Lloyd R, Pujol R, Puel JL. Physiology, pharmacology and plasticity at the inner hair cell synaptic complex. Hear Res. 2007;227:19–27. doi: 10.1016/j.heares.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepecky NB. Structure of the mammalian cochlea. In: Dallos P, Popper AN, Fay RR, editors. The cochlea. New York: Springer; 1996. pp. 44–129. [Google Scholar]
- Stankovic K, Rio C, Xia A, Sugawara M, Adams JC, Liberman MC, Corfas G. Survival of adult spiral ganglion neurons requires erbB receptor signaling in the inner ear. J Neurosci. 2004;24:8651–8661. doi: 10.1523/JNEUROSCI.0733-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara M, Murtie JC, Stankovic KM, Liberman MC, Corfas G. Dynamic patterns of neurotrophin 3 expression in the postnatal mouse inner ear. J Comp Neurol. 2007;501:30–37. doi: 10.1002/cne.21227. [DOI] [PubMed] [Google Scholar]
- Weisz C, Glowatzki E, Fuchs P. The postsynaptic function of type II cochlear afferents. Nature. 2009;461:1126–1129. doi: 10.1038/nature08487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler EF, Bothwell M, Schecterson LC, von Bartheld CS. Expression of BDNF and NT-3 mRNA in hair cells of the organ of Corti: quantitative analysis in developing rats. Hear Res. 1994;73:46–56. doi: 10.1016/0378-5955(94)90281-x. [DOI] [PubMed] [Google Scholar]
- Wise AK, Richardson R, Hardman J, Clark G, O'Leary S. Resprouting and survival of guinea pig cochlear neurons in response to the administration of the neurotrophins brain-derived neurotrophic factor and neurotrophin-3. J Comp Neurol. 2005;487:147–165. doi: 10.1002/cne.20563. [DOI] [PubMed] [Google Scholar]
- Ylikoski J, Pirvola U, Moshnyakov M, Palgi J, Arumäe U, Saarma M. Expression patterns of neurotrophin and their receptor mRNAs in the rat inner ear. Hear Res. 1993;65:69–78. doi: 10.1016/0378-5955(93)90202-c. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Liu Q, Davis RL. Complex regulation of spiral ganglion neuron firing patterns by neurotrophin-3. J Neurosci. 2005;25:7558–7566. doi: 10.1523/JNEUROSCI.1735-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]