

Abstract
Shock syndromes are of three types: cardiogenic, hemorrhagic and inflammatory. Hemorrhagic shock has its initial deranged macro-hemodynamic variables in the blood volume and venous return. In cardiogenic shock there is a primary pump failure that has cardiac output/mean arterial pressure as initial deranged variables. In Inflammatory Shock it is the microcirculation that is mainly affected, while the initial deranged macrocirculation variable is the total peripheral resistance hit by systemic inflammatory response.
Keywords: Physiology, physiopathology, shock
INTRODUCTION
The three main shocks (cardiogenic, hemorrhagic and inflammatory) differ in the primum movens i.e., the initial circulation variable that becomes deranged: HS is a failure of the peripheral circulation that has its initial deranged variables in the blood volume and venous return, and to follow, cardiac output/mean arterial pressure; in CS is the pump that fails and has its initial deranged variables in cardiac output/mean arterial pressure; in IS the microcirculation is affected while the initial deranged macrocirculation variable is the total peripheral resistance hit by systemic inflammatory response (SIR) or by I-R phenomenon [Figure 1] [Schemes 1–3].
Figure 1.
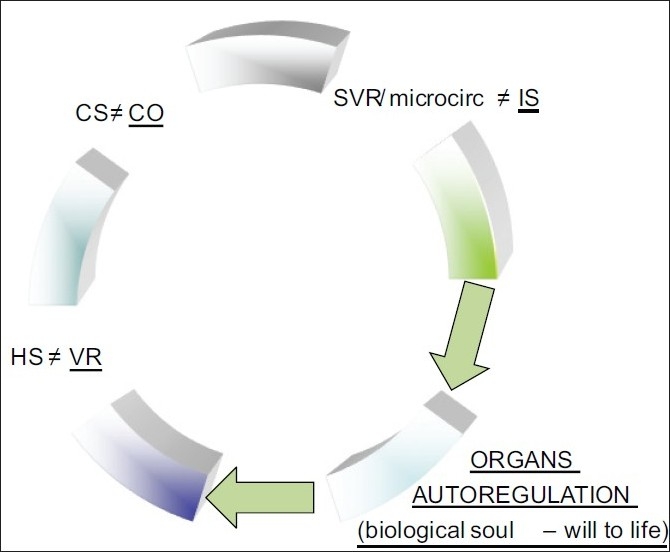
Primary variable derangement in abnormal macrocirculation
Scheme 1.
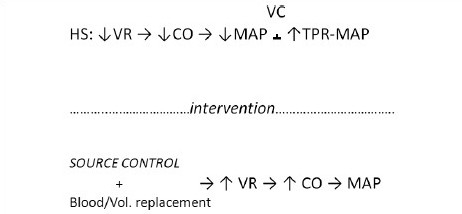
Variables interaction in reversible HS
Scheme 3.
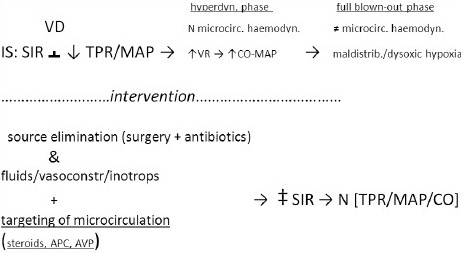
Variables interaction in reversible IS
Scheme 2.
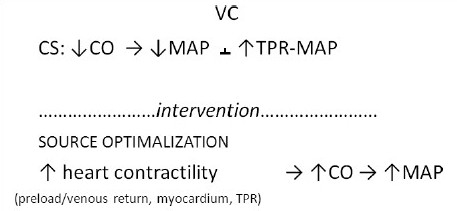
Variables interaction in reversible CS
All shocks in full-blownout phase are effectively vasoconstrictors, including the IS [Schemes 1–3]. What makes SS different from the other variants of IS is the initial hyperdynamic response of SS, lipopolysaccharide (LPS) and other bacterial toxins mediated, that is responsible of the hyperdynamic, physiologically compensatory, vasodilatory phase.
OXYGEN DELIVERY
The delivery of oxygen to cells (DO2) depends on the content of oxygen in the blood mainly combined to hemoglobin (Hb), which is regulated by the respiratory system, and from its transport in the blood, which is function of the cardiac output/mean arterial pressure/peripheral vascular resistance inter-relationships and of the oxygen extraction/consumption rate at cellular/tissues level, the latter being the main driver, controller and modulator of the oxygen delivery system. [Appendix A] Because oxygen has a low solubility in plasma, it is oxygen carried by red blood cells (RBC) specifically, and not ‘blood’ (plasma and RBCs) in general, which determines arterial oxygen content (CaO2) in the oxygen delivery equation. O2 loose in the blood which is not combined to Hb accounts instead for the component of partial pressure of oxygen (PaO2) that can be increased. Once Hb is completely saturated with oxygen (HbO2) oxygenation is increased either by increasing its concentration on air (fraction inspired oxygen-FiO2) or by increasing the pressure into the alveoli with which is administered in the air-oxygen mixture (intra-alveolar pressure). Hypoxia, which is low oxygen to tissues, is classified into three main categories: scarce oxygen offer from macrocirculation, maldistribution of offer and incapacity of consumption. Hypoxemia or respiratory failure, is a frequent cause of hypoxia pertaining to the ingress/egress of gases in and out the body; shock, a cause of hypoxemia or dysoxia, pertains instead to their exchange in the target organs and tissues. There is an indissoluble inter-relation and overlapping between shock and hypoxia in biological and conceptual terms: etiology and treatment often overlap.
Appendix A.

Correlation between macro-hemodynamic and biochemical vital functions and factors
Chronic anemia is practically irrelevant as cause of hypoxia. In the absence of pathological Hb or acute RBCs loss (acute hemorrhage), hypoxia is never observed to be symptomatic or clinically significant until Hb goes at around 3 g/dL.[1] The author has seen few patients with chronic anemia of 3 g/dL of Hb living normal life, maybe not classifiable as such for an athlete, but enough to allow them going shopping normally for quite some time complaining only of slight dizziness. Therefore, there must be some circulation variables and factors other than the cardiac output-mediated oxygen content increase, such as mitochondria adaptation speed and speed of erythropoietin’ synthesis, that with all likelihood make difference between clinically significant hypoxia as seen in a drop between 14 and 12 g/dL of an acute hemorrhage and the almost asymptomatic status of chronic anemia even at the lowest level like 3-5 g/dL.
WHAT IS CARDIAC RESERVE?
Cardiac reserve is the maximum quantity of blood that can be pumped above basal normal level during exercise or to compensate basic deficits within physiological limits. The lower is or becomes the myocardial functional reserve i.e., the healthy part of myocardium capable to answer to venous return variations during diastole with an increase of contractility according to Frank-Starling law, higher the chances of shock overlapping failure of the pump. This occurs because decreased compliance leads to increased left/right-ventricular end-diastolic pressures (L/R-VEDP) causing increased wall tension and increased oxygen consumption (VO2), which in turn decreases the cardiac output. Any failure of half of the heart will eventually affect all myocardium for different reasons, not the least the pressure that a dilated ventricle maintains on the other yet normal during diastole with consequential dysfunctional systolic phase. When LV dysfunction accompanies RV ischemia/ infarction, the RV becomes further compromised because of increased RV afterload and reduction in stroke volume. In such circumstances, the use of afterload-reducing agents or an intra-aortic counterpulsation device is often necessary to unload the LV and benefit the RV. Same result occurs in the situation of increased compliance of the myocardium in excess of the reflex capacity of its fibers to stretch and pump away the R-VEDV. The functionality of myocardium is therefore affected not only by venous return but also by myocardial contractility, wall tension and by afterload, which is the sum of the resistance of all arterioles as manifested by aortic wall tension during systole against which the heart has to produce a stroke volume. Myocardial contractility is affected by structural myocardium damage, rhythm disorders and valve diseases.
ACUTE REAL-TIME HEMODYNAMIC RESPONSE TO HEMORRHAGE AND HYPOXEMIA
The cardiovascular system responds to hypotension and hypovolemic shock by increasing the heart rate, increasing myocardial contractility and constricting peripheral blood vessels as result of the direct stimulation via the sympathetic system on heart and vessels by the cardiac and vasomotor centers in the reticular activating substance of lower pons and medulla oblongata. The cardiac and vasomotor centers via parasympathetic vagal nerves modulate heart rate and via sympathetic spinal cord-peripheral nerves control the basal tone of the entire circulation (heart, arterioles and venules), indirectly affecting macrocirculation and microcirculation driving pressures as consequence of upstream flows and pressures regulation [Appendix B].
Appendix B.
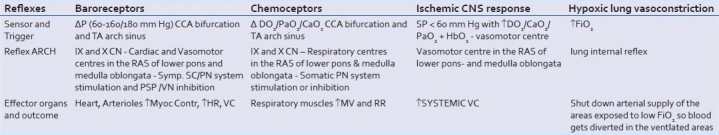
The four acute life-saving and maintaining hemodynamic reflexes
The sympathetic system releases catecholamines (noradrenaline and adrenaline) acting on α1-receptors, with a concomitant decrease of acetylcholine release from the vagi: tachycardic/inotropic and vasoconstrictor actions are mediated, respectively, by β1- and α1-receptors in heart and blood vessels;[2] muscarinic receptors mediate the action of acetylcholine released by the vagi. Endogenous acetylcholine is effective on heart but in-influent on vessels contrarily to exogenous acetylcholine. The adrenal medulla also participates as well to the two catecholamines release as a target organ of the overall sympathetic system stimulation.
This reflex arc is regulated by the baroreceptors in the common carotids bifurcation sinus and aortic arch that are sensitive to pressure variations peaks. The baroreceptor reflexes discharge continuously via vagal and glossopharyngeal nerves afferent sensory pathways, to the cardiac and vasomotor centers. The receptors discharge basally and continuously at pressures within a range between 50-60 and 160-180 mmHg, and do not respond above and below those values. After 1-2 days the baroreceptors stabilize the pressure to the new values. This is relevant in elderly people who are hypertensive. Arterioles and venulae are the target organs of the sympathetic nervous system vasoconstrictor tone. These reflexes are inhibitory in that more the afferent are stimulated by an increase of pressure more the vasomotor center is inhibited and the vasodilator-bradycardia vagal-mediated effect prevail, and vice versa.
Other areas in the brain like the hypothalamus and the cerebral cortex also affect the vasomotor centers and the baseline vasoconstrictor tone following psychological and physical stimuli like pain, cold or emotional shock.
The simple vasovagal syncope or any other syncope as a temporary hypotension and loss of consciousness and bradycardia followed by tachycardia on awakening is a result of intense stimulation of β2-receptors in striated muscles. Emotional stimuli from the hypothalamus directly abut to the cardiovascular centers in the midbrain, inhibiting the sympathetic center and stimulating the vagal controlling center to the heart, in the same time stimulating β2-receptors to the muscles with result of vasodilatation and bradicardia.
Arginine vasopressin (AVP) hormone levels are also increased in hypotensive hemorrhagic shock[3] stimulated by low pressure and low atrial filling. However, their impact is not as immediate or real time as cathecolamines because the antidiuretic hormone (ADH) as independent factor peaks in 2 hours and holds blood pressure for max 6 hours. The aldosterone/renin-angiotensin (A/RA) system is much slower in reaching its effects at full extent within 12-24 hours.
Fluids readjustments between the three compartments (intravascular, extracellular and intracellular) shift water in the circulation by osmosis within hours in association with the A/RA system.
Blood is shifted, literally squeezed, by the mechanism of vasoconstriction from skin and soft tissues first and from visceral organs to follow (gut, liver and lungs with kidney dysfunction) toward heart and brain due to the ingenious adrenergic distribution in the body organs that makes the brain the most protected organ. The different distribution of inducible nitric oxide synthase (iNOS) is likely also to play a role, though still to be defined.
Decompensation in HS occurs when natural endogenous catecholamines fail to maintain compensatory vasoconstriction and manifests as hypotension. Hyporeactivity to catecholamines has been seen as mediated by an enhanced release of calcium-dependent constitutive cNOS or eNOS and iNOS at a late stage,[4] and to a release of endothelin by the endothelium.[5] Endothelin is a vasoconstricting substance normally released by the damaged endothelium in crush injury. This finding indicates a possible damage by the endothelium by hypoxia as a contributing factor for decompensation in adjunct to ADH depletion/failure and constitutive NOS release.
Vascular reactivity and calcium sensitivity are increased in early shock (immediately and 30 min afterwards) and decreased in late shock (1 and 2 h after shock) linearly with noradrenaline (NE) and calcium cations (Ca++) concentrations. Rho-kinase activity goes also in parallel and in synchronicity with calcium and NE levels and positively correlates with the changes of vascular reactivity and calcium sensitivity. Rho-kinase is involved in the biphasic change of vascular reactivity and calcium sensitivity after hemorrhagic shock; it is possible that the enzyme regulates vascular reactivity through the regulation of calcium sensitivity. Vascular smooth muscle is desensitized to calcium after hemorrhagic shock, contributing to the development of vascular hyporeactivity. Rho-kinase-regulating agents, therefore, can be used on shock-induced vascular hyporeactivity.[6] Angiotensin II also may act via Rho-kinase intermediation while insulin exerts the opposite effect. AVP, alias ADH, can also constrict blood vessels by the activation of Rho-kinase.[7] A Protein-Kinase C has also been associated with the mechanism of action of AVP.[8] Calcium influxes, either from extracellular fluid or from the endoplasmic reticulum, are determinant for the final pathway to vascular contraction.
Moreover, atria and pulmonary artery have low-pressure baroreceptors responding to an increase of pressure with an increase of heart rate and decrease of ADH secretion by the hypothalamus; other atrial receptors directly stimulate the sinus node to increase rate on increase of pressure. These reflexes turn out useful on readjustments after volume overload in normal or pathological situations.
Hypoxemia triggers the same alarm reaction of hypotension by direct stimulation of the vasomotor center in the medulla oblongata (ischemic CNS response) with a powerful increase of blood pressure and in the same time with an increase of ventilation by direct triggering of the peripheral PaO2-sensitive chemoreceptors in the carotid bodies at the bifurcation of the common carotid artery offering to the medulla and pons respiratory centers. The vasomotor center therefore can discharge autonomously, directly stimulated by brain hypoxia only, independently on peripheral baroreceptors stimulation. This powerful blood pressure rise caused by intense stimulation of the vasomotor center occurs at pressures below 60 mmHg as a last ditch to increase total blood pressure and by extension brain perfusion pressures too in the same time, avoiding deleterious effects of direct brain arterioles stimulation thanks to the ingenious distribution of α1- and α2-receptors in the body.
Chemoreceptors, present in the very same carotid and aortic bodies where baroreceptors lie, respond to hypoxemia stimulus (PaO2 decrease) secondary to decreased CaO2 from decreased DO2 so do get triggered normally until pressures <80 mmHg and the pulse start becoming impalpable. Their effect is to increase DO2, the respiratory center stimulated by hypoxemia increases ventilation and minute volume.
Hypoxic vasoconstriction in the lungs is another reflex protective mechanism that shuts down arterial supply in areas of the lung exposed to low FiO2 and has the aim to divert blood to better ventilated areas. Nitric oxide (NO) inhalation can be used to open these vasoconstrictors area and decrease the physiological dead space.
The metaphysical aim of these hemodynamic responses is to maintain perfusion to the noble organs, which is absolute guarantee of the patient still being alive or capable to stay so. The hemodynamic aim of the response is governed by the imperative to safeguard the noblest organs of brain and heart by diverting blood from skin and soft tissues first and then from visceral organs too in the attempt to prevent brain and heart hypoxic damage.[9] This priority pattern has been engineered by nature according to whether the organ tissues are replaceable and at which extent and speed that can occur. The absence of direct sensitivity in the brain vessels to vasoconstricting catecholamines, due to the preponderance in humans of α2- and β2-receptors instead of α1 as in systemic circulation arterioles,[10] but to pH/PCO2 levels is a further example of the ingeniousness of nature in preserving brain blood supply with a double mechanism, a direct one, the metabolic, and an indirect one, the systemic pressure control. This explains why during any systemic sympathetic response blood is squeezed toward the brain by the vasoconstriction of the other organs and not from the brain, which effectively is the most protected organ. Two further observations can be made at glance from the distribution of receptors: the noteworthy and conspicuous absence of adrenergic innervations in the arteries, whose tone or tension is controlled by mechanical factors, namely pressure and flow and elastic compliance; the absence of β2 in the skin/mucosae, where vasodilatative action would deregulate temperature control and delicate functions like nutrient absorption, in the same time making both tissues the first victims of the sympathetic vasoconstriction during hemorrhage, aiming to divert by squeeze the blood toward more important organs and safeguard prioritized functions. Nothing in nature is without sense, or purpose, and no creation is more wonderful, unique sophisticated and higher than the human body. It is not by chance that the most pleasant physiological activities, nutrition and sexuality, are aimed to preserve the body and its continuation as bridge to infinite from creation.
MICROCIRCULATORY DISORDERS IN SEPSIS
Sepsis produces structural damages,[11–12] derangements and dysfunction in the microcirculation ecosystem, more exactly: a persistent and exaggerated LIR; a coagulopathy in the direction of procoagulant state; endothelial dysfunction in the sense of deficient or impaired reactivity and vasomotion that when evolves in failure heralds an irreversible state; a overproduction of NO, oxygen maldistribution and capillary stop flow, resulting in dysoxic-hypoxia.[13–20]
The body acute response to an insult is ontogenic and biologically universal. Inflammation, as reaction to insult, is universal in Man and animals and so the immune response with immunoglobulins and immune cells. The localized inflammatory response (LIR) is omnipresent and omnirespondent except on hemorrhage first hit where its actors are lost out circulation through the solution of continuity. It is an obligatory phenomenon characterized by microvascular and cellular changes aimed to bring neutrophils plus cellular and humoral factors into the damaged or injured area. These cellular and humoral factors (inflammatory mediators) have the property of amplifying in situ the inflammatory phenomenon and to reproduce it distally in systemic fashion [Appendix C]. The vascular changes go usually mainly in the direction of vasodilatation of the arterioles and initial formation of trasudate as ultrafiltrate of plasma constituted by water and few small diameter proteins following the increase in hydrostatic pressure. Three types of microvascular responses have been identified: an immediate transient response involving only venulae due to endothelial cells contraction; an immediate persistent response involving all the microcirculation components (arterioles, capillaries and venulae) due to direct endothelial damage; a delayed persistent response involving capillaries and venulae due to slow onset endothelial cells damage.[21–23] The stasis of flow and the fluid shift increases concentration (hematocrit) and facilitates migration of leukocytes and monocytes out of capillaries membranes toward the triggering situation through a consequential series of phenomena: margination from the laminar flow, rolling/loose bonding, adhesion, transmigration/diapedesis via contractility and pseudopods formation changes. The job is completed with other steps: chemotaxis to target, recognition of target via opsonines or lymphocytes, engulfment of the prey into phagosomes by macrophages, lisosomes enzymatic degradation and killing through the ‘oxidative respiratory burst’ by oxygen radicals/superoxide anions or their products like hydrogen peroxide. In lung and liver leukocytes adhesion can occur independently on adhesion molecules; this may explain why acute onset pneumonia and hepatitis are more frequent and easier to catch than myositis or myocarditis for example. Cells and bacteria can also been killed in absence of oxygen by several substances present in the lysosomes. Any inflammatory process damages cells and tissue via the action of oxygen radicals on lipids membrane and terminates or in resolution or in regeneration or in scarring according to the hit organ regenerative capacity and the rapidity of onset of the hypoxic effects and body counteracting systems. The inflammatory reaction becomes life-threatening when protracts beyond the local interaction with the causa prima or by becoming systemic. It takes around 6 hours for activation. Reduction or failure of neutrophil migration to infection sites is associated with a poor outcome in sepsis.[24] Classical examples are the secondary and tertiary peritonitis.
Appendix C.
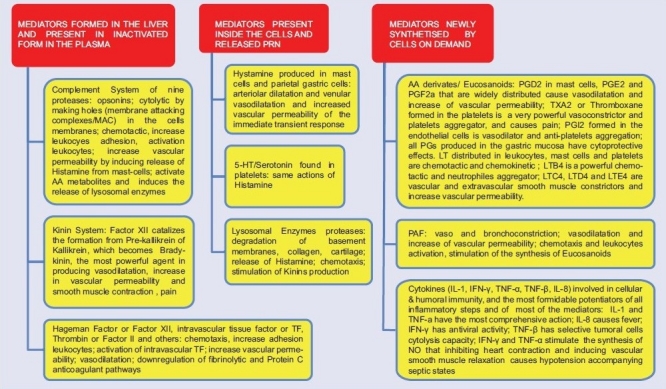
Effects of the inflammatory mediators
Some immune cells and immunoglobulins are structured part of the inflammatory response but the immune response main task is that all its components are the main actors during the flow catabolic and in anabolic phase to contain or slow infection progression or second hit.[25,26] If the immune system is cofactor in the innate immune response, it may also be in fact in competition with it by immunosuppressing inflammatory response opening a motorway to unabated not counteracted SIR from bacteriemia and in particular endotoxemia. Apoptosis of lymphocytes is strongly associated with sepsis progression.[27–29] The structural damage certainly accounts for the deranged, exaggerated and self destructive LIR.[30] Leukocytes activated by septic inflammation generate reactive oxygen species (ROS) that directly disrupt microcirculatory structures. These and other inflammatory mediators alter barrier function in the microcirculation, including junctions between cells and possibly the endothelial glycocalyx, leading to tissue edema and further oxygen extraction deficit.[31] The intensity and persistence correlate with the consequences of sepsis (i.e., multiple organ failure and death).[32] Why LIR becomes SIR, it can only be postulated on the basis of two realistic assumptions: 1) different individual susceptibility to counteract an inflammatory insult with the innate immune inflammatory response, and 2) the entity and duration of the damage by the insult on the structure and function of the microcirculation.
Coagulation derangement always accompanies any hemorrhage or inflammation.[33] The relationship between any bodily insult, external – traumatic or infective – or at lesser extent internal – spontaneous, degenerative, neoplastic, inflammatory, toxic – and coagulation, is a phylogenetical one. Localized activation of the coagulation system, alongside the innate inflammatory/immune response and local and systemic hemodynamic reflexes serves to protect the body against any insult with the metaphysical aim to prevent blood loss and damage from alien substances.[34] Platelets aggregation and fibrin deposition throughout the microcirculation form microthrombi, which eventually lead to ischemia and contributes to MOD/MOF;[35,36] moreover, as consequence of the consumption of platelets and fibrinogen, a generalized capillary oozing in mucosae or through wounded or open tissues also occurs (disseminated intravascular coagulation or DIC). Coagulopathy in sepsis ensues in concomitance and as result of the deranged inflammatory response. Normal EC help maintaining an anticoagulant state necessary to avoid death by systemic total microcoagulation. Exposure to inflammatory and/or septic stimuli rapidly leads to an increase of procoagulant behavior due to loss or decrease of the anticoagulation properties. Factor XII or Hageman Factor formation triggers three pathways, two pertaining coagulation of which one is procoagulant (intrinsic coagulation cascade with end formation of, in order, thrombin or factor II, fibrinogen, fibrinopeptideds and fibrin), and the other is anticoagulant (fibrinolytic system activation with end formation of, in order, plasminogen, the fibrinolytic plasmin and fibrin degradation products), and a third one (the Kinin System) pertaining the inflammatory response. Without this self-balancing of the pro- and anti-coagulation systems, any insult would result in death from generalized microcirculation clotting. Factor XII is formed in the liver and becomes activated among others by contact with collagen, basement membranes, trypsin, kallikrein, factor XI and bacterial endotoxins.
Thrombin, in turn, can promote inflammatory response and so can, though in a lesser amount, some other factors. The release of endothelium-derived factors such as NO and prostacyclin (PGI2) is impaired in sepsis; NO and PGI2 not only control vascular tone but also have antiadhesive and profibrinolytic property (tissue plasminogen activator-tPa release). Their loss facilitates leucocyte and platelet aggregation with aggravation of coagulopathy. Various membrane-associated components with anticoagulant properties expressed in the outer membrane of ECs, namely tissue factor pathway inhibitor (TFPI), protein C (PC), protein S, thrombomodulin (TM) and heparan sulfate (HS), are also damaged by sepsis. The activation of coagulation factors, concomitant to impaired fibrinolysis, is associated with fibrin deposition, microclots formation and DIC, resulting in ischemia and multiple generalized microhemorrhages by coagulation factors consumption . DIC with diffuse paradoxical oozing due to consumption coagulopathy by the disseminated microthrombi is a prognostic marker of sepsis indicating a late-stage critical illness, and becomes a sign of irreversibility if persists despite treatment All coagulation increase vascular permeability and produce vasodilatation; are chemotactic and enhance leucocytes adhesion by increasing the expression of adhesion molecules and platelet activating factor on intravascular cells; activate the intravascular tissue factor expression and downregulate the fibrinolytic and PC anticoagulant pathways. Anticoagulants that inhibit any of these factors would be expected to dampen the inflammatory response. The three major natural anticoagulant mechanisms are antithrombin, activated PC and the TFPI.[37,38]
The term ‘endothelial dysfunction’ refers to decreased endothelial-dependent vascular relaxation, associated with a decrease of NO formation and release . Endogenous acetylcholine has scarce or hardly any vasodilatory effect, but when injected it does produce vasodilatation acting on muscarinic receptors of endothelial cells. Acetylcholine, likewise ATP, ADP, Bradykinin, histamine, serotonin act via NO increase of cyclic GMP mediated by guanylate cyclase, enzyme inactivated by superoxide, Hb and methylene blue.[39,40] Forearm blood flow responses to intra-arterial infusions of endothelium-dependent (i.e., acetylcholine) and endothelium-independent vasodilators (i.e., sodium nitroprusside, carbon monoxide, light) is a reliable indicator of EC function that can be studied using venous occlusion plethysmography combined with a rapid cuff inflator and hypoxic challenge. The underlying pathophysiological mechanism is simply a manifestation of the metabolic/oxygen driven flow regulation. The significance of endothelial dysfunction as mechanism and effect of a sepsis/inflammatory-induced damage is best highlighted by the phenomenon observed in practice of mottled cyanosis from stagnant hypoxia (no blanching and reflow by ‘reactive hyperemia’ of extremities capillaries on digital pressure due to impaired reactivity), an unexceptional and absolute sign of irreversibility in shock. Endothelium-dependent relaxation may be impaired for many days. This effect has been termed ‘endothelial stunning’. After recovery from the acute insult, the endothelium may remain dysfunctional (‘stunned’) for a long period of time before full recovery. This inability of endothelium and smooth muscle layer to counteract increased sympathetic tone may be the link between infection and inflammation to infarction. Impaired reactive hyperemia has been found during systemic inflammatory response.[41–44] Endotoxin impairs oxygen uptake in ECs.[45,46] Endothelium dysfunction is to be considered a reversible form of light structural damage that if persisting leads to refractory vasomotor paralysis, a sign of irreversibility heralding exitus.
NO maintains microvascular homeostasis by regulating arteriolar tone, RBC and leukocyte deformability, leukocyte-endothelial adhesion in mesenteric and skeletal muscle postcapillary venules, platelet adhesion and aggregation to endothelial cells, blood volume and mitochondrial respiration.[47,48] NO over-production is considered the main factor of refractory hypotension in sepsis. The underlying mechanism for its hemodynamic effects is the inhibitory role that NO plays on AVP release.[49] Synchronously to its effect on AVP release, during endotoxemia NO maintains and increases vascular permeability in the intestine, heart, liver and kidney. In the arterioles NO in response to vasodilatative stimuli diffuses down its concentration gradient, from endothelial cell to smooth muscle cell, where it opposes sympathetic or chemically mediated vasoconstriction by relaxing smooth muscle via a guanylate cyclase/cGMP-mediated reaction. The heterogeneous expression of iNOS in different areas of organ beds becomes accentuated during sepsis resulting in pathological shunting of flow. Areas that are lacking iNOS have less NO-induced vasodilation and become underperfused. The smooth muscle cells that line the arterioles and regulate perfusion lose their adrenergic sensitivity and tone in sepsis[50,51] due to NO contribution to abnormal reactivity by altering calcium influx in the maintenance of vessel basal tone.[52] Overproduction of NO decreases blood pressure but it also has protective effect on organs microcirculation: NO inhibition in fact increases blood pressure but does not increase O2ER and VO2, ending up actually decreasing CO and DO2 with risk of cardiac failure, coronary and intestinal hypoperfusion.[53,54] Over-production of NO during sepsis has also been associated with impaired microvascular reactivity, reduced RBC deformability, decreased functional capillary density, blocked leukocytes adhesion, scavenging ROSs and reduced oxygen consumption.[47,48]
Dysoxia in sepsis, as discrepancy between delivery and extraction of oxygen, is a derangement caused initially by flow maldistribution. It is observed and accounted by the phenomenon of ‘spatial and temporal hetereogenity of capillaries flow’. Spatial and temporal heterogeneity of capillaries flow refers to the observation with microscopic techniques in the same field on septic patients or animals of some capillaries being underperfused (reduced capillaries density) due sluggish or stopped or no flow, while other capillaries and venules have normal blood flow, and others have an abnormally high blood flow (increased capillaries density). The phenomenon is noticeable in an experimental sepsis model[55–58] and on humans[59–61] in skeletal muscle, intestinal villi, diaphragm, tongue and liver. ‘Capillary stopped-flow phenomenon’ can occur in normotensive situation and at the same level of hypotension endotoxemic animals become more underperfused than the ones with clinically significant hemorrhage.[62] The loss of perfusion in capillaries with stopped flow is accompanied by, and correlated to, an impaired oxygen extraction rate in the capillaries affected and an increased SVO2. In adjacent normal capillaries the oxygen extraction rate is instead increased and SVO2 decreased.
Some regions of tissue become over-supplied with oxygen– those supplied by capillaries with increased O2ERc – whereas other areas with capillary stop-flow and decreased extraction become under-supplied. Nevertheless, the overall oxygen consumption is decreased . Why is that so? As stopped flow increases, the remaining functionally normal capillaries end up offloading greater amounts of oxygen to an increased volume of surrounding tissue. Although tissues are still capable of extracting oxygen, oxygen is not being delivered where it is needed, and the overall oxygen's consumption is diminished. Maldistribution of RBC flow at the capillary level and mismatching between local oxygen supply and local oxygen demand de facto occur.[13]
Normal SaO2 at the capillary arteriolar end and a lower SaO2 at the capillary venular end (SvO2) is found in areas with increased functional density, high oxygen extraction rate and oxygen consumption; likewise, an increased SvO2 is found early in the derangement in areas low-flow/stop-flow and late when cytopathic hypoxia with impaired consumption by mitochondrial failure ensues. The loss of FCD in sepsis is associated to loss of RBC deformability and over-production of NO.[47] Heterogeneity of capillary blood flow in the end leads to impaired oxygen consumption, local tissue hypoxia, necrosis, inflammation and ultimately organ dysfunction. The loss of capillary blood flow in fact potentiates the effects of proinflammatory mediators by increasing their residence time in the microcirculation and tissue, further continuing the pathological cascade of inflammation. A combination of inflammatory and coagulation-mediated factors contributes to microvascular stasis, fibrin deposition, altered RBC deformability, altered platelets aggregation or adhesion properties, increased leukocyte adhesion, reduced leukocyte deformability, endothelial swelling and microthrombi formation. The end-result of these events is micro-occlusion of capillaries from microthrombi of fibrin and platelets and complete stop flow, causing localized hypoxia and direct cell death or by self-destructive inflammatory response. These observations indicate that microcirculation in sepsis loses its ability to regulate capillary blood flow as it is unable to redistribute RBCs to regions of low PO2 and increased oxygen demand. Another observation deducible by microscopic studies is that dysoxia in sepsis is not characterized by a uniform decrease in O2 delivery, rather by a patchy and disperse distribution of oxygen.[63] No capillary recruitment occurs in IS, likewise occurs instead in HS where dormant arteriolar and capillary beds are reopened by shunting blood from under-perfused or under-ventilated areas; no increase in capillaries vasomotion occurs either . Fluids resuscitation and normalization or even optimization of macrohemodynamic variables does not affect microcirculation dysfunction or capillary hetereogenity. This explains why all studies on supranormal oxygen delivery have failed against a maldistribution issue. Factual macro-microcirculation dissociation in fact ensues. Random/disperse/patchy tissues dysoxia/hypoxia effectively precedes hypotension and can persist in presence of normal blood pressure . Since the loss of capillaries in remote organs begins to occur several hours after the initial injury, and hence several hours after leukocyte activation that takes around 6 hours, it is likely that activation and/or injury of the microvascular endothelium in remote organs is the critical first step leading to capillary heterogeneity and loss.
CLINICAL CORRELATIONS
There is a gap, a missing link in our understanding between the phenomena observed in microcirculation during sepsis and the clinical picture as far as the timing and the correlation. The phenomenon of hyperdynamic phase of septic shock compounds the situation as discrepancy. The temporal order of events correlated in microcirculation during the evolution of sepsis to SS is not clear.
Two-thirds of oxygen is released from arteriole before reaching capillaries by diffusion into neighboring vessels and tissues. Nature does not want to take risks: as for the intrinsic morphological weakness and liability of delicate structures like the capillaries, the terminal exchangers and scavengers of tissues, it wants to make sure we live if something happens at capillary level in the core of microcirculation. We can think of, and define as “life-unit”, that ‘portion of alive tissue’ composed by tissue cells and capillaries supplied of oxygen – as substrates travel within vessels until the terminal capillaries where diffuse directly to target cells – by one specific arteriole i.e., without overlapping of supply by diffusion by other arterioles. Such ‘morphological and functional structure’ would be the minimal common denominator of life. It is nowadays a necessary concept to introduce in critical care following the knowledge of the precapillaries oxygen distribution in the microcirculation and the discovery of the random repetitive and cumulative nature of the inflammatory shocks.
The conceptualization of a morphofunctional unit, basic minimum common denominator of the living body is obviously difficult to demonstrate and this is expected to be so. This being the case, and it is very likely to be so, it would be the very core of human existence, whose synchronous fusion of form and energy could well be seen as our biological soul, our ‘will to life’ fortunately independent on our will. Such a physiological unit as minimum common denominator is not separable from the unit of the body, which is another reason why it is going to be difficult to demonstrate under our mental structure and function of partial and temporary capacity-limited human beings. The effects of stop-flow phenomenon are an indirect proof of its existence. If all or some tissues were supplied by adjacent pathways other than the arteriolar/life-unit complex, why then there is maldistribution and disoxia following capillary heterogenity? If there was no life-unit, any capillary stop-flow situation would not end up in hypoxia as the stop-flow affected tissue would be still supplied by ‘collateral oxygen supply’, likewise the collateral circulation of chronic arterial obstruction.
Following these considerations a plausible working hypothesis can be postulated by plotting the above known alterations with real-time relevant variables variations like SvO2 and lactate [Figure 2].
Figure 2.
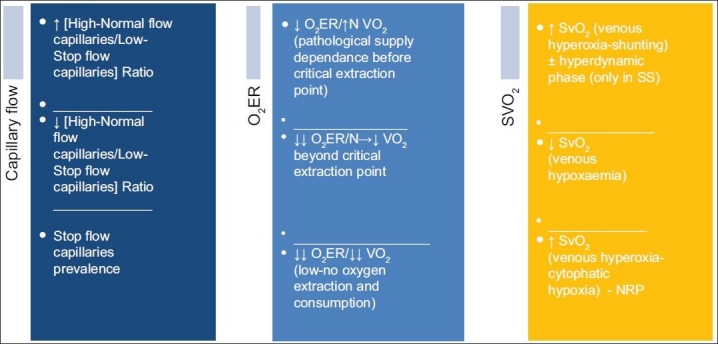
Clinical correlates and timing of occurrence of the microcirculation disorders in sepsis
Hetereogenity must be the first derangement and accounts for the hyperdynamic changes in the microcirculation.[64,65] Capillary low stop-flow is the primum movens with associated high SvO2 and low O2ER. This causes increased flow in some of adjacent capillaries within the same life-unit by diversion and as result of the increased need of perfusion in the surrounding territory with an increased area to supply which now includes the unsupplied areas with decreased capillary density and low stop-flow.[66] These shunted areas exhibit increased SvO2 and an increased oxygen extraction more than it is needed locally.[67] The overall oxygen consumption, however, remains high as long as the number of low-flow capillaries does not overwhelm a correspondent number of high-flow capillaries. At systemic level this phase of overall low extraction and high consumption is described as hyperdynamic phase, characterized by elevated cardiac output, increased oxygen delivery (DO2), decreased systemic vascular resistance (with or without decreases in mean arterial blood pressure, increased tissue oxygen consumption, but impaired oxygen extraction capacity and early lactic acidosis.[68–71] The temporary hyperdynamic phase must be seen as a compensatory response to the complessive low oxygen extraction momentary phase as the prevailing overall result in the computation of all ‘life-units’ metabolism. This phase of ‘pathological supply dependence’ terminates when the critical extraction rate is reached.[72] At a SvO2 of 50% metabolism starts becoming anerobic.
A proper shock phase with prevalence of decreased SvO2 and O2ER and hypoxia as a matter of fact follows with progression of the insult. This is likely to occur in synchrony with decreasing endothelial ability to relax and with the prevalence of low/no flow capillaries (decreased FCD) on the normal/high flow capillaries in the same life-unit. Once the endothelial cell injury persists, the dysfunction becomes structural damage, with worsening of the LIR, accentuated leucocytes adhesiveness and platelets aggregation, impaired fibrinolysis, resulting in an aggravation of the situation of dysoxic-hypoxia by mere stagnant-hypoxia from occlusion. The irreversible ability of the arteriolae endothelium to respond to vasomotor factors and stimuli (stagnant hypoxia/mottled cyanosis) finally heralds exitus; hyperoxia (↑SvO2) then reappears and indicates mitochondrial failure (cytophatic hypoxia)[73] and terminal stage [Figure 3].
Figure 3.
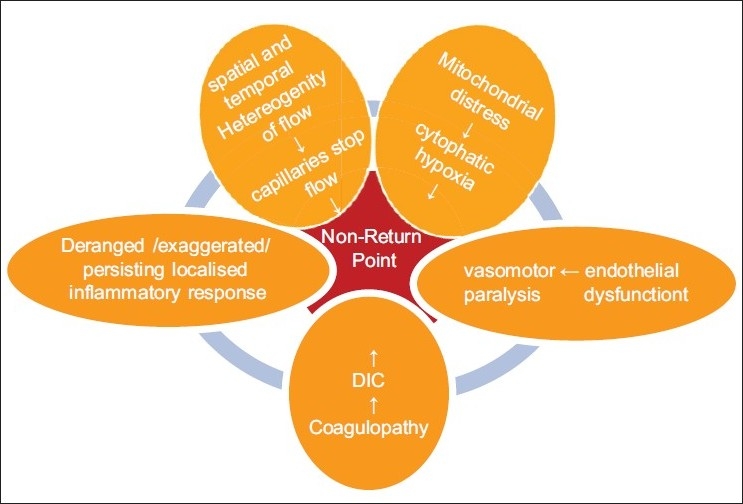
Microcirculation disorders in sepsis effects of the damage by lps and other toxins
It is likely that in the initial phase of sepsis there is a predominance of microcirculation dysfunction and later mitochondria fail out indicating a failure of compensation of the microcirculation derangement. The level of respiratory dysfunction in the mitochondria correlates with patient outcome.[74] As interpreted and proposed by Singer et al, sepsis and other critical illnesses in its natural unmitigated evolution produce a biphasic inflammatory, immune, hormonal, and metabolic response of MOD eventually evolving to MOF. The acute phase is marked by an abrupt rise in the secretion of so-called stress hormones and inflammatory mediators with an associated increase in mitochondrial and metabolic activity. This would correspond to the flow-catabolic phase of the stress-response. The prevalence of the inflammation on the endocrine wave ends up diminishing energy production, metabolic rate, and normal cellular processes, leading to multiple organs dysfunction. MOD could be a protective mechanism, because reduced cellular metabolism actually increases the chances of survival as a resistance against the insult. It is possible that progression from MOD to MOF is initially a functional abnormality that eventually becomes structural with decrease in mitochondrial activity and oxidative phosphorylation, leading to reduced cellular metabolism and disoxia.[75]
APPENDICES
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Grocott MP, Martin DS, Levett DZ, McMorrow R, Windsor J, Montgomery HE. Caudwell Extreme Everest Research Group. Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med. 2009;360:140–9. doi: 10.1056/NEJMoa0801581. [DOI] [PubMed] [Google Scholar]
- 2.Ahlquist RP. Present state of alpha- and beta-adrenergic drugs I. The adrenergic receptor. Am Heart J. 1976;92:661–4. doi: 10.1016/s0002-8703(76)80086-5. [DOI] [PubMed] [Google Scholar]
- 3.Hock CE, Su JY, Lefer AM. Role of AVP in maintenance of circulatory homeostasis during hemorrhagic shock. Am J Physiol. 1984;246:H174–9. doi: 10.1152/ajpheart.1984.246.2.H174. [DOI] [PubMed] [Google Scholar]
- 4.Thiemermann C, Szabó C, Mitchell JA, Vane JR. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Natl Acad Sci U S A. 1993;90:267–71. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma AC, Singh G, Gulati A. Decompensation characterized by decreased perfusion of the heart and brain during hemorrhagic shock: role of endothelin-1. J Trauma. 2002;53:531–6. doi: 10.1097/00005373-200209000-00022. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Liu L, Xu J, Yang G, Ming J. Changes of Rho kinase activity after hemorrhagic shock and its role in shock-induced biphasic response of vascular reactivity and calcium sensitivity. Shock. 2006;26:504–9. doi: 10.1097/01.shk.0000228796.41044.41. [DOI] [PubMed] [Google Scholar]
- 7.Yang G, Liu L, Xu J, Li T. Effect of arginine vasopressin on vascular reactivity and calcium sensitivity after hemorrhagic shock in rats and its relationship to Rho-kinase. J Trauma. 2006;61:1336–42. doi: 10.1097/01.ta.0000197928.99745.22. [DOI] [PubMed] [Google Scholar]
- 8.Yang G, Li T, Xu J, Liu L. PKC plays an important mediated effect in arginine vasopressin induced restoration of vascular responsiveness and calcium sensitization following hemorrhagic shock in rats. Eur J Pharmacol. 2010;628:148–54. doi: 10.1016/j.ejphar.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 9.Haljamäe H. Microcirculation and hemorrhagic shock. Am J Emerg Med. 1984;2:100–7. doi: 10.1016/0735-6757(84)90117-7. [DOI] [PubMed] [Google Scholar]
- 10.Nakai K, Ikatura T, Naka Y, Nakakita K, Kamei I, Imai H, et al. The distribution of adrenergic receptors in cerebral vessels: An autoradiographic study. Brain Res. 1986;381:148–52. doi: 10.1016/0006-8993(86)90703-1. [DOI] [PubMed] [Google Scholar]
- 11.Abe R, Oda S, Sadahiro T, Nakamura M, Hirayama Y, Tateishi Y, et al. Gram-negative bacteremia induces greater magnitude of inflammatory response than Gram-positive bacteremia. Crit Care. 2010;14:R27. doi: 10.1186/cc8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandraki I, Palacio C. Gram-negative versus Gram-positive bacteremia: What is more alarming? Crit Care. 2010;14:161. doi: 10.1186/cc9013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis CG, Bateman RM, Sharpe MD, Sibbald WJ, Gill R. Effect of a maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am J Physiol Heart Circ Physiol. 2002;282:H156–64. doi: 10.1152/ajpheart.2002.282.1.H156. [DOI] [PubMed] [Google Scholar]
- 14.Vallet B. Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: A role in organ dysfunction? Critical Care. 2003;7:130–8. doi: 10.1186/cc1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levi M, de Jonge E, van der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. 2003;16:43–7. doi: 10.1023/B:THRO.0000014592.27892.11. [DOI] [PubMed] [Google Scholar]
- 16.Doerschug KC, Delsing AS, Schmidt GA, Haynes WG. Impairment in microvascular reactivity are related to organ failure in human sepsis. Am J Physiol Heart Circ Physiol. 2007;293:H1065–71. doi: 10.1152/ajpheart.01237.2006. [DOI] [PubMed] [Google Scholar]
- 17.Bolon ML, Peng T, Kidder GM, Tyml K. Lipopolysaccharide plus hypoxia and reoxygenation synergistically reduce electrical coupling between microvascular endothelial cells by dephosphorylating connexin-40. J Cell Physiol. 2008;217:350–9. doi: 10.1002/jcp.21505. [DOI] [PubMed] [Google Scholar]
- 18.Luo TH, Wang Y, Lu ZM, Zhou H, Xue XC, Bi JW, et al. The change and effect of endothelial progenitor cells in pig with multiple organ dysfunction syndromes. Crit Care. 2009;13:R118. doi: 10.1186/cc7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 20.Engelberger RP, Pittet YK, Henry H, Delodder F, Hayoz D, Chioléro RL, et al. Acute endotoxaemia inhibits microvascular nitric oxide-dependent vasodilation in humans. Shock. 2011;35:28–34. doi: 10.1097/SHK.0b013e3181ec71ab. [DOI] [PubMed] [Google Scholar]
- 21.Cotran RS, Majno G. The delayed and prolonged vascular leakage in inflammation. I. Topography of the leaking vessels after thermal injury. Am J Pathol. 1964;45:261–81. [PMC free article] [PubMed] [Google Scholar]
- 22.Ryan GB, Majno G. Acute inflammation.A review. Am J Pathol. 1977;86:183–276. [PMC free article] [PubMed] [Google Scholar]
- 23.Majno G. Maude Abbott lecture-1991. The capillary then and now: An overview of capillary pathology. Mod Pathol. 1992;5:9–22. [PubMed] [Google Scholar]
- 24.Alves-Filho JC, De Freitas A, Spiller F, Souto FO, Cunha FQ. The role of neutrophils in severe sepsis. Shock. 2008;30:3–9. doi: 10.1097/SHK.0b013e3181818466. [DOI] [PubMed] [Google Scholar]
- 25.Christou NV, McLean AP, Meakins JL. Host defense in blunt trauma: Interrelationships of kinetics of energy and depressed neutrophil function, nutritional status and sepsis. J Trauma. 1980;20:833–41. doi: 10.1097/00005373-198010000-00003. [DOI] [PubMed] [Google Scholar]
- 26.Flohé SB, Flohé S, Schade FU. Invited review: deterioration of the immune system after trauma: Signals and cellular mechanisms. Innate Immun. 2008;14:333–44. doi: 10.1177/1753425908100016. [DOI] [PubMed] [Google Scholar]
- 27.Gogos C, Kotsaki A, Pelekanou A, Giannikopoulos G, Vaki I, Maravitsa P, et al. Early alterations of the innate and adaptive immune statuses in sepsis according to the type of underlying infection. Crit Care. 2010;14:R96. doi: 10.1186/cc9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rolli J, Loukili N, Levrand S, Rosenblatt-Velin N, Rignault-Clerc S, Waeber B, et al. Bacterial flagellin elicits widespread innate immune response defense mechanisms, apoptotic signalling, and a sepsis-like systemic inflammatory response in mice. Crit Care. 2010;14:R160. doi: 10.1186/cc9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turrel-Davin F, Guignant C, Lepape A, Mougin B, Monneret G, Venet F. Upregulation of the pro-apoptotic genes BID and FAS in septic shock patients. Crit Care. 2010;14:R133. doi: 10.1186/cc9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller AM, Cronen C, Muller KM, Kirkpatrick CJ. Heterogeneous expression of cell adhesion molecules by endothelial cells in ARDS. J Pathol. 2002;198:270–5. doi: 10.1002/path.1186. [DOI] [PubMed] [Google Scholar]
- 31.Fink MP. Intestinal epithelial hyperpermeability: Update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. 2003;9:143–51. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Sessler CN, Windsor AC, Schwartz M, Watson L, Fisher BJ, Sugerman HJ, et al. Circulating ICAM-1 is increased in septic shock. Am J Respir Crit Care Med. 1995;151:1420–7. doi: 10.1164/ajrccm.151.5.7735595. [DOI] [PubMed] [Google Scholar]
- 33.Pottmeyer E, Vassar MJ, Holcroft JW. Coagulation, inflammation and response to injury. Crit Care Clinics. 1986;2:683–703. [PubMed] [Google Scholar]
- 34.Opal SM. Phylogenetic and functional relationships between coagulation and the innate immune response. Crit Care Med. 2000;28:S77–80. doi: 10.1097/00003246-200009001-00017. [DOI] [PubMed] [Google Scholar]
- 35.Vincent JL, De Backer D. Does disseminated intravascular coagulation lead to multiple organ failure? Crit Care Clin. 2005;21:469–77. doi: 10.1016/j.ccc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Gando S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med. 2010;38:S35–42. doi: 10.1097/CCM.0b013e3181c9e31d. [DOI] [PubMed] [Google Scholar]
- 37.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 38.Levi M, de Jonge E, van der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. 2003;16:43–7. doi: 10.1023/B:THRO.0000014592.27892.11. [DOI] [PubMed] [Google Scholar]
- 39.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 40.Furchgott RF, Cherry PD, Zawadzki JV, Jothianandan D. Endothelial cells as mediators of vasodilation of arteries. J Cardiovasc Pharmacol. 1984;6:S336–43. doi: 10.1097/00005344-198406002-00008. [DOI] [PubMed] [Google Scholar]
- 41.Astiz ME, DeGent GE, Lin RY, Rackow EC. Microvascular function and rheologic changes in hyperdynamic sepsis. Crit Care Med. 1995;23:265–71. doi: 10.1097/00003246-199502000-00011. [DOI] [PubMed] [Google Scholar]
- 42.Wang P, Ba ZF, Chaudry IH. Endothelium-dependent relaxation is depressed at the macro- and microcirculatory levels during sepsis. Am J Physiol. 1995;269:R988–94. doi: 10.1152/ajpregu.1995.269.5.R988. [DOI] [PubMed] [Google Scholar]
- 43.Bhagat K, Moss R, Collier J, Vallance P. Endothelial ‘stunning’ following a brief exposure to endotoxin: A mechanism to link infection and infarction?? Cardiovasc Res. 1996;32:822–9. [PubMed] [Google Scholar]
- 44.Hingorani AD, Cross J, Kharbanda RK, Mullen MJ, Bhagat K, Taylor M, et al. Acute systemic inflammation impairs endothelium-dependent dilation in humans. Circulation. 2000;102:994–9. doi: 10.1161/01.cir.102.9.994. [DOI] [PubMed] [Google Scholar]
- 45.Motterlini R, Kerger H, Green CJ, Winslow RM, Intaglietta M. Depression of endothelial and smooth muscle cell oxygen consumption by endotoxin. Am J Physiol. 1998;275:H776–82. doi: 10.1152/ajpheart.1998.275.3.H776. [DOI] [PubMed] [Google Scholar]
- 46.Karimova A, Pinsky DJ. The endothelial response to oxygen deprivation: Biology and clinical implications. Intensive Care Med. 2001;27:19–31. doi: 10.1007/s001340000790. [DOI] [PubMed] [Google Scholar]
- 47.Bateman RM, Sharpe MD, Ellis CG. Bench-to-bedside review: Microvascular dysfunction in sepsis-hemodynamics, oxygen transport, and nitric oxide. Crit Care. 2003;7:359–73. doi: 10.1186/cc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hauser B, Matejovic M, Radermacher P. Nitric oxide, leukocytes and microvascular permeability: Causality or bystanders? Crit Care. 2008;12:104. doi: 10.1186/cc6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giusti-Paiva A, De Castro M, Antunes-Rodrigues J, Carnio EC. Inducible nitric oxide synthase pathway in the central nervous system and vasopressin release during experimental septic shock. Crit Care Med. 2002;30:1306–10. doi: 10.1097/00003246-200206000-00025. [DOI] [PubMed] [Google Scholar]
- 50.Baker CH, Wilmoth FR. Microvascular responses to E. coli endotoxin with altered adrenergic activity. Circ Shock. 1984;12:165–76. [PubMed] [Google Scholar]
- 51.Price SA, Spain DA, Wilson MA, Harris PD, Garrison RN. Subacute sepsis impairs vascular smooth muscle contractile machinery and alters vasoconstrictor and dilator mechanisms. J Surg Res. 1999;83:75–80. doi: 10.1006/jsre.1998.5568. [DOI] [PubMed] [Google Scholar]
- 52.Chen SJ, Li SY, Shih CC, Liao MH, Wu CC. NO contributes to abnormal vascular calcium regulation and reactivity induced by peritonitis-associated septic shock in rats. Shock. 2010;33:473–8. doi: 10.1097/SHK.0b013e3181bea334. [DOI] [PubMed] [Google Scholar]
- 53.Broccard A, Hurni JM, Eckert P, Liaudet L, Schaller MD, Lazor R, et al. Tissue oxygenation and hemodynamic response to NO synthase inhibition in septic shock. Shock. 2000;14:35–40. doi: 10.1097/00024382-200014010-00007. [DOI] [PubMed] [Google Scholar]
- 54.Hollenberg SM, Cinel I. Bench-to-bedside review: Nitric oxide in critical illness-update 2008. Crit Care. 2009;13:218. doi: 10.1186/cc7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lam C, Tyml K, Martin C, Sibbald W. Microvascular perfusion is impaired in a rat model of normotensive sepsis. J Clin Invest. 1994;94:2077–83. doi: 10.1172/JCI117562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellis CG, Wrigley SM, Groom AC. Heterogeneity of red blood cell perfusion in capillary networks supplied by a single arteriole in resting skeletal muscle. Circ Res. 1994;75:357–68. doi: 10.1161/01.res.75.2.357. [DOI] [PubMed] [Google Scholar]
- 57.Nevière R, Mathieu D, Chagnon JL, Lebleu N, Millien JP, Wattel F. Skeletal muscle microvascular blood flow and oxygen transport in patients with severe sepsis. Am J Respir Crit Care Med. 1996;153:191–5. doi: 10.1164/ajrccm.153.1.8542115. [DOI] [PubMed] [Google Scholar]
- 58.Humer MF, Phang PT, Friesen BP, Allard MF, Goddard CM, Walley KR. Heterogeneity of gut capillary transit times and impaired gut oxygen extraction in endotoxemic pigs. J Appl Physiol. 1996;81:895–904. doi: 10.1152/jappl.1996.81.2.895. [DOI] [PubMed] [Google Scholar]
- 59.De Backer D, Creteur J, Preiser JC, Dubois MJ, Vincent JL. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002;166:98–104. doi: 10.1164/rccm.200109-016oc. [DOI] [PubMed] [Google Scholar]
- 60.Sakr Y, Dubois MJ, De Backer D, Creteur J, Vincent JL. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit Care Med. 2004;32:1825–31. doi: 10.1097/01.ccm.0000138558.16257.3f. [DOI] [PubMed] [Google Scholar]
- 61.Creteur J, De Backer D, Sakr Y, Koch M, Vincent J. Sublingual capnometry tracks microcirculatory changes in septic patients. Intensive Care Med. 2006;32:516–23. doi: 10.1007/s00134-006-0070-4. [DOI] [PubMed] [Google Scholar]
- 62.Nakajima Y, Baudry N, Duranteau J, Vicaut E. Microcirculation in intestinal villi: A comparison between hemorrhagic and endotoxin shock. Am J Respir Crit Care Med. 2001;164:1526–30. doi: 10.1164/ajrccm.164.8.2009065. [DOI] [PubMed] [Google Scholar]
- 63.Ellis CG, Jagger J, Sharpe M. The microcirculation as a functional system. Crit Care. 2005;9:S3–8. doi: 10.1186/cc3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Astiz M, Rackow EC, Weil MH, Schumer W. Early impairment of oxidative metabolism and energy production in severe sepsis. Circ Shock. 1988;26:311–20. [PubMed] [Google Scholar]
- 65.Walley KR. Heterogeneity of oxygen delivery impairs oxygen extraction by peripheral tissues: Theory. J Appl Physiol. 1996;81:885–94. doi: 10.1152/jappl.1996.81.2.885. [DOI] [PubMed] [Google Scholar]
- 66.Krogh A. The number and distribution of capillaries in muscle with calculation of the oxygen pressure head necessary for supplying the tissue. J Physiol. 1919;52:409–15. doi: 10.1113/jphysiol.1919.sp001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ince C, Sinaappel M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med. 1999;27:1369–77. doi: 10.1097/00003246-199907000-00031. [DOI] [PubMed] [Google Scholar]
- 68.Rackow EC, Kaufmann BS, Falk JL, Astiz ME, Weil MH. Hemodynamic response to fluid repletion in patients with septic shock: Evidence for early depression of cardiac performance. Circ Shock. 1987;22:11–22. [PubMed] [Google Scholar]
- 69.Samsel RW, Nelson DP, Sanders WM, Wood LD, Schumacker PT. Effect of endotoxin on systemic and skeletal muscle O2 extraction. J Appl Physiol. 1988;65:1377–82. doi: 10.1152/jappl.1988.65.3.1377. [DOI] [PubMed] [Google Scholar]
- 70.Anning PB, Sair M, Winlove CP, Evans TW. Abnormal tissue oxygenation and cardiovascular changes in endotoxemia. Am J Respir Crit Care Med. 1999;159:1710–5. doi: 10.1164/ajrccm.159.6.9801124. [DOI] [PubMed] [Google Scholar]
- 71.Yang S, Cioffi WG, Bland KI, Chaudry IH, Wang P. Differential alterations in systemic and regional oxygen delivery and consumption during the early and late stages of sepsis. J Trauma. 1999;47:706–12. doi: 10.1097/00005373-199910000-00015. [DOI] [PubMed] [Google Scholar]
- 72.Schumacker PT, Samsel RW. Oxygen delivery and uptake by peripheral tissues: Physiology and pathophysiology. Crit Care Clin. 1989;5:255–69. [PubMed] [Google Scholar]
- 73.Fink MP. Bench-to-bedside review: Cytopathic hypoxia. Crit Care. 2002;6:491–9. doi: 10.1186/cc1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–23. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 75.Singer M, De Santis V, Vitale D, Jeffcoate W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364:545–8. doi: 10.1016/S0140-6736(04)16815-3. [DOI] [PubMed] [Google Scholar]