


Abstract
We attempted to devise a transcription system in which a particular DNA sequence of interest could be inducibly expressed under the control of a modified polymerase III (pol III) promoter. Its activation requires a mutated transcription factor not contained endogenously in human cells. We constructed such a promoter by fusing elements of the β-lactamase gene of Escherichia coli, containing a modified TATA-box and a pol III terminator, to the initiation region of the human U6 gene. This construct functionally resembles a 5′-regulated pol III gene and its transcribed segment can be exchanged for an arbitrary sequence. Its transcription in vitro by pol III requires the same factors as the U6 gene with the major exception that the modified TATA-box of this construct only interacts with a TATA-binding protein (TBP) mutant (TBP-DR2) but not with TBP wild-type (TBPwt). Its transcription therefore requires TBP-DR2 exclusively instead of TBPwt. In order to render the system inducible, we fused the gene coding for TBP-DR2 to a tetracycline control element and stably transfected this new construct into HeLa cells. Induction of such a stable and viable clone with tetracycline resulted in the expression of functional TBP-DR2. This system may conceptually be used in the future to inducibly express an arbitrary DNA sequence in vivo under the control of the above mentioned promoter.
INTRODUCTION
A primary goal of this project was to design an RNA polymerase III (pol III)-dependent promoter which was dissociated from the cellular transcription machinery and could be activated only by the expression of a transcription factor absent from a normal cell. This promoter would then form the basis for an inducible pol III system in mammalian cells.
In higher eukaryotic cells, three general types of pol III promoters have been distinguished (1). The type 1 promoter (5S rRNA genes) comprises gene internally located A- and C-boxes as well as an intermediate element. Transcription from this promoter requires TFIIIA, TFIIIC1, TFIIIC2, TFIIIBβ and pol III. Type 2 promoters (e.g. tRNA genes, VA RNA genes and Alu-sequences) are characterised by gene internal A- and B-boxes. Expression of these genes requires the same transcription factors as the type 1 promoter with the exception of TFIIIA (2–5). Type 3 promoters are located 5′ of the transcription initiation site and are composed of a TATA-box, a proximal sequence element (PSE) and a distal sequence element (DSE) (6). Specific initiation of transcription from type 3 promoters depends on the PSE binding protein (PBP) (7), also designated as PTF (8–10) or SNAPc (11,12), TFIIIBα (5), TFIIIU (13), pol III and the TATA-binding protein (TBP) (7,14), which in this case is not part of a TBP–TAF complex.
As described previously, hTFIIIB can be separated into two functionally different forms, TFIIIBα and TFIIIBβ. hTFIIIBβ is predominantly active on genes with intragenic promoters and contains TBP and associated proteins, whereas TFIIIBα is a TBP-free entity, which must be supplemented with TBP for in vitro transcription of the U6 snRNA gene (5).
Strubin and Struhl (15) examined mutants of the TBP molecule from yeast and human, which show a modified DNA binding specificity due to an exchange of three amino acids. The three substitutions are located in a region of 12 amino acids within the first or second repeat (direct repeat 1 and direct repeat 2, respectively) in the highly conserved C-terminal part of the protein. Based on the numbering in yeast, the amino acids at positions 194 (Ile), 203 (Val) and 205 (Leu) were replaced by Phe, Thr and Val. Both the human and the yeast TBP mutants were able to promote RNA synthesis from promoters containing a TGTAAA-box instead of the wild-type TATA-box sequence (consensus sequence TATAAA), while these TATA-box mutants were not transcribed in a system containing only the wild-type TBP protein. The high specificity of these TBP mutants and similar mutants of the Arabidopsis TBP-isoform for the TGTAAA sequence could also be demonstrated in plant cells (16).
We initiated studies to analyse whether these TBP mutants would also recognise various analogous TATA-box mutants of the human U6 gene in a reconstituted human pol III transcription system in vitro.
The long term aim of these studies was to establish an in vivo system in which the expression of a mutated variant of TBP could be induced, thereby leading to the modulated RNA synthesis of a particular sequence (for example, antisense RNA or double-stranded RNA possibly involved in post-transcriptional gene silencing) under the control of such a mutated TATA-box. The advantages of the RNA polymerase III expression system are the high rate of synthesis with multiple rounds of initiation and the efficient termination of pol III catalysed RNA synthesis. We report here on the first steps toward this goal.
MATERIALS AND METHODS
Plasmids
The plasmids pUVAI and pUhU60.35 were as previously described (7,17) and contained single copies of the genes coding for VAI RNA and human U6 snRNA respectively.
Oligonucleotides
17mer reverse sequencing-primer (-26-M13/pUC) and 17mer sequencing-primer (-20-M13/pUC) (Roche Diagnostics, Mannheim, Germany). PDR2467–490, 5′-GTCCTCCGATCCCGTTCTTCGGGG-3′; PDR2-5′, 5′-GCATTTTGAATTCCTGTTTGAGCTCACCCAG-3′; hU6-5′, 5′-GGACGAAACACCGTGCACGCTTCGGC-3′; hU60.35 –29<<, 5′-AGATGTAAAAAGCCAAGAAATCGAAATACT-3′.
Buffers
Buffer A contained 20 mM HEPES pH 7.9, 10% (v/v) glycerol, 3 mM DTT and 0.2 mM PMSF. Buffer B contained 20 mM Tris–HCl pH 7.9, 10% (v/v) glycerol, 5 mM MgCl2, 3 mM DTT and 0.2 mM PMSF. All fractions employed were dialysed against buffer B supplemented with 60 mM KCl before use, except for the polymerase fraction (see below).
Preparation of cytoplasmic extract (HeLa S100) and purification of transcription factors
HeLa cytoplasmic extract (HeLa S100) was prepared as described by Weil et al. (18). The S100 extract was dialysed against buffer A supplemented with 100 mM KCl and was subsequently chromatographed through phosphocellulose as described previously (19).
The PCC fraction (350–600 mM KCl) was used to prepare human TFIIIC1, TFIIIC2 and PBP as described previously (20) with the following modifications.
PBP. The PCC fraction was dialysed against buffer B supplemented with 60 mM KCl, loaded onto a Mono Q column (‘MQ’; Amersham-Pharmacia) at 5 mg protein/ml bed volume. After washing the column with the same buffer, bound proteins were eluted with a linear gradient of 60–450 mM KCl. Fractions eluting at 300–330 mM KCl contained PBP and were devoid of TFIIIC1 and TFIIIC2, as well as of TFIIIBα and RNA pol III.
hTFIIIC1. Fractions eluting at 250–290 mM KCl in buffer B from Mono Q (20) were diluted to 200 mM KCl in buffer B and applied to an EMD-SO3– Fractogel column (‘ESF’; Merck, Darmstadt, Germany) at 5 mg protein/ml bed volume. After washing the column with 200 mM KCl in buffer B, bound proteins were eluted with a linear gradient of 200–600 mM KCl and a 1 M KCl step. hTFIIIC1 activity eluted at 450–500 mM KCl.
TFIIIU. Fractions eluting at 150–190 mM KCl in buffer B from the MQ gradient (TFIIIC0) (20) were analysed in in vitro transcription assays and were applied to an EMD-SO3– Fractogel column at 5 mg protein/ml bed volume. After washing the column with 200 mM KCl in buffer B, bound proteins were eluted with a linear salt gradient of 200–600 mM KCl. The activity, which stimulates U6 transcription (TFIIIU) eluted at 360–420 mM KCl.
The PCB fraction (350 mM KCl) was used to prepare human TFIIIBα, TFIIIBβ and RNA polymerase III (pol III) by EMD DEAE Fractogel chromatography as described previously (20,21). TFIIIBβ was further purified by EMD-SO3– Fractogel chromatography as described (20). The polymerase fraction was also applied to EMD-SO3– Fractogel and was step-eluted. The polymerase activity eluted in the 400–650 mM KCl fraction and was free of TFIIIBβ, TFIIIC1 and TFIIIC2 activities. BSA (0.1 mg/ml) was added and the polymerase fraction was diluted with glycerol to a final concentration of 50% (v/v) glycerol. All fractions were stored at –80°C.
Recombinant human TBP (rhTBP) was expressed in Escherichia coli and purified as described (22).
In vitro transcription
The in vitro transcription mixtures contained the appropriate protein fractions, 1 µg plasmid DNA, 600 µM ATP, CTP, UTP, 30 µM GTP and 3 µCi [α-32P]GTP (Amersham-Pharmacia), 20 U RNase Block Ribonuclease Inhibitor (Stratagene), 60 mM KCl, 20 mM Tris–HCl pH 7.9, 10% glycerol and 5 mM MgCl2 in a final volume of 50 µl. After 90 min incubation at 30°C, the RNA was purified and loaded onto a denaturing 7 M urea–6% polyacrylamide gel. The gel was analysed by autoradiography for at least 1 day at –80°C with intensifying screens.
Primer extension assay
In vitro transcription using the primer extension assay was performed as described (23). For the primer extension analysis of the PDR2 gene, a specific primer (PDR2467–490) hybridising against the RNA between position 62 and 85 (relative to the first G of the ‘Ini’ sequence) was used.
Western blotting
For the screening of the HeLa TBP-DR2 clones, cells were harvested and counted 48 h after doxycycline induction. Cells (5 × 105) were resuspended in 50 µl 1× SDS sample buffer, treated with 250 U Benzonase™ (Merck) and subsequently fractionated on SDS–polyacrylamide gels (12.5%). Proteins were electrophoretically transferred to a PVDF membrane (Immobilon-P; Millipore) using semi-dry electroblotting as described by Kyhse-Andersen (24). After transfer, the membrane was stained with Ponceau S solution (Serva), destained and blocked with 10% skimmed milk powder in PBS/Tween. For detection of TBP containing a histidine tag, a monoclonal anti-histidine antibody (anti-RGS-His; Qiagen) was employed. Antigen–antibody complexes were detected by the ECL system (Amersham-Pharmacia).
DNase I footprint analysis
DNA fragments were generated by PCR amplification of pUPDR2-Expression with 5′-end-labelled 17mer reverse sequencing-primer and unlabelled 17mer sequencing-primer (M13/pUC; Roche Diagnostics). Individual protein fractions were preincubated with 1 µg of an equimolar mixture of the 110 and 147 bp pUC/HpaII fragments, as non-specific competitor, in a total volume of 50 µl for 15 min at 30°C. Subsequently, 15 000 c.p.m. of the labelled fragment was added and incubation continued for a further 45 min at 30°C. Samples were digested with 40 ng DNase I for 1 min at room temperature. After termination of the reaction by addition of an equal volume of 900 mM ammonium acetate, the DNA was purified as described (20,25) and finally electrophoresed on 6% (w/v) denaturing sequencing gels. After the run, urea was eluted from the gel, which was dried and autoradiographed overnight at –80°C with an intensifying screen.
For an exact determination of the DNase I protection pattern, a sequencing reaction using the 17mer reverse sequencing-primer (M13/pUC) was conducted in parallel.
Stable transfection of HeLa Tet-on cells
Transfection of HeLa Tet-on cells (Clontech) was accomplished using the ‘Effectene’ transfection reagent (Qiagen) according to the manufacturer’s instructions. In order to be able to select positive clones, cells were co-transfected with a hygromycin-resistance plasmid serving as a selection marker.
Cloning of PDR2 constructs
pUPDR2. A 43 bp long Sau3A fragment of the β-lactamase gene, containing a pol III termination signal was isolated from pBR322 and subcloned into a BamHI linearised pUC18 plasmid. The resulting clones were verified for the correct orientation of the insert by restriction analysis. The plasmid containing a pol III termination sequence, was designated as pUTV and was used as vector for the subcloning of the TBP-DR2 dependent promoter. For this, the 400 bp long EcoRI–XmnI fragment of pBR322 containing the TBP-DR2 dependent promoter was cloned into the ‘termination’-vector (pUTV) which had been digested with EcoRI/SmaI. This two-step cloning procedure was required to shorten the intervening sequence between the PDR2 promoter and the pol III termination signal (Fig. 2).
Figure 2.

Structure of the TBP-DR2-dependent promoter in the region of the β-lactamase gene in pBR322 and constructs derived therefrom. (A) Schematic restriction map of the EcoRI–PstI fragment of pBR322 containing the assumed origin of the TBP-DR2-dependent transcript. (B) Restriction map of the EcoRI/HindIII fragments of the three different TBP-DR2-dependent promoter constructs named pUPDR2, pUPDR2/hU6 and pUPDR2-Expression. The regions homologous to known RNA polymerase III promoter elements like the PSE element (?PSE?), the U6 initiation region (‘Ini’) and the pol III termination signal (TTTTT) are indicated.
pUPDR2/hU6. Both sequence parts were amplified in two separate reactions by PCR. The two plasmids pUPDR2 and pUhU60.35 were used as templates for this reaction. The amplification of the PDR2 promoter was accomplished using the two oligonucleotides PDR2-5′ and PDR2467–490. Two point mutations were inserted into the amplified promoter fragment using the PDR2-5′ primer, resulting in the creation of an EcoRI restriction site in this region. Subsequently, the internal hU6 sequence was amplified with the oligonucleotides hU6-5′ and pUC/M13 sequencing. By using a point mutation within the oligonucleotide hU6-5′ the U6 initiation region was transferred into the ‘Ini’ sequence of the PDR2 promoter thereby creating an Alw44I recognition sequence. After amplification, both fragments were cut by Alw44I in the initiation region and subsequently ligated by the resulting sticky ends. Thus, the PDR2 initiation sequence was restored and now contained the coding sequence and the terminator of the hU6 gene in its 3′ region. Subsequently, the resulting fragment was digested with EcoRI/HindIII and cloned into the vector plasmid, which had also been linearised by EcoRI and HindIII digestion.
PDR2-Expression. This cloning was also initiated from a promoter fragment amplified with the PDR2-5′ and PDR2467–490 oligonucleotide. Subsequently, the resulting fragment was digested with Alw44I in the proximity of the initiation region. The recessed 3′ terminus of the double-strand was filled-in with DNA polymerase I large fragment (Klenow fragment) resulting in a DNA fragment with a blunt end. Subsequently, this fragment was cut in the 5′ region of the promoter at the EcoRI restriction sequence inserted by the two point mutations of the PDR2-5′ oligonucleotide as described above. This fragment was inserted into the pUTV terminator plasmid (see above), which had been linearised with EcoRI and Ecl136I (blunt). The short sequence from the pUC18 multiple cloning site, which is located between the Ecl136I restriction sequence and the termination signal of the vector (pUTV), lies in the so-called PDR2-Expression construct between promoter (PDR2) and terminator (TV).
Site-directed mutagenesis
Different mutants of the human U6 gene were created in the region of the TATA-box by site specific mutagenesis using asymmetric PCR with a single mutant oligonucleotide (26).
RESULTS
Transcription of different TATA box mutants of the hU6 gene
In order to establish a transcription system independent of endogenous wild-type TBP (TBPwt), we used mutated forms of TBP (TBP-DR2 or TBP-DR1) instead of TBPwt to transcribe the human U6 gene in vitro. For this purpose, the sequence of the human U6 TATA-box was accordingly changed to TGTAAA by PCR in vitro mutagenesis. We created various constructs containing the TGTAAA-mutation in different orientation and/or distance with respect to the initiation region and examined whether these TATA-box mutants led to a reduction of the transcription efficiency of their associated gene sequences. Figure 1 (lanes 15–21) shows one of the TATA-box mutants with the sequence TGTAAA in the orientation corresponding to that of the wild-type U6 gene. We found that the mutation of the wild-type TATA-box leads to a significant reduction of the transcription efficiency (compare lanes 8–14 with 15–21) and, in some cases, no hU6 RNA could be detected. This loss of transcription was independent from the orientation of the TGTAAA-box (data not shown).
Figure 1.

In vitro transcription of a U6 gene containing a variant TATA-box. In vitro transcription systems based on 10 µl phosphocellulose fraction B and 20 µl phosphocellulose fraction C (lanes 1–21) containing either pUC18 (lanes 1–7), pUhU60.35 (lanes 8–14) or pUhU60.35 –29<< (lanes 15–21) as template, were supplemented either with increasing amounts (5–20 ng) of recombinant human TBPwt (lanes 2–4, 9–11 and 16–18) or recombinant human TBP-DR2 (lanes 5–7, 12–14 and 19–21), respectively. Lanes 1, 8 and 15 contained no recombinant TBP. Lanes 2, 5, 9, 12, 16 and 19 were supplemented with 5 ng, lanes 3, 6, 10, 13, 17 and 20 with 10 ng, and lanes 4, 7, 11, 14, 18 and 21 with 20 ng recombinant TBPwt or TBP-DR2 respectively. In vitro transcription was carried out using 1 µg plasmid DNA as template. The position of the U6 transcripts is indicated and the position of the TBP-DR2 dependent transcript is marked with an asterisk.
At the same time, we analysed whether these mutations of the TATA-box could be suppressed by addition of one of the mutated TBP variants, DR1 (data not shown) or DR2. Addition of either recombinant TBPwt (Fig. 1, lanes 16–18) or recombinant TBP-DR2 (Fig. 1, lanes 19–21), to a transcription system containing phosphocellulose fractions PCB/PCC, was unable to activate the transcription of this mutated U6 gene. This failure was not due to inactive preparations of the recombinant proteins, because both forms of TBP were able to activate transcription of the hU6 wild-type gene (Fig. 1, lanes 8–14). Therefore, the expected suppression of this TATA-box mutation by co-mutation of the TBP molecule does not occur in the human pol III system, as was shown for yeast and plants.
Because of the failure of the reported TATA-box mutations to respond to the co-mutated TBP variants, we attempted to identify other sequence variants of the TATA-box that could fulfil this requirement. We fortuitously discovered such a sequence within the plasmid pUC18. We found that the supplementation of the PCB/PCC reaction mixture with TBP-DR2 (Fig. 1, lanes 5–7, 12–14 and 19–21) resulted in an ∼320 bp transcript, the synthesis of which was strictly TBP-DR2 dependent. This transcript (shown by an asterisk in Fig. 1) was synthesised independently of the pol III promoter subcloned into the polylinker of the pUC-plasmid and could also be detected using only pUC18 (Fig. 1, lanes 1–7) or pBR322 (data not shown) as template for the transcription reaction in vitro.
Identification and cloning of a promoter sequence that depends on TBP-DR2 for its expression
Subsequently, we identified the DNA sequence in the pBR322 plasmid, from which the TBP-DR2 dependent transcript was synthesised. For this purpose, sequence sections at least 320 bp (the length of the transcript; Fig. 2A) upstream of at least four thymidine residues (pol III termination signal), were examined for homologies to the promoter sequences of the hU6 and mU6 gene respectively.
In the region of the β-lactamase gene of pBR322, which conveys ampicillin resistance, a 7 bp long sequence was identified, in which six nucleotides were identical to the hU6 and mU6 initiation region (Fig. 2A). This sequence is located ∼320 nt upstream of a typical pol III termination signal. Additionally, a sequence section, homologous to the PSE-element of the hU6 and mU6 gene, is located 23 bp upstream of this region (Fig. 2A).
To examine the TBP-DR2 dependence of the transcribed sequence and its promoter in more detail, this DNA segment was subcloned into the polylinker of pUC18. At the same time, the sequence between the initiation region and the terminator was shortened, in order to be able to differentiate the vector transcript (shown by an asterisk in Fig. 1) from the RNA synthesised from this subcloned sequence. This was achieved in two separate steps, as described in Materials and Methods.
This plasmid (Fig. 2B) contained the promoter and the termination signal at a distance of ∼100 bp from each other and it was designated as pUPDR2 (PDR2 = DR2 dependent promoter).
Functional investigations of the cloned PDR2 construct
Since we were dealing with an artificial promoter construct, it was necessary to verify that it would respond to TBP-DR2 in a predictable and functionally meaningful manner. In particular it was of fundamental importance to verify by DNase I protection assays that the mutated TBP-DR2, but not TBPwt, would bind to this artificial promoter.
Comparison of the binding of TBP-DR2 and TBPwt to the non-coding strand of the PDR2 promoter
The analysis of the TBP-DR2 binding to the non-coding strand of the PDR2 promoter is shown in Figure 3A. Samples 1 and 5 served as control reactions and contained no protein, while samples 2–4 contained increasing amounts of TBP-DR2.
Figure 3.

Footprint of TBP-DR2 and TBPwt on the non-coding strand of the PDR2 promoter. For the investigation of the non-coding strand, a DNA fragment was used, which was amplified by PCR with the M13 sequencing-primer and the 5′-labelled M13 reverse-sequencing-primer using the PDR2-Expression plasmid (Fig. 2B) as template. To be able to identify the region protected by TBP-DR2 a sequencing reaction was conducted in parallel (lanes A, C, G and T). (A) Binding of TBP-DR2 to the PDR2 promoter. Increasing amounts of TBP-DR2 (10, 20 and 40 ng; lanes 2–4) were incubated with the 5′-labelled PDR2 fragment. The control reactions containing no TBP-DR2 (free DNA) are shown in lanes 1 and 5. Lanes 2–5 additionally contained NP-40 at a very low concentration (0.0025%). By this procedure, DNase I protection by TBP-DR2 can be detected with smaller protein quantities and the digestion pattern of DNA is not affected even by higher NP-40 concentrations (data not shown). (B) Comparison of TBPwt and TBP-DR2. Either TBP-DR2 (lane 2, 20 ng; lane 3, 40 ng) or TBPwt (lane 4, 20 ng; lane 5, 40 ng) were incubated as described in (A) but contained no NP-40. Lanes 1 and 6 served as control reactions without protein. (C) Summary of the TBP-DR2 binding to the coding and non-coding strand of the PDR2 promoter sequence. The sequence sections homologous to the hU6 gene (?PSE? and Ini) are written in bold characters. Filled circles and asterisks signify hypersensitive sites in the footprint and schematic representations respectively.
The TBP-DR2 footprint on the non-coding strand of the PDR2 promoter extends over a length of at least 15 (from –20 to –34) and maximally 23 nt (from –20 to –42) and is bordered by hypersensitive sites at –14 and –43/–45 of the promoter sequence.
Figure 3B shows the comparative binding of TBP-DR2 and TBPwt to the same DNA fragment. The samples without TBP are shown in lanes 1 and 6. While TBP-DR2 (lanes 2 and 3) exhibits the DNase I protection pattern already shown in Figure 3A, no protection occurred with TBPwt (lanes 4 and 5). Also, no hypersensitive sites, which could reflect the binding of a protein, appeared.
We also performed footprint analyses on the coding strand of the PDR2 promoter (data not shown). The binding region of TBP-DR2 on this strand spans a sequence of 15 nucleotides from –20 to –34 relative to the transcription start point. For TBPwt, again no binding to this promoter could be detected.
In summary (Fig. 3C), a corresponding sequence of 15 nucleotides on both strands of the PDR2 promoter represents the binding region of TBP-DR2. This extends from position –20 to –34 relative to the transcription start point. The sequence region protected by TBP-DR2 against DNase I digestion possibly extends up to nucleotide –42 on the non-coding strand. For TBPwt, no interaction with the PDR2 promoter DNA could be observed.
In vitro transcription from the PDR2 promoter
In addition to the binding studies reported above, it was essential to prove by in vitro transcription that the strict TBP-DR2 dependency of the reported constructs had been retained.
As shown in Figure 4A, the dependence of the PDR2 transcription on TBP-DR2 was not abolished as a consequence of the subcloning. A specific signal could be detected only in the presence of TBP-DR2 (lanes 5–7). In contrast, PDR2 RNA was not detectable, either without (lane 1) or with rising quantities of TBPwt (lanes 2–4), whereas an efficient RNA synthesis occurred even with the smallest quantity of TBP-DR2 (lane 5). The PDR2 transcript shows the expected length of ∼100 nt. At the top of the gel shown in Figure 4A, one can again recognise the vector transcript of ∼320 nt length, which is likewise synthesised in a TBP-DR2 dependent fashion as already discussed.
Figure 4.

Functional investigations of the cloned PDR2 construct. The sequence section of the pBR322 plasmid with homologies to the mammalian U6 promoter was subcloned into the multiple cloning site of pUC18 and subsequently analysed by transcription experiments in vitro. (A) Dependence of the PDR2 clone on TBP-DR2. A transcription system, based on phosphocellulose fractions PCB and PCC (lanes 1–7), corresponding to the reconstituted transcription system shown in Figure 1, was supplemented either with TBPwt (lanes 2–4; 5, 10 and 20 ng) or TBP-DR2 (lanes 5–7; 5, 10 and 20 ng). Lane 1 shows the control sample without additional recombinant human TBP. The asterisk indicates the TBP-DR2 dependent vector transcript of ∼320 nt. The transcript resulting from the subcloned promoter is marked by PDR2. (B) α-Amanitin characterisation of the PDR2 transcription in vitro. Three identical transcription samples contained 10 µl PCB, 20 µl PCC, 10 ng recombinant human TBP-DR2 and 1 µg pUPDR2 as template. The control sample without the inhibitor α-amanitin is shown in lane 1. The samples shown in lanes 2 and 3 contained α-amanitin at a concentration of 2 and 300 µg/ml respectively. The PDR2 transcript is appropriately indicated. The signal at the bottom of the gel represents tRNA contained as a carrier.
In order to ensure that the PDR2 RNA is a transcript of RNA polymerase III, three transcription samples containing fraction PCB, PCC and TBP-DR2 were analysed in the presence of the inhibitor α-amanitin (Fig. 4B). The result shows that the low α-amanitin concentration (2 µg/ml; Fig. 4B, lane 2) had no influence on the PDR2 signal and the long vector transcript (shown by an asterisk in Fig. 4B) in comparison to the control reaction (lane 1). With the high α-amanitin concentration (300 µg/ml; Fig. 4B, lane 3), however, both specific signals (PDR2, and as shown by an asterisk) disappeared, thus proving that the examined transcripts are synthesised by RNA polymerase III (27).
Determination of the initiation nucleotide
On the basis of the sequence homology to the U6 initiation region and its distance to the terminator, which corresponds to the length of the PDR2 transcript, it was assumed that the sequence section, homologous to the U6 initiation region is the transcription start point of PDR2 RNA. In order to prove this assumption and to determine the starting nucleotide (‘+1’) exactly, primer extension experiments were conducted (Fig. 5). All samples were treated with DNase I before the hybridisation reaction, in order to digest the template DNA. Transcription samples containing either PCB/PCC or PCC/TFIIIBα fractions were supplemented with TBP-DR2 in lanes 3 and 5. Sample 1 contained no protein but DNA, nucleotides and RNase-inhibitor and served as a control for non-specific primer/plasmid–DNA hybrids.
Figure 5.
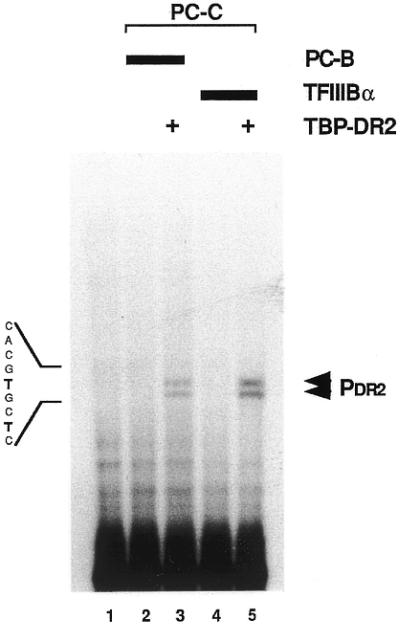
Primer extension analysis of the PDR2 RNA. Transcription samples containing either PCB/PCC (5/15 µl; lanes 2 and 3) or TFIIIBα/PCC (20/15 µl; lanes 4 and 5) were tested with (lanes 3 and 5) or without (lanes 2 and 4) recombinant TBP-DR2 respectively. After purification of the resulting RNA the samples were hybridised with a radioactively labelled oligonucleotide (PDR2467–490) complementary to the PDR2 RNA. After the reverse transcription reaction the resulting cDNAs were separated on a 6% denaturing PAA gel in comparison with a sequencing reaction conducted with the same radioactively labelled oligonucleotide.
The reverse transcript (RT) reveals a double band. These two specific RT signals arise in all samples containing TBP-DR2 (Fig. 5, lanes 3 and 5). The signal strength depends on the transcription system used and shows a higher transcription efficiency with the TFIIIBα fraction (lane 5) than with the PCB fraction (lane 3).
The alignment of the two RT signals with a sequencing reaction conducted in parallel shows that both 3′ ends of the RTs correspond to a thymidine residue in the PDR2 sequence of the coding strand. It can be assumed that the longer of both signals reflects the complete reverse transcript. It is very unlikely that these two signals were caused by an alternative initiation process, because in the in vitro transcription reaction only one signal could be detected (Fig. 4A, lanes 5–7).
Construction and functional characterisation of additional TBP-DR2-dependent vector variants
One objective of the investigations concerning the PDR2 promoter was to establish an inducible, pol III-catalysed transcription system suitable for the expression of an RNA sequence of interest. Therefore, it was necessary to determine whether an exchange of the transcribed region downstream of the PDR2 promoter would show any influence on the transcription efficiency. We therefore replaced the artificial PDR2 coding sequence by the coding sequence of the human U6 gene (PDR2/hU6). Additionally, a vector was designed which enabled any sequence between the PDR2 promoter and terminator to be cloned, with the aim of expressing such a sequence in vivo only in the presence of TBP-DR2 (PDR2-Expression). A schematic representation of the resulting constructs PDR2/hU6 and PDR2-Expression is shown in Figure 2B.
These new constructs were subsequently examined particularly with respect to their strict dependence on TBP-DR2. In Figure 6, the in vitro transcription of pUPDR2, pUPDR2-Expression and PDR2/hU6 were compared to each other. In the presence of TBP-DR2, all templates were transcribed efficiently and specifically. As expected, the length of the specific transcripts varied according to the size of the individual constructs used (lanes 2, 5 and 8). Without addition of TBP-DR2 to the transcription reaction, or in the presence of TBPwt, none of these new constructs yielded a specific signal. This result clearly demonstrated that the strict requirement for TBP-DR2 for transcription from the examined promoter is not influenced by sequences located in its 3′ region.
Figure 6.

In vitro transcription of pUPDR2, pUPDR2/hU6 and pUPDR2-Expression in the presence of TBPwt or TBP-DR2 respectively. In vitro transcription was performed and the positions of the individual transcripts are indicated. Three transcription samples of each template (lanes 1–3, pUPDR2; lanes 4–6, pUPDR2-Expression; lanes 7–9, pUPDR2/hU6) were analysed in a transcription system based on 10 µl phosphocellulose fraction B plus 20 µl phosphocellulose fraction C (lanes 1, 4 and 7) and were supplemented with either 20 ng TBP-DR2 (lanes 2, 5 and 8) or 20 ng TBPwt (lanes 3, 6 and 9).
Transcription factors required for expression from the TBP-DR2-dependent promoter
To determine which transcription factors besides TBP-DR2 are required for expression from the PDR2 promoter, it was necessary to replace the relatively crude phosphocellulose fractions with more purified transcription factors. As a first step the PCB fraction was replaced by TFIIIBα- or TFIIIBβ-containing fractions and transcription was compared to the hU6 gene as a control. Both the hU6- and the PDR2 genes were progressively transcribed by increasing amounts of TFIIIBα (Fig. 7A, lanes 2–4 and 9–11) but not TFIIIBβ (lanes 5–7 and 12–14).
Figure 7.

Transcription factors required for the efficient transcription from the TBP-DR2-dependent promoter. (A) Transcription of pUhU60.35 and pUPDR2 in vitro in the presence of TFIIIBα or TFIIIBβ. Increasing amounts of a TFIIIBα fraction (lanes 2 and 9, 10 µl; lanes 3 and 10, 20 µl; lanes 4 and 11, 30 µl) or a TFIIIBβ fraction (lanes 5 and 12, 2.5 µl; lanes 6 and 13, 5 µl; lanes 7 and 14, 10 µl) were reconstituted with 15 µl phosphocellulose fraction C (lanes 1–14). The samples with pUhU60.35 (lanes 1–7) were supplemented with 20 ng recombinant TBPwt (lanes 1–7) while the samples with pUPDR2 (lanes 8–14) contained 20 ng TBP-DR2. Control reactions without TFIIIB activity are shown in lanes 1 and 8 respectively. (B) Replacement of phosphocellulose fraction C by PBP, TFIIIC1 and TFIIIU in the in vitro transcription of pUPDR2 and pUPDR2/hU6. Individual components of the PCC fraction were added in different combinations as indicated above each lane: PBP, 2 µg MQ-fraction; TFIIIC1, 0.4 µg ESF-fraction; TFIIIU, 1.8 µg ESF-fraction. The reactions were incubated and processed as described, using 1 µg of pUPDR2 (lanes 1–7) or 1 µg pUPDR2/hU6 (lanes 8–14) as template. All reactions were supplemented with 20 ng recombinant TBP-DR2 and 20 µl TFIIIBα fraction. Lanes 1 and 8 show the positive control with 15 µl PCC fraction containing the TFIIIC1, pol III, PBP and TFIIIU activity. All other samples (lanes 2–7 and 9–14) contained 2 µl purified pol III.
To examine which components of the PCC fraction were necessary for transcription of the PDR2 promoter, the PCC fraction was replaced by more purified factors contained therein. The participation of TFIIIC1, TFIIIU and PBP, purified by EDF and MonoQ chromatography was investigated by testing these factors in different combinations in a transcription system supplemented with TFIIIBα, pol III and TBP-DR2 (Fig. 7B). In a reconstituted in vitro transcription assay (data not shown) it was shown that TFIIIC2 does not play a role in the transcription of the PDR2 construct.
As was expected, the pol III/TFIIIBα basal-system alone led to no specific RNA transcript (Fig. 7B, lanes 2 and 9 respectively). The individual addition of TFIIIU- (lanes 3 and 10), TFIIIC1- (lanes 4 and 11) or PBP-containing fractions (lanes 5 and 12) likewise resulted in no efficient RNA-synthesis. The combination of TFIIIC1/PBP (lanes 6 and 13) resulted in only a low transcription activity from both genes. Efficient RNA synthesis was only observed after the combination of all three fractions (TFIIIU, TFIIIC1 and PBP; Fig. 7B, lanes 7 and 14).
From this, it can be concluded that all constructs containing the PDR2 promoter are expressed independently of their transcribed sequence and require the same factors (TFIIIBα, TFIIIU, TFIIIC1 and PBP) as the mammalian U6 gene.
Inducible expression of TBP-DR2 in HeLa cells
To establish an inducible RNA polymerase III expression system in vivo, a system for the modulated expression of TBP-DR2 was developed in HeLa cells. For this purpose a tetracycline (doxycycline) inducible mammalian expression system was used.
The DNA sequence coding for histidine tagged TBP-DR2 was cloned into a suitable expression plasmid under the control of the tetracycline responsive element (TRE). After transfection of HeLa Tet-on cells (28), positive cell clones expressing TBP-DR2 in the presence of doxycycline, were identified by anti-histidine antibodies in a western blot.
Figure 8A shows that in the uninduced status no TBP with histidine tag could be detected (lane 1), while it could clearly be recognised after induction (lane 2). In the sample with recombinant histidine tag-TBP, serving as a positive control, a signal of identical molecular mass could be found (Fig. 8A, lane 3), whereas it was missing in the samples with HeLa whole-cell extract and those containing TBP without a histidine tag (lanes 4 and 5).
Figure 8.

Tetracycline inducible expression of TBP-DR2 in HeLa cells. (A) Western blot analysis. Cellular extracts from uninduced (lane 1) and doxycycline-induced cells (lane 2) of one of the selected clones were probed with anti-histidine antibodies. Recombinant TBP-DR2 with a histidine tag (10 ng; lane 3), 25 µg HeLa whole-cell extract (WCE; lane 4) and 10 ng recombinant TBPwt without histidine tag (lane 5) served as controls. (B) Functional investigation of TBP-DR2 expressed in HeLa cells. To analyse whether the expressed TBP-DR2 was functionally active, 50 µg S100 from HeLa cells expressing TBP-DR2 was used to transcribe the PDR2 promoter (lanes 4–6) in vitro before (lane 5) and after (lane 6) doxycycline induction of these HeLa cells. Additionally, 50 µg S100 from untransfected HeLa Tet-on cells was used as a negative control (lane 4). As a control, all three extracts were simultaneously analysed by in vitro transcription of pUhU60.35 (lanes 1–3) and pUVAI DNA (lanes 7–9).
When testing for the entire TBP content with monoclonal anti-TBP antibodies, a specific signal could be detected in both conditions, i.e. in positive and negative controls (data not shown).
Finally the question was addressed whether the expressed TBP-DR2, containing the histidine tag, was functionally active (Fig. 8B). Cytoplasmic extracts from uninduced and induced HeLa-DR2 cells were analysed in an in vitro transcription assay using the PDR2 plasmid as template. Previous experiments had documented (see above) that this construct is transcribed only in the presence of active TBP-DR2. At the same time, a transcription reaction with the mU6 (Fig. 8B, lanes 1–3) or the VAI gene (lanes 7–9) was conducted. S100 from untransfected HeLa Tet-on cells was used as a negative control for transcription of the PDR2 construct.
With S100 from uninduced cells and the PDR2 gene as template, the specific transcript was barely visible (Fig. 8B, lane 5). Also, no transcript from the PDR2 gene was found in the absence of TBP-DR2 with HeLa-S100 as negative control (lane 4) while with S100 from induced HeLa-DR2 cells a transcript of the PDR2 gene could be detected (lane 6). The transcription with pUVAI and pUmU6 also served to prove that the three examined extracts were comparably active and differences observed in the transcription efficiency of the PDR2 gene were not attributable to different activities of the extracts. This result clearly demonstrated that the TBP-DR2 protein, which is synthesised in HeLa-DR2 cells after doxycycline induction, is functionally active. A stable HeLa-DR2 clone was thus isolated, which is able to synthesise TBP-DR2 after doxycycline induction, and therefore can initiate transcription from the PDR2 promoter in vitro.
DISCUSSION
Identification of a promoter sequence which is exclusively controlled by TBP-DR2
In order to develop an inducible RNA polymerase III system we first constructed a pol III promoter, which can exclusively be activated by mutated TBP variants but not by TBPwt. We initially concentrated our efforts on the promoter of the human U6 gene, which offers several advantages. It represents a pol III gene showing a high rate of synthesis with multiple rounds of initiation and efficient termination. More importantly, it is entirely controlled by an upstream promoter containing a TATA-box, and does not pose the problems associated with gene internal pol III promoters, which could conceivably limit the expression of arbitrary DNA sequences under the control of such a promoter.
Based on the work of Strubin and Struhl (15) and Heard et al. (16) we attempted to construct an RNA polymerase III promoter which operated independently from TBPwt. In these reports, mutants of the TBP molecule from yeast and human were analysed, and showed an altered DNA binding specificity due to an exchange of three amino acids. In the yeast system (Saccharomyces cerevisiae; 15) as well as in plant cells (Nicotiana plumbaginifolia; 16), two variants of the TATA-box binding protein (DR1 or DR2) exhibited an altered specificity for several mutations of the TATA-box. The most efficient of these mutations was the sequence TGTAAA. The described mutants of TBP were able to suppress the TGTAAA mutation in vivo.
In order to obtain a promoter controlled by TBP-DR2, we constructed TATA-box mutants of the human U6 snRNA gene. Since an inverse orientation of the TATA-box has been described for the hU6 gene (29), one of these mutants contained a reverse TGTAAA-box (hU60.35 –29<<) corresponding to the orientation of the authentic TATA-box in the wild-type U6 gene. As expected, functional investigation of these TATA-box mutants by in vitro transcription showed a strong reduction of the transcription efficiency. However, none of these mutations could be suppressed by supplementation of the transcription reactions with either recombinant human TBP-DR1 or TBP-DR2.
A possible explanation for the differences between our results and those of Strubin and Struhl (15) and Heard et al. (16) may be related to the fact that the experiments were conducted in different species. For example, the distances of the TATA-box to other promoter elements and to the transcription start point vary between these different organisms (30). Because of the differences in the experimental systems, the functional characterisation of the TATA-box mutants described here cannot be compared directly with those previously described (15,16).
In spite of the failure to exploit the established TATA-box mutants for our purpose, we continued our search for other variant TATA-box sequences. We were fortuitously led to a promoter sequence contained in the plasmid pUC18, which is efficiently transcribed by RNA polymerase III only in the presence of TBP-DR2 but which is completely refractory even to high concentrations of TBPwt. This transcript originates from a sequence located in the β-lactamase gene of pBR322, which shows a high degree of homology to the promoter of the mammalian U6 gene. Six of seven nucleotides in the initiation region of the U6 snRNA and the TBP-DR2 dependent promoter are identical and primer-extension analyses have confirmed that initiation of the DR2-RNA occurs in this sequence section.
The region of the PDR2 promoter which is homologous to the PSE element is divided into the typical two halves, previously described for the mammalian U6 genes by Simmen et al. (31) and Wanandi et al. (32). They are, however, separated by six nucleotides in the construct described here, and the potential PSE and the variant TATA-box partially overlap. We hence shifted the PSE to the ‘wild-type’ distance relative to the TATA-box but this had no effect on transcription efficiency (data not shown). Since the DSE has little effect on the in vitro transcription of the human U6 gene (7), the effect of this element was not further investigated in this context.
Functional characterisation of the PDR2 promoter by DNase I protection and in vitro transcription
The observed homologies led us to the assumption that the DR2-dependent promoter, although representing an artificial construct, may function as a quasi 5′-regulated RNA pol III promoter and at the same time may fulfil the prerequisites essential for our approach.
As a first step to verify its functional significance, the region of the PDR2 promoter to which TBP-DR2 binds was determined by footprint analyses. The section of the PDR2 promoter protected by TBP-DR2 spans 15 nucleotides on both strands of the PDR2 DNA and is bordered by nucleotides at –20 and –34 relative to the transcription start point. This binding region of TBP-DR2 lies in an AT-rich sequence, which is characteristic for the binding sequence of TBP (consensus sequence: TATAAA). The sequence in the middle of the identified binding site is AGTAAA. This differs only in one nucleotide from the recognition sequence published for TBP-DR2 (TGTAAA) in yeast and plant cells (15,16) and therefore could represent the specific recognition sequence for the interaction of TBP-DR2 with the artificial promoter outlined here. However, this still requires further investigation. The distance of this presumed TATA-box to the starting nucleotide of transcription is 24 nucleotides, which agrees with the distance of the TATA-box to the starting nucleotide in the hU6 promoter (30,33). It should be stressed, however, that binding of TBPwt to the PDR2 promoter could not be detected in any of the footprint experiments.
Besides the binding of TBP-DR2 to the PDR2 promoter it was shown that this protein also binds to the hU6 TATA-box and that the boundaries of DNase I protection are identical to those of the TBPwt footprint (data not shown). These data agree with those of Strubin and Struhl (15) showing the binding of the mutated yeast TBP to the wild-type TATA-box of S.cerevisiae (TATAAA) and they support the observation that TBP-DR2 can replace the wild-type protein in the transcription of the hU6 gene in vitro (Fig. 1, lanes 12–14).
The transcription factors TFIIIBα, TFIIIU, TFIIIC1 and PBP were found to be essential for the in vitro expression of the PDR2 constructs, which is in accordance with the factors required for the expression of the mammalian U6 gene (13).
These data support the conclusion that the PDR2 promoter, although representing an artificial construct, behaves like a type 3 pol III promoter. It employs the same transcription factors, except that it can only be activated by TBP-DR2 but not by wild-type TBP. It can therefore be concluded that one of the prerequisites initially postulated, namely the construction of a promoter allowing the specific expression of a desired sequence only in the presence of TBP-DR2, was fulfilled.
Transcription efficiency does not depend on sequences located downstream of the initiation region. This fact is of crucial importance for the potential employment of this promoter in vivo to synthesise a copy RNA from short DNA sequences corresponding to the length of a typical pol III gene and lacking an internal pol III termination signal. This system may be of particular interest for the expression of a short copy RNA, complementary to the sequence for the initiation of translation of a specific mRNA (antisense RNA; 34). Although requiring experimental verification, it is also conceivable to synthesise a short double-stranded RNA in vivo which corresponds to a sense and antisense sequence of an endogenous mRNA. The cognate mRNA is then degraded and the gene is silenced. This type of post-transcriptional gene silencing is called RNA interference (35,36). This sequence specific degradation of mRNA could prove to be useful for the analysis of gene function and/or the employment as an anti-viral or anti-tumor agent (37).
Expression of TBP-DR2 in HeLa cells after induction by tetracycline
For the modulated expression of TBP-DR2 in mammalian cells in vivo, a tetracycline-controlled system was used. The gene coding for TBP-DR2 was cloned into the TRE-plasmid directly behind the tetracycline-dependent CMV promoter, so that its expression could be regulated by the addition of tetracycline or doxycycline (pTRE/TBP-DR2). The cloned TBP-DR2 insert contained a translation start point as well as a histidine tag, enabling one to discriminate by western blot between the recombinant TBP-DR2 and the endogenous TBPwt. The subcloned vector construct (pTRE/TBP-DR2) was transfected into HeLa Tet-on cells expressing the reverse Tet-activator upon induction with tetracycline.
The selection of clones inducibly expressing TBP-DR2 was achieved in a western blot with antibodies against the histidine tag. Figure 8A shows one of the clones which synthesised TBP-DR2 after the induction with doxycycline, while this protein was barely detectable in uninduced cells. The functional analysis of this clone revealed that the induced expression of TBP-DR2 in HeLa cells yielded an intact protein. This was verified by in vitro transcription in cellular extracts from such cells proving that TBP-DR2 was capable of correctly recognising and activating the PDR2 promoter. The fact that transcription from the hU6 and VAI genes was not altered after induction shows that TBP-DR2 does not exert a negative effect on the expression of these genes and supports the conclusion that it specifically activates the PDR2 promoter.
In the meantime a system has been developed, which will be used in future experiments to further minimise the expression level of the recombinant protein in uninduced cells. One of these systems contains a tetracycline-controlled hybrid protein as a silencer, which actively represses the transcription of the desired gene (38).
Future experiments, aimed at the in vivo application of the model system described here, will involve the fusion of potentially interesting DNA sequences to the PDR2 promoter and the stable integration of such constructs in cell lines. An important prerequisite for such experiments was to prove that the expression of TBP-DR2 had no influence on the viability of the transfected cells. We have shown that the stably transfected cells, inducibly expressing TBP-DR2, could be maintained for several months, hence proving that TBP-DR2 has no toxic effect on these cells.
In summary, we were able to generate a HeLa cell clone in which a modified form of the TBP (TBP-DR2) could be expressed upon induction. This transcription factor is essentially required for the RNA polymerase III catalysed expression governed by the PDR2 promoter. In the future, this system can be potentially employed for the regulated in vivo expression of a short RNA sequence (for example, antisense or double-stranded RNA), under the control of the PDR2 promoter.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Ulla Kopiniak, Frauke Seifart and Sarah Fehl for expert technical assistance and Stephan Weser for helpful discussions. This work was supported by grants of the Deutsche Forschungsgemeinschaft and the European Union.
References
- 1.Willis I.M. (1993) RNA polymerase III: genes, factors and transcriptional specificity. Eur. J. Biochem., 212, 1–11. [DOI] [PubMed] [Google Scholar]
- 2.Yoshinaga S.K., Boulanger,P.A. and Berk,A.J. (1987) Resolution of human transcription factor TFIIIC into two functional components. Proc. Natl Acad. Sci. USA, 84, 3585–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paule M.R. and White,R.J. (2000) Transcription by RNA polymerases I and III. Nucleic Acids Res., 28, 1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z. and Roeder,R.G. (1996) TFIIIC1 acts through a downstream region to stabilize TFIIIC2 binding to RNA polymerase III promoters. Mol. Cell. Biol., 16, 6841–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teichmann M. and Seifart,K.H. (1995) Physical separation of two different forms of human TFIIIB active in the transcription of the U6 or the VAI gene in vitro. EMBO J., 14, 5974–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel G.R. (1991) RNA polymerase III transcription of genes that lack internal control regions. Biochem. Biophys. Acta, 1088, 1–9. [DOI] [PubMed] [Google Scholar]
- 7.Waldschmidt R., Wanandi,I. and Seifart,K.H. (1991) Identification of transcription factors required for the expression of mammalian U6 genes in vitro. EMBO J., 10, 2595–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy S., Yoon,J.-B., Gerster,T. and Roeder,R.G. (1992) Oct-1 and Oct-2 potentiate functional interactions of a transcription factor with the proximal sequence element of small nuclear RNA genes. Mol. Cell. Biol., 12, 3247–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon J.B., Murphy,S., Bai,L., Wang,Z. and Roeder,R.G. (1995) Proximal sequence element-binding transcription factor (PTF) is a multisubunit complex required for transcription of both RNA polymerase II- and RNA polymerase III-dependent small nuclear RNA genes. Mol. Cell. Biol., 15, 2019–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon J.-B. and Roeder,R.G. (1996) Cloning of two proximal sequence element-binding transcription factor subunits (γ and δ) that are required for transcription of small nuclear RNA genes by RNA polymerases II and III and interact with the TATA-binding protein. Mol. Cell. Biol., 16, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadowski C.L., Henry,R.W., Lobo,S.M. and Hernandez,N. (1993) Targeting TBP to a non TATA-box cis-regulatory element: a TBP-containing complex activates transcription from snRNA promoters through the PSE. Genes Dev., 7, 1535–1548. [DOI] [PubMed] [Google Scholar]
- 12.Henry R.W., Sadowski,C.L., Kobayashi,R. and Hernandez,N. (1995) A TBP-TAF complex required for transcription of human snRNA genes by RNA polymerases II and III. Nature, 374, 653–656. [DOI] [PubMed] [Google Scholar]
- 13.Oettel S., Kober,I. and Seifart,K.H. (1998) The activity binding to the termination region of several pol III genes represents a separate entity and is distinct from a novel component enhancing U6 snRNA transcription. Nucleic Acids Res., 26, 4324–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmen K.A., Bernués,J., Parry,H.D., Stunnenberg,H.G., Berkenstam,A., Cavallini,B., Egly,J.M. and Mattaj,I.W. (1991) TFIID is required for in vitro transcription of the human U6 gene by RNA-polymerase III. EMBO J., 10, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strubin M. and Struhl,K. (1992) Yeast and human TFIID with altered DNA binding specifity for TATA elements. Cell, 68, 721–730. [DOI] [PubMed] [Google Scholar]
- 16.Heard D.J., Kiss,T. and Filipowicz,W. (1991) Both Arabidopsis TATA binding protein (TBP) isoforms are functionally identical in RNA-polymerase II and III transcription in plant cells: evidence for gene-specific changes in DNA binding specifity of TBP. EMBO J., 12, 3519–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider H.R., Waldschmidt,R., Jahn,D. and Seifart,K.H. (1989) Purification of human transcription factor IIIC and its binding to the gene for ribosomal 5S RNA. Nucleic Acids Res., 17, 5003–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weil P.A., Segall,J., Ng,S.Y. and Roeder,R.G. (1979) Faithful transcription of eukaryotic genes by RNA polymerase III in systems reconstituted with purified DNA templates. J. Biol. Chem., 254, 6163–6173. [PubMed] [Google Scholar]
- 19.Jahn D., Wingender,E. and Seifart,K.H. (1987) Transcription complexes for various class III genes differ in parameters of formation and stability towards salt. J. Mol. Biol., 193, 303–313. [DOI] [PubMed] [Google Scholar]
- 20.Oettel S., Härtel,F., Kober,I., Iben,S. and Seifart,K.H. (1997) Human transcription factors IIIC2, IIIC1 and a novel component IIIC0 fulfil different aspects of DNA binding to various pol III genes. Nucleic Acids Res., 25, 2440–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teichmann M., Dieci,G., Huet,J., Rüth,J., Sentenac,A. and Seifart,K.H. (1997) Functional interchangeability of TFIIIB components from yeast and human cells in vitro. EMBO J., 16, 4708–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waldschmidt R. and Seifart,K.H. (1992) TFIIA is required for in vitro transcription of mammallian U6 genes by RNA polymerase III. J. Biol. Chem., 267, 16359–16364. [PubMed] [Google Scholar]
- 23.Düring F., Gerhold,H. and Seifart,K.H. (1990) Transcription factor USF from duck erythrocytes transactivates expression of the histone H5 gene in vitro by interacting with an intragenic sequence. Nucleic Acids Res., 18, 1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kyhse-Andersen J. (1984) Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J. Biochem. Biophys. Meth ., 19, 203–209. [DOI] [PubMed] [Google Scholar]
- 25.Seifart K.H., Wang,L., Waldschmidt,R., Jahn,D. and Wingender,E. (1989) Purification of human transcription factor IIIA and its interaction with a chemically synthesized gene encoding human 5S rRNA. J. Biol. Chem., 264, 1702–1709. [PubMed] [Google Scholar]
- 26.Perrin S. and Gilliland,G. (1990) Site-specific mutagenesis using asymmetric polymerase chain reaction and a single mutant primer. Nucleic Acids Res., 18, 7433–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seifart K.H. and Sekeris,C.E. (1969) ) α-Amanitin, a specific inhibitor of transcription by mammalian RNA polymerase. Z. Naturforsch [B], 24, 1538–1544. [DOI] [PubMed] [Google Scholar]
- 28.Gossen M., Freundlieb,S., Bender,G., Muller,G., Hillen,W. and Bujard,H. (1995) Transcriptional activation by tetracyclines in mammalian cells. Science, 268, 1766–1769. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y. and Stumph,W.E. (1995) RNA polymerase II/III transcription specifity determined by TATA box orientation. Proc. Natl Acad. Sci. USA, 92, 8606–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waibel F. and Filipowicz,W. (1990) U6 snRNA genes of Arabidopsis are transcribed by RNA polymerase III but contain the same two upstream promoter elements as RNA polymerase II-transcribed U-snRNA genes. Nucleic Acids Res., 18, 3451–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmen K.A., Bernués,J., Lewis,J.D. and Mattaj,I.W. (1992) Cofractionation of the TATA binding protein with the RNA polymerase III transcription factor TFIIB. Nucleic Acids Res., 20, 5889–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wanandi I., Waldschmidt,R. and Seifart,K.H. (1993) Mammalian transcription factor PBP.Characterization of its binding properties to the proximal sequence element of U6 genes. J. Biol. Chem., 268, 6629–6640. [PubMed] [Google Scholar]
- 33.Kunkel G.R., Maser,R.L., Calvet,J.P. and Pederson,T. (1986) U6 small nuclear RNA is transcribed by RNA polymerase III. Proc. Natl Acad. Sci. USA, 83, 8575–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohuczy D. and Phillips,M.I. (2000) Antisense inhibition of the renin-angiotensin system in brain and peripheral organs. Methods, 22, 197–209. [DOI] [PubMed] [Google Scholar]
- 35.Bass B.L. (2000) Double-stranded RNA as a template for gene silencing. Cell, 101, 235–238. [DOI] [PubMed] [Google Scholar]
- 36.Sijen T., Kooter,J.M. (2000) Post-transcriptional gene-silencing: RNAs on the attack or on the defense? Bioessays, 22, 520–531. [DOI] [PubMed] [Google Scholar]
- 37.Bosher J.M., Labouesse,M. (2000) RNA interference: genetic wand and genetic watchdog. Nat. Cell Biol., 2,E31––36.. [DOI] [PubMed]
- 38.Freundlieb S., Schirra-Müller,C. and Bujard,H. (1999) A tetracycline activation/repression system with increased potential for gene transfer into mammalian cells. J. Gene Med., 1, 4–12. [DOI] [PubMed] [Google Scholar]