

Abstract
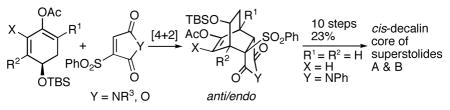
Regio, stereo and facial selective [4+2] cycloadditions between highly activated vinyl sulfones and 1,3-dienes derived from (R)-4-t-butyldimethyl-silyloxy-2-cyclohexen-1-one provide a powerful approach for the asymmetric synthesis of compounds containing the bicyclo[2.2.2]octanone carbon skeleton. This new methodology has been successfully applied to the asymmetric synthesis of the cis-decalin core structure of the potent anticancer marine natural products superstolides A and B.
Bicyclo[2.2.2]octanone derivatives have attracted considerable interests to organic chemists because the unique molecular architecture can be found in natural products1 and can serve as scaffolds in the design of therapeutic agents.2 In addition, the rigid bicyclo[2.2.2]-octanone structure can undergo versatile transformations to other molecular structures that are difficult to be constructed.3 Although a number of methods have been developed for the synthesis of racemic bicyclo[2.2.2]-octanone derivatives,4 asymmetric synthesis of highly functionalized bicyclo[2.2.2]octanone derivatives still poses a formidable synthetic challenge.5
We have recently reported for the first time that 1,3-dienes derived from (R)-4-t-butyldimethyl-silyloxy-2-cyclohexen-1-one can undergo stereo and facial selective asymmetric [4+2] cycloadditions with various activated symmetric dienophiles (Scheme 1).6 These reactions are exclusively endo selective and occur at the face syn to the bulky TBSO group to afford predominantly (or sometimes exclusively) syn/endo products. These controlled [4+2] cycloadditions increase the asymmetric complexity from one asymmetric center in the starting material to five asymmetric centers in the products in a single step, and provide a powerful approach for the asymmetric synthesis of compounds containing the bicyclo[2.2.2]octanone carbon skeleton.
Scheme 1.

[4+2] Cycloadditions with syn Facial Selectivity
To expand the scope of these new asymmetric [4+2] cycloadditions, we decided to investigate the possibility of employing unsymmetric dienophiles. We were particularly interested in highly activated unsymmetric dienophiles such as vinyl sulfone 6 (Scheme 2).7 Because of the steric hindrance between the alkyl (or aryl) sulfone moiety and TBSO group, 7-anti/endo should be formed predominantly.
Scheme 2.

[4+2] Cycloadditions with anti Facial Selectivity
In the event, 1,3-diene 5 reacted with dienophile 6a8 to provide 7a in 92% yield (entry1, Table 1). The reaction was highly regioselective and completely stereo- and facial selective. As expected, the [4+2] cycloaddition occurred at the face anti to the bulky TBSO group and 7-syn/endo was not detected. The facial selectivity observed in this reaction is opposite to that of those reactions employing symmetric dienophiles shown in Scheme 1.
Table 1.
[4+2] Cycloadditions with anti Facial Selectivitya
| entry | 1,3-diene 5 | dienophile 6 | 7-anti/endo | yieldb |
|---|---|---|---|---|
| 1 | 5 | 6a R = Ph, Y = NPh | 7a | 92% |
| 2 | 5 | 6b R = Ph, Y = NBn | 7b | 93% |
| 3 | 5 | 6c R = Ph, Y = NEt | 7c | 79% |
| 4 | 5 | 6d R = Ph, Y = O | 7d | 58% |
| 5 | 5 | 6e R = t-Bu, Y = NPh | 7e | 83% |
All reactions were run in CH2Cl2 at 25 °C for 1 day under argon.
These were isolated yields. All compounds were fully characterized.
Several highly reactive vinyl sulfones (6b–6e) were chosen to study the scope and limitation of this asymmetric [4+2] cycloaddition, and the results are summarized in Table 1. All reactions provided exclusive 7-anti/endo products in very good yields (entry 2–5, Table 1). These experiments showed that the N-substituent on the maleimide moiety of the dienophile had no effect on the stereo- and facial selectivity of the [4+2] cycloadditions (entry 1–3, Table 1). In addition, α-(phenylsulfonyl)maleic anhydride 6d reacted with 1,3-diene 5 in the same fashion. The relatively low yield of 7d was due to the decomposition of the anhydride moiety of the product on the silica gel during flash column chromatography.9 Furthermore, it was found that the phenylsulfonyl group could be replaced by the t-butylsulfonyl group and there was no change in the facial selectivity of the reaction (entry 5, Table 1).
We then turned our attention to the scope of chiral 1,3-dienes (Scheme 3). The results of asymmetric [4+2] cycloadditions between various chiral 1,3-dienes (8a–8e) and vinyl sulfone 6a are summarized in Table 2.10
Scheme 3.

[4+2] Cycloadditions with anti Facial Selectivity
Table 2.
[4+2] Cycloadditions with anti Facial Selectivitya
| entry | 1,3-diene | dienophile | 9-anti/endo | yieldb |
|---|---|---|---|---|
| 1 |
8a: R1 = Ac, R2 = Me R3 = X = H |
6a | 9a | 83% |
| 2 |
8b: R1 = Ac, R2 = n-Bu R3 = Me, X = H |
6a | 9b | 97% |
| 3 |
8c: R1 = CO2Me, R2 = H R3 = X = H |
6a | 9c | 87% |
| 4 |
8d: R1 = TIPS, R2 = H R3 = Me, X = H |
6a | 9d | 90% |
| 5 |
8e: R1 = TIPS, R2 = H R3 = Me, X = I |
6a | 9e | 91% |
All reactions were run in CH2Cl2 at 25 °C for 1 day under argon unless other stated.
These were isolated yields. All compounds were fully characterized.
Compound 9-anti/endo was the only product that was isolated from the reaction, and the yields (9a–9e) were uniformly excellent. Experimental results show that introducing an alkyl group at 1 and/or 4 position of the 1,3-diene had no effect on the stereochemical outcome of the reaction (entries 1 and 2, Table 2). It should be noted that among four newly created stereogenic centers in 9b three of them are quaternary carbons and two of them are bridgehead quaternary carbons, which are difficult to construct. In addition, the reactions were relatively faster when 1,3-diene 8c with a vinyl carbonate moiety and 1,3-dienes 8d and 8e with silyl enol ether moiety were employed (entries 3, 4 and 5, Table 2). Furthermore, entry 5 indicates that an iodo group at the 3 position of the 1,3-diene had no effect on the facial selectivity (entry 5, Table 2).
The enantiopure products of these highly controlled [4+2] cycloadditions contain the rigid bicyclo[2.2.2]-octanone carbon skeletons that are rich in both functionality and stereochemical complexity. These compounds are excellent scaffolds for further synthetic manipulations. To demonstrate the synthetic utility we decided to design a concise approach for the conversion of compound 7a to the cis-decalin core structure present in the highly potent anticancer marine natural products superstolides A (10) and B (11) that were isolated from the deep-water marine sponge Neosiphonia superstes collected off New Caledonia (Figure 1).11,12
Figure 1.
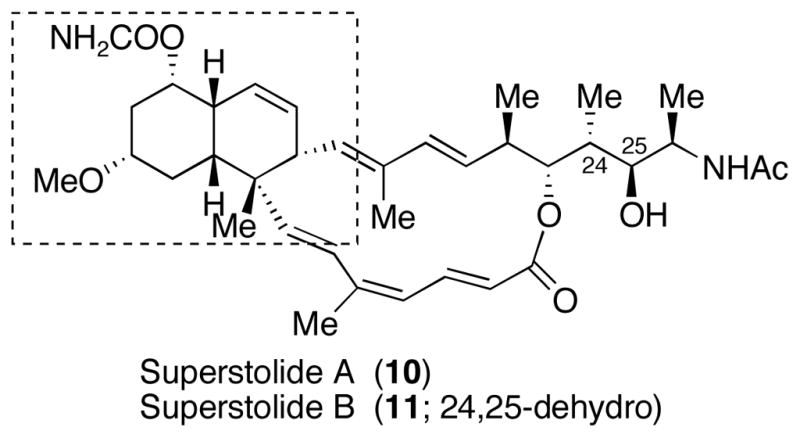
Anticancer Marine Natural Products Superstolides A and B
Compound 7a was chemo- and regioselectively reduced to give compound 12 in 94% yield. Hemiacetal 12 was protected by a TES group to afford compound 13, which was converted to a samarium enolate followed by the addition of MeI to provide compound 14 with the requisite stereochemistry of the quaternary carbon in 88% yield. Compound 14 was treated with 1N HCl in i-PrOH at 120 °C in a sealed tube to reach an equilibrium to give a mixture of 15 and 16 in about 1:3 ratio with a combined yield of 95% (compound 15 could be recycled to 16 under the same reaction conditions). Compound 16 was converted to olefin 17 in 84% yield under the standard conditions. Vinyl lithium 18, prepared from the corresponding vinyl stannane,13 underwent a stereospecific 1,2-addition to ketone 17 to afford tert-alcohol 19, which underwent an anionic oxy-Cope rearrangement to provide cis-decalin 20 with the five requisite stereogenic centers and the double bond at the desired positions. Stereoselective reduction of ketone 20 by Me4NBH4 followed by the methylation of the resulting alcohol gave compound 21 in 78% yield.
The conversion of compound 21 to 23 failed under various Tamao-Fleming oxidation conditions because of the quick cleavage of the isopropyl group of the hemiaminal ether moiety of compound 21 in the presence of typical strong acidic conditions of the Tamao-Fleming oxidation and the labile olefin moiety toward various electrophilic reagents such as Hg(OAc)2 and Br2.14 To solve these common problems in Tamao-Fleming oxidation it was imperative to develop a mild procedure that avoids the strong acidic conditions as well as those electrophilic reagents. We were delighted to observe that the phenyl group of the dimethylphenylsilyl moiety could be cleaved by TBAF in wet DMF at 65 °C to give compound 22, which was easily oxidized to alcohol 23 in 82% yield. It was discovered that the reaction temperature of 65 °C was critical. If the temperature was too low, the reaction was very slow. If the temperature was too high, then compound 22 further reacted with TBAF resulting in protiodesilylation.15,16 In 10 operations the [4+2] cycloaddition adduct 7a was converted to the cis-decalin core structure present in superstolides A (10) and B (11), and 2 fused rings, 6 stereogenic centers (including 1 quaternary carbon) and a double bond were established.
In conclusion, we have demonstrated for the first time that 1,3-dienes derived from (R)-4-t-butyldimethyl-silyloxy-2-cyclohex-en-1-one react with highly activated vinyl sulfones in a highly regio-, stereo- and facial-selective fashion. The facial selectivity of these cycloadditions is opposite to that of those [4+2] cycloadditions employing symmetric dienophiles. In addition, this new methodology has been successfully applied to the asymmetric synthesis of the cis-decalin core structure of the potent anticancer marine natural products Superstolides A and B. Furthermore, a mild procedure was developed to solve a long-standing problem in the Tamao-Fleming oxidation of dimethyl-phenylsilyl group. The scope and limitation of this protocol is currently under investigation and will be reported in due course.
Supplementary Material
Scheme 4.

Synthesis of the Core Structure of Superstolides A and B
Acknowledgments
This work was financially supported by a Grant (R01 CA109208) from the NIH.
Footnotes
Supporting Information Available: Experimental procedures and compound characterization. This material is available free of charge via Internet at http://pubs.acs.org.
References
- 1.(a) Nicolaou KC, Toh QY, Chen DYK. J Am Chem Soc. 2008;130:11292. doi: 10.1021/ja804588r. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Vassilikogiannakis G, Simonsen KB, Baran PS, Zhong YL, Vidali VP, Pitsinos EN, Couladouros EA. J Am Chem Soc. 2000;122:3071. doi: 10.1002/(sici)1521-3773(19991203)38:23<3555::aid-anie3555>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; (c) Ishiuchi K, Kubota T, Hayashi S, Shibata T, Kobayashi J. Tetrahedron Lett. 2009;50:6534. [Google Scholar]; (d) Huang JM, Yokoyama R, Yang CS, Fukuyama Y. J Nat Prod. 2001;64:428. doi: 10.1021/np0005715. [DOI] [PubMed] [Google Scholar]; (e) Srikrishna A, Satyanarayana G. Tetrahedron: Asymmetry. 2005;16:3992. [Google Scholar]; (f) Li D, Wang F, Cai S, Zeng X, Xiao X, Gu Q, Zhu W. J Antibiot. 2007;60:317. doi: 10.1038/ja.2007.40. [DOI] [PubMed] [Google Scholar]; (g) Magnus P, Gazzard L, Hobson L, Payne AH, Rainey TJ, Westlund N, Lynch V. Tetrahedron. 2002;58:3423. [Google Scholar]
- 2.(a) Schlapper C, Seebacher W, Kaiser M, Brun R, Saf R, Weis R. Bioorg Med Chem. 2007;15:5543. doi: 10.1016/j.bmc.2007.05.042. [DOI] [PubMed] [Google Scholar]; (b) Javanmard S, Deutsch HM, Collard DM, Kuhar MJ, Schweri MM. J Med Chem. 1999;42:4836. doi: 10.1021/jm990306k. [DOI] [PubMed] [Google Scholar]; (c) Deutsch HM, Gelbaum LT, McLaughlin GM, Fleischmann TJ, Earnhart LL, Haugwitz RD, Zalkow LH. J Med Chem. 1986;29:2164. doi: 10.1021/jm00161a006. [DOI] [PubMed] [Google Scholar]; (d) Moriarty RM, Enache LA, Zhao L, Gilardi R, Mattson MV, Prakash O. J Med Chem. 1998;41:468. doi: 10.1021/jm970059p. [DOI] [PubMed] [Google Scholar]; (e) Katz JL, Izenwasser S, Terry P. Psychopharmacology. 2000;148:90. doi: 10.1007/s002130050029. [DOI] [PubMed] [Google Scholar]; (f) Grunewald GL, McLeish MJ, Criscione KR. Bioorg Med Chem Lett. 2001;11:1579. doi: 10.1016/s0960-894x(01)00245-1. [DOI] [PubMed] [Google Scholar]; (g) Whitney JG, Gregory WA, Kauer JC, Roland JR, Snyder JA, Benson RE, Hermann EC. J Med Chem. 1970;13:254. doi: 10.1021/jm00296a021. [DOI] [PubMed] [Google Scholar]; (h) Yeh VSC, Kurukulasuriya R, Madar D, Patel JR, Fung S, Monzon K, Chiou W, Wang J, Jacobson P, Sham HL, Link JT. Bioorg Med Chem Lett. 2006;16:5408. doi: 10.1016/j.bmcl.2006.07.062. [DOI] [PubMed] [Google Scholar]; (i) Cannon JG, Yang KW, Rodriguez M, Buckley JP. J Pharm Sci. 1971;60:1535. doi: 10.1002/jps.2600601021. [DOI] [PubMed] [Google Scholar]
- 3.Miyaoka H, Yamada Y. Bull Chem Soc Jpn. 2002;75:203. and references cited therein Finet L, Dakir M, Castellote I, Arseniyadis S. Eur J Org Chem. 2007:4116.Hua DH, Gung WY, Ostrander RA, Takusagawa F. J Org Chem. 1987;52:2509.Lee TH, Rao PD, Liao CC. Chem Commun. 1990:801.
- 4.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chem Int Ed. 2002;41:1668. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. and references cited therein Liao CC. Pure Appl Chem. 2005;77:1221.and references cited therein Fukushima M, Morii A, Hoshi T, Suzuki T, Hagiwara H. Tetrahedron. 2007;63:7154.
- 5.(a) Tzvetkov NT, Schmoldt P, Neumann B, Stammler HG, Mattay J. Tetrahedron: Asymmetry. 2006;17:993. [Google Scholar]; (b) Friberg A, Johanson T, Franzen J, Gorwa-Grauslund MF, Frejd T. Org Biomol Chem. 2006;4:2304. doi: 10.1039/b603500k. [DOI] [PubMed] [Google Scholar]; (c) Luo Y, Carnell AJ. J Org Chem. 2010;75:2057. doi: 10.1021/jo9023705. [DOI] [PubMed] [Google Scholar]; (d) Dong S, Zhu J, Porco JA., Jr J Am Chem Soc. 2008;130:2738. doi: 10.1021/ja711018z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fisher C, Defieber C, Suzuki T, Carreira EM. J Am Chem Soc. 2004;126:1628. doi: 10.1021/ja0390707. [DOI] [PubMed] [Google Scholar]; (f) Almqvist F, Ekman N, Frejd T. J Org Chem. 1996;61:3794. doi: 10.1021/jo9601167. [DOI] [PubMed] [Google Scholar]; (g) Paquette LA, Tsui HC. J Org Chem. 1996;61:142. [Google Scholar]; (h) Hua Z, Yu W, Su M, Jin Z. Org Lett. 2005;7:1939. doi: 10.1021/ol050339w. [DOI] [PubMed] [Google Scholar]; (i) Kitahara T, Miyake M, Kido M, Mori K. Tetrahedron: Asymmetry. 1990;1:775. [Google Scholar]
- 6.Hua Z, Chen L, Jin Z. Tetrahedron Lett. 2009;50:6621. doi: 10.1016/j.tetlet.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) El-Awa A, Noshi MN, Mollat du Jourdin X, Fuchs PL. Chem Rev. 2009;109:2315. doi: 10.1021/cr800309r. [DOI] [PubMed] [Google Scholar]; (b) Simpkins NS. Tetrahedron. 1990;46:6951. [Google Scholar]
- 8.Compounds 6a–6e were prepared using the procedure for the preparation of 6d from the following article: Ramezanian M, Abdelkader M, Padias AB, Hall HK, Jr, Brois SJ. J Org Chem. 1989;54:2852.
- 9.The actual yield was much higher based on the analysis of the 1H NMR spectrum of the crude product.
- 10.Compounds 8a–8e were prepared using the procedures published in ref. 6.
- 11.For the isolation of superstolides A and B: D’Auria MV, Debitus C, Paloma LG, Minale L, Zampella A. J Am Chem Soc. 1994;116:6658.D’Auria MV, Paloma LG, Minale L, Zampella A, Debitus C. J Nat Prod. 1994;57:1595. doi: 10.1021/np50113a024.
- 12.Synthetic efforts toward the synthesis of the cis-decalin core structure of superstolide A: Roush WR, Champoux JA, Peterson BC. Tetrahedron Lett. 1996;37:8989.Tortosa M, Yakelis NA, Roush WR. J Am Chem Soc. 2008;130:2722. doi: 10.1021/ja710238h.Tortosa M, Yakelis NA, Roush WR. J Org Chem. 2008;73:9657. doi: 10.1021/jo801794s.(d) reference 5h.
- 13.Cunico RF, Clayton FJ. J Org Chem. 1976;41:1480. [Google Scholar]
- 14.Fleming I, Henning R, Plaut H. J Chem Soc, Chem Commun. 1984:29.Jones GR, Landais Y. Tetrahedron. 1996;52:7599. and references cited therein.
- 15.During the writing of this manuscript, we found a very similar protocol for the Tamao-Fleming oxidation of methyldiphenylsilyl and triphenylsilyl groups to alcohols was reported (one example): Knölker H-J, Wanzi G. Synlett. 1995:378.
- 16.Protiodesilylation by TBAF was reported by Roush. See: Heitzman C, Lambert WT, Mertz E, Shotwell JB, Tinsley JM, Va P, Roush WR. Org Lett. 2005;7:2405. doi: 10.1021/ol0506821.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.