

Abstract
The clinical aspects of shock syndromes are described from their inception as compensated physiology to a stage of decompensation. The clinical significance of hypotension, fluid-responsive and non fluid-responsive hypotension, is discussed. Untimely or inadequate treatment leads to persistent subclinical shock despite adjustments of the macrohemodynamic variables, which evolves in a second hit of physiological deterioration if not aggressively managed. Irreversible shock ensues as consequence of direct hit or as result of inadequate or delayed treatment and is characterized by drug-resistant hypotension.
Keywords: Assessment, cryptic shock, hypotension, irreversible shock, shock
INTRODUCTION
Shock is an acute or hyperacute physiological derangement, a systemic syndrome characterized by signs and symptoms, which are the response of different organs to a situation of hypoperfusion for their cells basic metabolic needs. Perfusion means oxygen and nutrients delivery via blood flow. There are practically four categories of shock: Cardiogenic (CS), hemorrhagic (HS) and inflammatory (IS), which can be subdivided in septic (SS) and toxic shock (TS). IS is an umbrella term for a shock situation caused by persistent accentuated acute or acute-on-chronic SIR caused by a localized inflammatory response (LIR) to sepsis or tissue damage that has not been controlled locally becoming systemic (SIR), or by ischemia-reperfusion (I-R) phenomenon secondary to tissue ischemic damage. SS is none other than a sustained SIR caused by infection characterized by non-fluid respondent hypotension and hypoperfusion, while toxic shock is the subtype of IS caused by burns, ANP, ischemic-necrosis/gangrene, persistent or complicated intestinal obstruction, crush injury, characterized by I-R phenomenon. It can be said that the difference between SIR I-R, both sharing the same ontogenic inflammatory events, is in the vasodilatory effect of the initial SIR. Eventually, with progression of the structural damage to the endothelium by the bacterial toxins, SIR becomes characterized by vasoconstriction like I-R phenomenon.
Whatever causes it, shock is a situation of relative hypoxaemia due to failure of the circulation in delivering and distributing enough oxygen for the oxidative processes leading to ATP formation.
Assessment of shock
Signs and symptoms of shock, which is syndrome, are related to the different organ- specific response to hypoperfusion in a clinical progression based on an ‘inverse priority pattern’ in the body economy for importance of functions (skin first, visceral organs to follow, and the noble organs of heart and brain as last)[1–2] [Table 1].
Table 1.
Clinical signs in the three main categories of shock
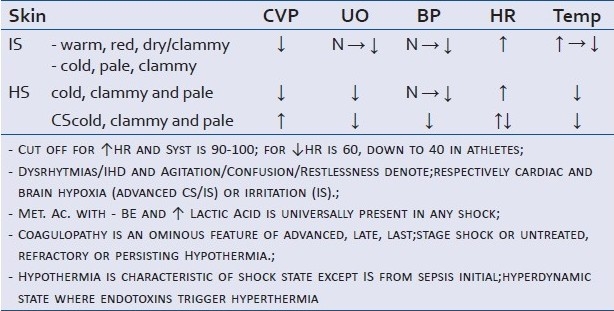
Organs are affected by hypoxia following hypoperfusion in HS and CS or by the direct toxic effect and higher oxygen demand of the IS. In the end shocks kill by generalized hypoxia, occurring at different speed and distribution: Slow, cumulative and randomly distributed in the body life units in IS and faster totalitarian in all life-units in HS/CS. Exception to this latter aspect is the immediate PTIR to trauma and or massive severe burns where can occur on the spot or within seconds to minutes.
The clinical manifestations of hypoxia from hypoxemic respiratory failure overlap the ones following hypoxemia from peripheral circulatory failure, and are directly related to i) the autonomic reflex responses of the respiratory and cardiac centers in the medulla oblongata and the peripheral chemoreceptors to oxygen variations (tachycardia, systolic increase, tachypnea, sweating); ii) the amount of desaturated hemoglobin present in the systemic or locoregional circulation blood (central and peripheral cyanosis respectively); iii) the different organs dependence to oxygen for their metabolism and their capacity of reproduction, substitution to losses and repair. Thus brain and heart are the two vital organs specifically always and absolutely to protect and safeguard from lack of oxygen. Signs such as restlessness, confusion, agitation, acute hypertension, or ischemic heart changes, arrhythmias and secondary cardiac failure/shock denote severe hypoxaemia with imminent respiratory, cardiac arrest. Semeiologically some diagnostic pitfalls must be kept in mind[3] [Table 2]. Sustained hypotension differentiates shock from fainting/syncope characterized by temporary limited hypotension and bradycardia.
Table 2.
Diagnostic pitfalls

Complications as such do not exist in shock. Multiple organs dysfunction (MOD) or failure (MOF) is a continuum of shock indicating multiple ‘single-organ’ unresolved hypoxias.
While consistent blood pressure (BP) drop together with HR shift indicates decompensated shock (unstably unstable), compensated shock is characterized by the presence of normalized BP and persisting HR shift (stably unstable). The cut-off values of systolic BP and HR is 90-100. Several people live well with a systolic of 90 mm Hg and others are hypotensive with a systolic of ≤ 100 mm Hg and elderly with atherosclerosis and hypertension can be hypotensive at a systolic of 120 mm Hg for example.[4] Similarly a cut- off of HR at low end is difficult to categorize as athletes for example can live well with HR of 40 or 50 bpm while others have low cardiac output (CO) and blood pressure (BP) at rhythms below 60 bpm. Physiological variables and clinical picture must both be considered and plotted together when assessing any critical patient.
Variable degrees of kidney dysfunction are inevitably present in shock.[5–6] Lungs are resistant to hypoxia and in HS shock do not get much damage - only impairment of ciliary activity and surfactant production. It is a prime and relevant target instead in any inflammatory process, local or systemic.[7]
Cardiac signs (arrhythmias, ischemia, failure and shock) occur early characterizing CS, at any stage toxins can hit the heart compromising CO in SS/IS[8–10] or at late stage and signifying last-stage compensation and ominous of imminent collapse in HS.
Cerebral signs (confusion, restlessness, agitation) in HS and CS are indicative of imminent circulatory collapse. Thanks to an ingenious distribution of α receptors in the body that leaves the brain auto-regulation unaffected by systemic mechanisms of protection until the end after having benefited indirectly by the maintenance of systemic pressures by systemic mechanisms, brain involvement in HS/CS is the last ditch, the last resort before collapse. In IS toxins and inflammatory mediators irritate the brain at any stage.[11–12]
Metabolic acidosis with lactate acidemia and negative base excess is universally present in shock and is a direct consequence of low perfusion in HS and CS or of the inflammation in IS; acidosis aggravates coagulopathy trend accompanying any hemorrhagic and inflammatory process and dampens catecholamines response. Liver disease, diabetes mellitus and chronic obstructive pulmonary disease are potential pitfalls to keep in mind when using arterial blood gases analysis as assessment and monitoring tool of PH and PCO2. Lactate acidemia usually signals cellular hypoxia caused by inflammation or cellular hypoxia caused by hypovolaemia.
Hyperthermia is present only in IS and is related to several mechanisms (toxins, hypermetabolism, central). Hypothermia accompanies the two vasoconstrictive types of shock and is result of vasoconstriction, hemorrhage, and perioperative heat loss; when it persists or becomes refractory to treatment is an ominous sign of irreversibility and the most obvious clinical marker of reduced metabolism typical of end-stage shock of any etiology.
Coagulopathy is caused by hypoxia, therapeutical hemodilution and hypothermia in HS; it becomes a sign of irreversibility in HS if it persists out of control. It can predictably be assumed that a hypoxic and inflamed liver contributes to it too. In IS it is a sign of advanced late stage and is caused by consumption; in post-traumatic inflammatory response (PTIR) following massive blunt trauma to multiple organs it preludes to imminent death (mouth gush). In trauma hypothermia and acidosis precede coagulopathy.[13]
Temperature of 34°C, PTT more than twice control/INR of 2 or above and pH below 7.4 with negative > 2meq/L of base excess define respectively hypothermia, coagulopathy and metabolic acidosis.
While HS, the other shock characterized by scarce perfusion has an identifiable clinical progression of signs which in its severe form overlaps most of the clinical features of CS at any time, CS is a life-threatening and unpredictable condition ab initio, and treatment must be in any case the most rapid as possible: it cannot be classified in mild/moderate/severe or in hyper or hypodynamic like HS and SS respectively.
In HS a previously normal heart is involved only in the final stages due to hypoperfusion of the coronaries and in IS its involvement depends on the quantity and quality of the action of toxins on myocardium.
Blood loss can be obvious in case of external wound or gastrointestinal loss (hemathemesis, melaena) or evidentiated on chest (hemothorax) or abdominal X-ray (pelvic fracture) or via peritoneal lavage and/or sonar (hemoperitoneum) or CT scan (mediastinal or retroperitoneal). Decision-making is, however, often required without auxiliary investigations and based on clinical grounds only. Visible thigh unilateral hematoma can easily give shock picture; in absence of pelvic fracture by clinical examination (inspection and palpation) or on X-ray and in presence of normal chest examination or X-ray, and of negative peritoneal lavage or sonar, a retroperitoneal bleeding is the cause of shock especially if the heart examination and an ECG do not exhibit alterations.[14] A sonar can only confirm such a sound clinical thinking.
Owing to similarity of the peripheral signs in the two low perfusion shocks, CS must be distinguished by HS presenting without obvious external hemorrhage. This is rather problematic in an unconscious patient with shock signs for example found on a street. A patient with CS from AMI developing significant gastro-intestinal hemorrhage from stress gastric ulcer or a reacutization with hemorrhage from a preexistent gastric ulcer, or a patient with gastrointestinal hemorrhage developing cardiac failure are not uncommon, and with either or both conditions causing fainting or mere cardiovascular collapse. These complex pictures take often a straight path to exitus.
A history of fainting plus rapid onset abdominal pain before shock may point to several pre-hospital differential diagnosis such as abdominal aorta aneurysm, acute necrotizing pancreatitis (ANP), acute myocardial infarction (AMI), sudden gynecological catastrophe, while history of chest pain plus minus syncope points towards acute myocardial infarction (AMI), thoracic aorta aneurysm (TAA), massive pulmonary embolism. TAA has a posterior irradiation of pain down to back where AMI can mimic an acute abdomen. Special investigations further than the basic chest X-Ray and electrocardiogram are often required for screening, specifically a two-dimensional echocardiography with color flow Doppler as trans-esophageal echography or trans-thoracic ecography (TTE), and sometimes contrast- enhanced chest computed tomographic, magnetic resonance imaging and scintigraphy to discriminate the different causes of CS.
Several conditions cause CS: cardiac tamponade, massive pulmonary embolism, hemodynamically significant dysrrhytmias, massive hypertensive pneumothorax, thoracic aorta dissection, left ventricular failure (LVF), acute mitral valve regurgitation (AMVR), ventricular septal defects (VSD), right ventricular failure (RVF). AMI is the most common cause of LVF (75-80% cases) and can be caused by acute coronary syndrome from a primary thrombus, a ruptured or erosed plaque with subsequent thrombus and fibrosis, spasm, stenosis, systemic hypoperfusion, hypoxemia, tachycardia, hypotension and acute anaemia. Hypoperfusion or high oxygen demand both can cause ischemic necrosis.
Congestive cardiac failure (CCF) describes the situation in which pump failure does not compromises perfusion yet. Any situation of IS or HS where increase or normalization of venous return with or without need for inotrops can technically be considered a form of cardiac failure. Generalized and pulmonary congestion occurs respectively in RVF or LVF. Any failure of half of the heart will eventually affect all myocardium for different reasons, not the least the pressure that a dilated ventricle maintains on the other yet normal during diastole with consequential dysfunctional systolic phase. When CO is low enough to cause organs reaction to hypoperfusion, the situation is defined as CS. CCF is usually a low CO scenario except in Beri Beri and artero-venous fistula (AVF) where there is a high CO due respectively to decreased TPR vitamin B deficiency-related and to excess of venous return (VR). It is compensated normally by fluid adjustments, myocardial adaptation according to the Frank Starling law manifesting only generalized edema. When compensatory mechanisms do not control CO and cause hypotension, CF becomes decompensated as acute-on-chronic condition, and when hypoperfusion installs, it becomes CS.
CS i.e. cardiac failure plus hypotension and hypoperfusion, is defined as the combination of sustained (>half an hour) hypotension < 90 mm Hg, signs of hypoperfusion, a PCWP > 18 mm Hg and a cardiac index (CI) < 2.2 L/min/m2.
Measurement of hemodynamic and metabolic variables via arterial lines, Swan-Ganz catheters (SGCs) with continuous/frequent CO monitoring, esophageal Doppler sonography (ODS), arterial line and peripheral tissues oximetry is required in ICU to diagnose shock, to give certainty on the type of shock as for the multiplicity of interfering and overlapping factors confusing the mere clinical picture and to monitor the patient's response to therapeutic manipulations[15–17] [Table 3].
Table 3.
Variables differences in the three main shock states

The metabolic variables changes occurring in IS follow the progression of the sepsis; special and temporal flow heterogeneity with capillary stop-flow phenomenon, the first pathological change, causes decreased extraction of oxygen (O2ER) and increased extraction in normal adjacent capillaries with high flow as result of shunting/diversion. This phenomenon accounts for maldistribution of perfusion and disoxia. At systemic level this status coincides with what it has been described as hyperdynamic phase or pathological supply dependence [(elevated CO, increased oxygen delivery (DO2), decreased systemic vascular resistance with or without decrease in mean arterial blood pressure, increased tissue oxygen consumption, overall increased SvO2 but impaired oxygen extraction capacity and early lactic acidosis]. Once the compensatory increase of flow and consumption reaches the critical extraction point, a proper shock phase with prevailing anaerobic metabolism and decreased SvO2 installs. [Table 3]
Microcirculation nowadays can more easily be visualized on bedside with orthogonal polarization spectral (OPS) imaging[18] and Side-stream Dark Field (SDF) imaging[19] and may be integrated by sublingual capnography.[20] OPS/SDF can also be integrated by vascular occlusion tests: the former evaluate the actual state of the microcirculation, whereas the vascular occlusion test evaluates the capacity of the arterioles to respond to intrinsic or extrinsic administered vasomotor agents.[21]
Oxygen tension (PO2) is assessed by oxygen microelectrodes inserted into the tissue of patients and animal models or indirectly by generic aspecific hypoxic markers. Bioenergetic of tissues is determined by ATP analysis or by nicotinamide adenine dinucleotide reduced form (NADH) fluorescence studies, by indirect calorimetry or by near-infrared spectroscopy.[22–26]
Failure of ‘hemorrhage’ and ‘infection/inflammation’ control
At some stage, after the compensatory mechanism will have lost their grip in controlling or modulating the derangement because of causa prima or primum movens protraction from inadequate or untimely management, a situation of hypotension and tachycardia, or bradycardia in some CS, plus MOD, ensues in any shock. As a matter of fact there is no such a thing as complications in shock. Shock is not a disease but a syndrome and, by definition and nature, a continuum systemic derangement ab initio, in extension, progression and outcome. By definition shock already comprehends what is described as MOD. What from a semantic point of view it is erroneously described in all textbooks as complication or MOD/MOF, is none other than the protraction or progression of the damage to organs-tissues by relative hypoxia. Notable examples are the frequent kidney and lung involvement as dysfunction or failure, and so must be considered the hepatic, adrenal and intestinal dysfunction/failure. In hemorrhage the progression of blood preservation towards organs with high physiological priority cannot occur without sending the ‘blood-lending’ organs in dysfunction; and in septicaemia, or severe sepsis, several organs become dysfunctional ab initio because of the systemic character of the disorder. MOD, therefore, and shock, are already conceptually a latent form of MOF about to occur, if the underlying derangements are not arrested and reversed! What is in fact MOF if not a shock that has not been arrested and reversed?
Hypotension
Hypotension is a cardinal sign of crucial significance in clinical scenarios of hemorrhage in that it signals the giving in of the compensatory neural and vascular system in its attempt to maintain blood flow to the noble organs (brain and heart) after having diverted it from the less noble (skin, muscles, soft tissues, kidney, liver, gut, lung) as last ditch defense mechanism. It represents the beginning of a possible pathway to exitus. The significance of the presence of hypotension in a clinical scenario with heart and brain signs of ischemia is that both these vital and essential organs will be the next ones to be affected by the scarcity of blood, and that they are about to be seriously damaged with a potentially fatal cerebro-vascular accident or heart attack. Sustained hypotension, which characterizes any shock, occurs as effect of arteriolar dilatation and is indication of the initial endothelial dysfunction at microcirculation/arteriolar level signaling the beginning of a still reversible decompensation. It is not by chance but with reason that reiterated consensus statements by ‘international consensus fashioners’ always stress and define as significant or shock-defining hypotension the hypotension not responding to fluids load.[27–28] This fits with the defining aspects of what is described as the ‘hyperdynamic phase of septic shock’, responding instead to fluids and needing fluids to maintain microcirculation integrity and its main function of perfusion, on an otherwise low-resistance circulation.[22–23,29–33] In other words, fluids are needed to prevent the passage from the compensated vasodilatory stage to a decompensated vasoconstricting form, which is the proper form of septic shock. Hypotension therefore must be persisting for definition of shock; fluids responding hypotension is not that different physiopathologically by a simple syncope or vaso-vagal faint.
Hypotension is signal to decompensation as in hemorrhage as in sepsis- scenario, where characterizes the definition of SIR and SS/IS. The other signs that have been included in the definition of SIR are actually aspecific and irrelevant for characterization as potentially due to other reasons and not being physio-pathologically characterizing SIR but merely its side-effects.[34–37] For example reflex tachycardia can be caused by hypotension but also by anxiety, hypoxia, hypercapnia, drugs, pain, temperature and direct irritating effect on the heart. Leukocytosis, pyrexia and tachypnoea have multiple causes too.
Thus SIR is the term that should be given to any inflammatory response to infection or tissue damage that from a localized source being acted upon by the philo and ontogenic localized inflammatory response (LIR) for different reasons the body fails to contain or counteract, becoming systemic, or to a I-R systemic damage. SIR is characterized by local and systemic release of inflammatory cells, coagulation factors, systemic factors and hormones, and toxic substances (endotoxins, bacterial exo-toxins, oxygen radicals, cytokines, nitric oxide (NO), leucotrienes, interleukins, interferons, tumour necrosis factor, complement, histamine, bradykinin, lysosomal and other proteolytic enzymes, arachidonic acid derivates or eucosanoids, platelet-activating factor and others. What characterizes SIR pathogenically, which is not a syndrome, is the systemic spread of the localized ontogenic inflammatory response, and clinically the hypotension, due to arteriolar vasodilation, and the microcirculation derangement with vasodilation, increased capillary permeability, oedema and pericapillary inflammation, and disoxia. A main specific target is the lung mucosa where it produces morphological and functional changes [acute lung injury (ALI) and pneumonia].
The type and amount of inflammatory mediators and/or toxic substances depends on the primary source. Besides the standard inflammatory mediators some sites release specific factors. A suppurative type of infection is standard of many bacterial infections; the pancreas for example releases among others a high content of elastase and proteolitic enzymes; a necrotic bowel is the spring platform for a high content of endotoxins and Gram-negative bacteria; a burnt tissue releases a great quantity of toxins and radicals; a gangrenous soft tissue lesion spreads a great quantity of toxins and anaerobs and a crush injury site a high quantity of myoglobins and endothelins particularly damaging kidneys. The effect of SIR on the macrocirculation is an indirect one of vasodilatation with relative hypovolemia by decrease of total peripheral resistance (TPR) and of hypotension following arteriolae and venulae dilatation, but it is on the microcirculation that its effects are more relevant producing microvascular and cellular derangements, with capillary fluid and inflammatory cells extravasation in the interstitial tissues and disoxia.[38–42]
Any shock in its established form is de facto specifically characterized by vasoconstriction. All toxic-inflammatory shocks (burns, ANP, ischemic-necrosis/gangrene, persistent or complicated intestinal obstruction, crush injury) are vasoconstricting, likewise HS and CSs, due to the prevailing effects of vasoconstricting factors e.g., endothelins and others, on the normally prevailing vasodilatory ones occurring in SIR. What then does make difference between a vasodilatory and a vasoconstricting effect when the mainstream of inflammatory response is similar in both the toxic and the septic derangements? The only plausible explanation, on the light of the knowledge acquired so far, is the role of the I-R phenomenon in the toxic-inflammatory derangements, where it represents the predominant and decisive physio-pathological mechanism.
Despite its predominant and characterizing vasodilatory effect, once SIR is not counteracted and allowed to protract in sustained form, it will inevitably damage microcirculation. The endothelium of the arterioles becomes itself oxygen-supply-dependant first and deficient later, and vasoconstricts (endothelial stunning). With progression or protraction of the structural damage endothelial dysfunction becomes failure, with no responsiveness to endogenous or exogenous vasoconstricting substances. In the end vasoparalysis signals impending death with stagnant hypoxia as signaled by no blanching and reflow to finger pressure. The progression of the endothelial crucial role in sepsis, as seen in late stages of endothelial hypoxia in CS and HS too, can be elegantly demonstrated by reactive hyperemia responses.[21,43–48]
Vasoconstrictor factors’ effect is initially overcome by the prevalent effect from the vasodilatative agents, this being particularly evident and accentuated in intensity and duration in the hyperdynamic preluding variant of the septic form of IS. Other IS situations by not infections-LPS toxins have a brief and less conspicuous hyperdynamic phase, giving rise soon to the hypodynamic situation characteristic of all shocks.
As for the ontological response to any body insult, SIR does not ensue as primary hit in HS because the same factors that would initiate it at the site of injury or damage are lost together with the blood from the injured spot.[49] There has been too much emphasis on SIR as autonomous concept. SIR should be defined as any infective or inflammatory process with hypotension and microcirculation derangement. As a matter of fact it is only hypotension that makes difference among the other four signs proposed for its definition (heart rate, fever, leucocytosis and tachypnea), which are non-specific and possibly due to a multiplicity of reasons, not necessarily to the hemodynamic derangement of arterioles dilatation and decreased systemic vascular resistance that is what SIR is mainly about. IS is an optimal umbrella term for describing shock features caused by sepsis or tissue damage, as result of persistent, accentuated, acute or acute-on-chronic SIR, with hypotension non-fluid respondent as result of inadequate compensatory vasoconstriction.[49–50] “Hypodynamic phase” is the name given to this decompensated phase, likewise in full-blown CS and HS, characterized by persisting hypotension and inadequate vasoconstriction.
The crucial question is why and how it happens that a LIR is not contained in situ but spreads all over the circulation. Ontogenic and philogenetical variations, possibly modulated by baseline nutritional status, explain variability to response of human body to LIR, which we should not forget was not engineered to defend the body against foreign pathogenic bacteria aggression and internal derangements damage or external injuries in an absolute way.
Cryptic (subclinical, refractory, persisting, unresolved) shock
Stabilization of shock as normalization of BP with or without normocardia, however, does not mean or is guarantee of adequate treatment.
Cryptic (subclinical, persisting, unresolved, refractory) shock is an untreated or inadequately treated shock. It is what kills in ICU patients with normal macro-hemodynamic variables and clinically elicitable peripheral perfusion.[51–52] While inadequate treatment of HS despite normal macro-dynamic variables shows itself with MOD/MOF by I-R phenomenon,[53] cryptic IS is more subtle and insidious and carries mortality between around 50-60%.[51]
In HS, tissue hypoperfusion also occurs long before the manifestation of hypotension.[13] MOD is concomitant and synchronous to hemorrhagic shock following ischemia or hypoperfusion[53–54] and translates clinically in persistent cryptic shock as manifestation of I-R. Whether it manifests as multiple organs dysfunction or failure depends on the level of ischemic damage and on the different organs physiological reserve in terms of hemodynamics and metabolic capacity. Organs in chronic compensated failure decompensate soon at minimal degrees of HS.
In IS, tissue hypoperfusion occurs long before the manifestation of hypotension. As for the macro-microcirculation dissociation, global oxygen transport parameters fail to measure or assess the status of the microcirculation in sepsis.
Recognition and management of cryptic subclinical IS still persisting after normalization of macro-hemodynamics is crucial for reduction of mortality [Table 4].
Table 4.
Cryptic shock

Global oxygen transport parameters like pH, lactate, Neg BE, SvO2, oxygen extraction index or ratio and the classical hemodynamic and metabolic variables can be relied as monitoring parameters in HS and CS but fail to measure or assess the status of the microcirculation in sepsis.[55]
Despite adequate fluid resuscitation and PO2, ATP remains low and lactate remains elevated,[22–23] suggesting that tissue PO2 is not a reliable indicator of bioenergetic status during sepsis, or conceivably of anaerobic metabolism. Fluid resuscitation in the animal model does not prevent loss of functional capillary density nor restores microvascular regulation or the VO2/DO2 mismatch in the tissue.[42] Serum lactate is an indicator of tissue hypoperfusion, decreased tissue oxygenation, and anaerobic metabolism, but altered pyruvate dehydrogenase and Na+, K+-ATPase activity, and increased glycolysis rate can also give high lactate levels. Limitations must be kept in mind: causes of lactic acidaemia other than decrease oxygen consumption from decreased delivery such as diabetic acidosis and cirrhosis must be excluded. Regardless of etiology, lactate elevation in severe sepsis and persistence of elevated lactate over time identifies a patient with a high risk of death and need aggressive resuscitation to achieve as soon as possible shock treatment end-points.[56]
The persistence of elevated lactic acid levels > 4 mmol/L however is a reliable and relevant sign, indicating unresolved hypoxaemia and high risk of death.[56] In patients with lactic acidaemia between 4 and 7mmol/L, bicarbonate and base deficit were singularly found to be normal in 20-25% and combined in 10% of cases. Only at lactate > 10 mmol/L there was never found normal bicarbonate or lactate.[56] Base deficit and lactate are the most sensitive factors in quantifying oxygen debt[57] and base deficit has been found to be a sensitive marker in hypoxia from hypovolaemia.[58–59] Low mixed venous oxygen saturation (SvO2), a global parameter that represents the oxygen saturation of pooled blood from all the postcapillary venules in the body, also identifies anaerobic metabolism and global tissue hypoxia. Central venous oxygen saturation (ScvO2) has a 7% higher value than SvO2 and is convenient when SGC are not used. Regardless of the oxygen saturation on the arteriolar side of capillaries, a markedly low SvO2 identifies that the venular ends of capillaries likely contain deoxygenated blood. Finding a normal or high SvO2 value (i.e. venous hyperoxia) at the bedside does not exclude tissue dysoxia from occurring as it may be a sign of capillary shunting in the early stages[60] and of impaired cellular oxygen utilization (i.e. cytopathic hypoxia) in late stages.[61]
Cirrhosis is a dreadful pitfall that by the mechanism of shunting has abnormal O2 extraction rate, A-V DO2 and SvO2 similar to IS initial trends (hyperdynamic phase, capillaries shunting) and must be kept in mind in the assessment of alterations of those variables and pH.
Hyperdynamic septic shock can therefore be defined in different ways such as partial/incomplete, compensated and cryptic shock.
Tachycardia in presence of normalized pressures is another sign of cryptic compensated shock (stably unstable) once other concomitant factors or causes (pain, anxiety, high temperature, hypoxia, hypercapnia, drugs) are excluded and managed. Persistent elevated A-V DO2 and decreased SvO2 are also indications of untreated persisting tissues oxygen deficit (dysoxia). Tissue tonometry when refined and made more reliable may solve the problem of early detection of cryptic persistent shock. The presence of concomitant MOD or MOF confirms the not-optimal or delayed resuscitation.
The two hits of physiological deterioration in Shock/MOD-MOF
Two hits of physiological deterioration can be clinically recognized in many non-survivors. In addition to the direct pathway to exitus (one hit model) by irreversible progressive shock due to untreated, inadequately treated or untimely treated shock, each of the two peripheral types of shock (HS and IS), if not treated properly or in time, can lead to death by enhancing each other mechanisms despite macro-hemodynamics reversal before a no-return point (two-hit model of clinically detected physiological deterioration). This second hit can occur anytime after the primary insult, from hours to days later. There is overwhelming evidence that HS has a second downfall mainly and mostly for its ischemic effect on gut mucosa in a sort of localized I-R phenomenon responsible of inflammatory cells activation for a second hit SIR especially targeting the lung mucosa.[62–84] The majority of deaths in intensive care follow usually a rapid second hit deterioration by refractory ALI and/or pneumonia.[85–86] There is now enough direct experimental and indirect clinical evidence in literature that when the gut barrier function is compromised or lost, due to ischemic damage to the mucosa, bacterial flora changes or antibiotics manipulation, the release in the systemic circulation of bacteria, bacterial toxins, endotoxins, cytokines, inflammatory cells and tissues cellular humoral mediators, nitric oxide and oxygen radicals, can lead to SIR/IR and MOF even in absence of proven or clinically suspected bacterial sepsis. The contribution to MOD/MOF by I-R as result of disruption of the gut mucosal barrier function, compared to the potential contribution by other ischemic organs, is mainly due to Gram-negative bacteria translocation and endotoxemia, specific of the gut and rarely triggered by other organs, released in circulation in addition to toxins and mediators of the I-R phenomenon. It is irrelevant whether inadequate perfusion in the fragile gut mucosa with a highly energy-dependent metabolism and functions is caused by the macro-hemodynamic decreased perfusion by hemorrhagic shock or increased intra-abdominal pressure, instead of by the micro-hemodynamic derangement with increased oxygen demand of an IS or SIR or I-R. In the end, once the gut barrier function is lost, the gut becomes the spring platform for the secondary hit insult by releasing bacteria and cells toxic products enough to produce a secondary more refractory shock/MOD/MOF. Under experimental conditions, IR phenomenon in the gut itself or from any other remote site, although it has as main target the mucosa of the lung, causes also splanchnic vasoconstriction, increased permeability and decreased intestinal mucosa thickness, bacterial translocation and endotoxemia.[72–73,87–90] I-R phenomenon from other organs like the liver may also contribute to second hit SIR.
Tissue hypoxia activates a large variety of vascular and inflammatory mediators that trigger local inflammation - endothelial dysfunction is an important early phenomenon in virtually all forms of I-R and may lead to a SIR that in many cases culminates in MOD or MOF.[53–54,91–92]
Leukocytes, in particular macrophages, are activated by translocated bacterial endotoxin and hypoxia/reoxygenation. Activated Kupffer cells release pathologically active substances such as inflammatory cytokines, reactive oxygen species (ROS), and NO. The hemorrhagic insult itself results in bacterial translocation from the gut in most animals and treatment with normal saline (NS), or hypertonic saline (HTS) or blood reduces early mortality but does not alter significantly the translocation rate. Only the combination of HTS and blood results in reduced bacterial translocation from gut to distant sites due to their beneficial effects on viscosity and reduced vasodilatation. This means that bacterial translocation on its own is not an essential pre-requisite for SIR/IR rather a pathogenic component like endotoxins or liposaccharides (LPS), exotoxins, and other inflammatory cells and factors.[93]
IS does not have a second hit as such, likewise HS, due to the random cumulative character of the sepsis and inflammatory disorders. Any involvement of lung, kidney and adrenals as dysfunction first and as failure to follow is the clinical threshold of their involvement ab initio by SIR in a continuum that passes through a phase of cryptic shock.
It has been suggested that SIR does not ensue as primary hit in HS because the same factors that initiate it are lost together with the blood loss from the injured spot.[49]
ALI, contrarily to what occurs in HS where is a late secondary hit event, in IS it occurs often as component of the first hit.[85–86]
Irreversible shock: Non-return-point
Organs are affected by hypoxia following hypoperfusion in HS and CS or by the direct toxic effect and higher oxygen demand of the IS.
Sepsis-related coagulopathy, endothelial dysfunction, structural damage and exaggerated persistent LIR, overlap the progression of sepsis systemic-random damage caused by the maldistribution of perfusion and disoxia following flow heterogeneity and capillaries stop-flow.[47,94–97] In the end when mitochondria cannot process oxygen any longer for intrinsic failure, then SvO2 increases again this time signaling cytopathic hypoxia and irreversibility[98] [Table 3].
Whether is hemorrhage or infection, the end result of both mechanisms, when not corrected, reversed or arrested, is the same: in the end all shocks kill with different aetiologies and at different speeds as effect and result of generalized hypoxia. Whether we call it shock, MOD or MOF, according to the view point of analysis, the determinant biological function, the funnel to exitus, is and remains the progressive loss of the capacity to produce energy. No or inadequate intervention during the stage of decompensation leads to the so called ‘no-return point’ (NRP), where all shocks enter a predeath agonic stage and exhibit universal hypodynamic features of unresponsiveness to catecholamines, reduced metabolism, hypothermia, and reduced oxygen consumption with SvO2 independently on shock etiology . Irreversible shocks in the end will enter a predeath agonic stage and exhibit universal hypodynamic features of unresponsiveness to catecholamines, reduced metabolism, hypothermia, and reduced oxygen consumption independently on shock etiology[99–100] [Figure 1]. Coagulopathy kills by occluding the microcirculation (arterioles, capillaries, venulae) and causing generalized hypoxia, hypothermia and acidosis by freezing and impairing life indispensable enzymatic processes and by dampening life-saving responses. Terminal primary hypothermia cannot be reversed, is accompanied by terminal coagulopathy and indicates energy-production failure; terminal coagulopathy manifests with bleeding distant to any possible injury or tissue damage site e.g. mouth gush, and irreversible acidosis.
Figure 1.
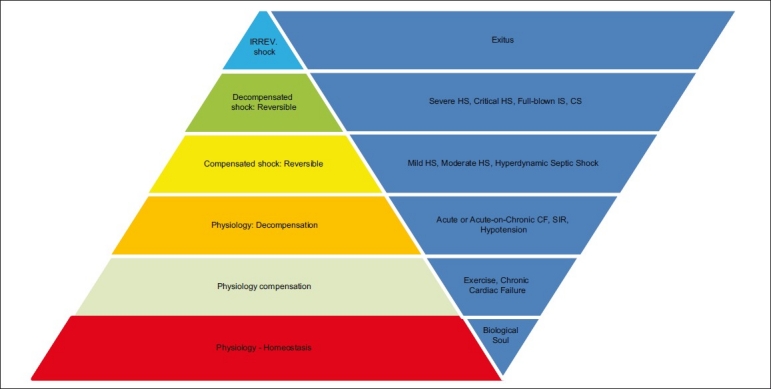
Unifying concept – Circulation from normal physiology to cessation of life
The passage from reversible to irreversible shock occurs at two NRPs: arteriolae and mitochondria. By non-return-point (NRP) is meant the point beyond which the arterioles-capillaries system or/and the cellular mitochondria cease functioning as result of an acute or persistent hypoxia. The first system cannot respond anymore to the oxygen/pressures/electric signals variations - stagnant hypoxia with no capillary blanching and reflow to digital pressure in superficial body areas displaying peripheral mottled cyanosis is a known sign of irreversibility and imminent death - and the second one cannot produce energy.
Adrenal insufficiency has also been associated with vasomotor paralysis and may represent a contributing factor probably linked to cortisone role in maintaining cells membranes stabilization.[101]
NRP is independent and not correlated to macrocirculation. This is why macrocirculation known variables cannot be relied upon for monitoring and prevention purposes. This is why patients suddenly die, as soon NRP is reached, despite seemingly normal or normalizing macrocirculation. Because of the impossibility of manipulating the energy factory at this stage, microcirculation integrity has a vital role from preventive and therapeutic point of view in critical illness. The microcirculation-ecosystem failure is the conditio sine qua non for irreversibility in all shocks.
CONCLUSIONS
Shock is a syndrome characterised by signs and symptoms, which are the result of the different organs response to a situation of low perfusion for their basic metabolic needs. The temporal sequence of the manifestations follows a pattern of inverse priority in the economy of human body physiology. Cryptic shock and two-hit clinical model of physiological deterioration are nowadays established concepts that need to be kept in focus if we want to prevent shock from reaching a NRP.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Haljamäe H. Microcirculation and hemorrhagic shock. Am J Em Med. 1984;2:100–7. doi: 10.1016/0735-6757(84)90117-7. [DOI] [PubMed] [Google Scholar]
- 2.Nakai K, Ikatura T, Naka Y, Ikatura T, Imai H, Komai N, et al. The distribution of adrenergic receptors in cerebral vessels: an autoradiographic study. Brain Res. 1986;381:148–52. doi: 10.1016/0006-8993(86)90703-1. [DOI] [PubMed] [Google Scholar]
- 3.Bruce CJ, Livingston DH, Schneider CA, Loder PA, Siegel JH. The effect of cocaine on the physiological response to hemorrhagic shock. Surgery. 1993;114:429–35. doi: 10.1097/00005373-199301000-00042. [DOI] [PubMed] [Google Scholar]
- 4.Rice IR, Kirkeby OJ. Effect of cerebral ischemia on the cerebrovascular and cardiovascular response to haemorrhage. Acta Neurochir (Wien) 1998;140:699–705. doi: 10.1007/s007010050165. [DOI] [PubMed] [Google Scholar]
- 5.Gettings LG, Reynolds HN, Scalea T. Outcome in post-traumatic acute renal failure when continuous renal replacement therapy is applied early vs late. Intensive Care Med. 1999;25:805–13. doi: 10.1007/s001340050956. [DOI] [PubMed] [Google Scholar]
- 6.Bagshaw SM, George C, Bellomo R. the ANZICS Database Management Committee. Early acute kidney injury and sepsis: a multicenter evaluation. Crit Care. 2008;12:R47. doi: 10.1186/cc6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tranbaugh RF, Elings VB, Christensen J, Lewis FR. Determinants of pulmonary interstitial fluid accumulation after trauma. J Trauma. 1982;22:820–6. doi: 10.1097/00005373-198210000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, et al. Tumor necrosis factor-alpha and interleukn-1 beta synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–18. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- 9.Kumar A, Brar R, Wang P, Dee L, Skorupa G, Khadour F, et al. Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999;276:R265–6. doi: 10.1152/ajpregu.1999.276.1.R265. [DOI] [PubMed] [Google Scholar]
- 10.Lee MA, Yatani A, Sambol JT, Deitch EA. Role of gut-lymph factors in the induction of burn-induced and trauma-shock-induced acute heart failure. Int J Clin Exp Med. 2008;1:171–80. [PMC free article] [PubMed] [Google Scholar]
- 11.Taccone FS, Su F, Pierrakos C, He X, James S, Dewitte O, et al. Cerebral microcirculation is impaired during sepsis: an experimental study. Crit Care. 2010;14:R140. doi: 10.1186/cc9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharshar T, Polito A, Checinski A, Stevens RD. Septic-associated encephalopathy - everything starts at a microlevel. Crit Care. 2010;14:199. doi: 10.1186/cc9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cosgriff N, Moore EE, Sauaia A, Kenny-Moynihan M, Burch JM, Galloway B. Predicting life-threatening coagulopathy in the massively transfused trauma patient: hypothermia and acidosis revisited. J Trauma. 1997;42:857–61. doi: 10.1097/00005373-199705000-00016. [DOI] [PubMed] [Google Scholar]
- 14.Trunkey DD. Initial treatment of patients with extensive trauma. N Engl J Med. 1991;324:1259–63. doi: 10.1056/NEJM199105023241806. [DOI] [PubMed] [Google Scholar]
- 15.Durham R, Neunaber K, Vogler G, Shapiro M, Mazuski J. Right ventricular end-diastolic volume as a measure of preload. J Trauma. 1995;39:218–23. doi: 10.1097/00005373-199508000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Singer M, Bennett ED. Non invasive optimization of left ventricular filling using oesophageal Doppler. Crit Care Med. 1991;19:1132–37. doi: 10.1097/00003246-199109000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Trof RJ, Danad I, Reilingh MW, Breukers RM, Groneveld J. Cardiac filling volumes versus pressures for predicting fluid responsiveness after cardiovascular surgery: the role of systolic cardiac function. Crit Care. 2011;15:R73. doi: 10.1186/cc10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groner W, Winkelman JW, Harris AG, Ince C, Bouma GJ, Messmer K, et al. Orthogonal polarization spectral imaging: a new method for study of the microcirculation. Nat Med. 1999;5:1209–12. doi: 10.1038/13529. [DOI] [PubMed] [Google Scholar]
- 19.Ince C. Sidestream dark field (SDF) imaging: an improved technique to observe sublingual microcirculation. Crit Care. 2005;8(suppl 1):72. [Google Scholar]
- 20.Creteur J, De Backer D, Sakr Y, Koch M, Vincent J. Sublingual capnometry tracks microcirculatory changes in septic patients. Intensive Care Med. 2006;32:516–23. doi: 10.1007/s00134-006-0070-4. [DOI] [PubMed] [Google Scholar]
- 21.De Backer D, Ospina-Tascon G, Salgado D, Favory R, Creteur J, Vincent JL. Monitoring the microcirculation in the critically ill patient: current methods and future approaches. Intensive Care Med. 2010 doi: 10.1007/s00134-010-2005-3. [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 22.Astiz M, Rackow EC, Weil MH, Schumer W. Early impairment of oxidative metabolism and energy production in severe sepsis. Circ Shock. 1988;26:311–20. [PubMed] [Google Scholar]
- 23.Anning PB, Sair M, Winlove CP, Evans TW. Abnormal tissue oxygenation and cardiovascular changes in endotoxemia. Am J Respir Crit Care Med. 1999;159:1710–15. doi: 10.1164/ajrccm.159.6.9801124. [DOI] [PubMed] [Google Scholar]
- 24.Siegemund M, Van Bommel J, Ince C. Assessment of regional tissue oxygenation. Intensive Care Med. 1999;25:1044–60. doi: 10.1007/s001340051011. [DOI] [PubMed] [Google Scholar]
- 25.Caille V, Squara P. Oxygen uptake-to-delivery relationship: a way to assess adequate flow. Crit Care. 2006;10(S):S4. doi: 10.1186/cc4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tweddell JS, Ghanayem NS, Hoffman GM. Pro: NIRS is “standard of care” for postoperative management.Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2010;13:44–50. doi: 10.1053/j.pcsu.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Intensive Care Med. 2004;30:536–55. doi: 10.1007/s00134-004-2210-z. [DOI] [PubMed] [Google Scholar]
- 28.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock. Intensive Care Med. 2008;34:17–60. doi: 10.1007/s00134-007-0934-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rackow EC, Kaufmann BS, Falk JL, Astiz ME, Weil MH. Hemodynamic response to fluid repletion in patients with septic shock: evidence for early depression of cardiac performance. Circ Shock. 1987;22:11–22. [PubMed] [Google Scholar]
- 30.Samsel RW, Nelson DP, Sanders WM, Wood LD, Schumacker PT. Effect of endotoxin on systemic and skeletal muscle O2 extraction. J Appl Physiol. 1988;65:1377–82. doi: 10.1152/jappl.1988.65.3.1377. [DOI] [PubMed] [Google Scholar]
- 31.Schumacker PT, Samsel RW. Oxygen delivery and uptake by peripheral tissues: physiology and pathophysiology. Crit Care Clin. 1989;5:255–69. [PubMed] [Google Scholar]
- 32.Walley KR. Heterogeneity of oxygen delivery impairs oxygen extraction by peripheral tissues: theory. J Appl Physiol. 1996;81:885–894. doi: 10.1152/jappl.1996.81.2.885. [DOI] [PubMed] [Google Scholar]
- 33.Yang S, Cioffi WG, Bland KI, Chaudry IH, Wang P. Differential alterations in systemic and regional oxygen delivery and consumption during the early and late stages of sepsis. J Trauma. 1999;47:706–12. doi: 10.1097/00005373-199910000-00015. [DOI] [PubMed] [Google Scholar]
- 34.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. ACCP/SCCM Consensus Conference Committee: Definitions for severe sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 35.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS: International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530–8. doi: 10.1007/s00134-003-1662-x. [DOI] [PubMed] [Google Scholar]
- 36.Baue AE. MOF, MODS, and SIRS: what is in a name or in an acronym? Shock. 2006;26:438–49. doi: 10.1097/01.shk.0000228172.32587.7a. [DOI] [PubMed] [Google Scholar]
- 37.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. ACCP/SCCM Consensus Conference Committee: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 2009;136(5 Suppl):e28. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 38.Taylor AE, Moore TM. Capillary fluid exchange. Am J Physiol. 1999;277:S203–10. doi: 10.1152/advances.1999.277.6.S203. [DOI] [PubMed] [Google Scholar]
- 39.Dellinger RP. Cardiovascular management of septic shock. Crit Care Med. 2003;31:946–55. doi: 10.1097/01.CCM.0000057403.73299.A6. [DOI] [PubMed] [Google Scholar]
- 40.Ellis CG, Wrigley SM, Groom AC. Heterogeneity of red blood cell perfusion in capillary networks supplied by a single arteriole in resting skeletal muscle. Circ Res. 1994;75:357–68. doi: 10.1161/01.res.75.2.357. [DOI] [PubMed] [Google Scholar]
- 41.Nakajima Y, Baudry N, Duranteau J, Vicaut E. Microcirculation in intestinal villi: a comparison between hemorrhagic and endotoxin shock. Am J Respir Crit Care Med. 2001;164:1526–30. doi: 10.1164/ajrccm.164.8.2009065. [DOI] [PubMed] [Google Scholar]
- 42.Ellis CG, Bateman RM, Sharpe MD, Sibbald WJ, Gill R. Effect of a maldistribution of microvascular blood flow on capillary O2 extraction in sepsis. Am J Physiol Heart Circ Physiol. 2002;282:156–64. doi: 10.1152/ajpheart.2002.282.1.H156. [DOI] [PubMed] [Google Scholar]
- 43.Bhagat K, Moss R, Collier J, Vallance P. Endothelial ‘stunning’ following a brief exposure to endotoxin: a mechanism to link infection and infarction? Cardiovasc Res. 1996;32:822–29. [PubMed] [Google Scholar]
- 44.Hingorani AD, Cross J, Kharbanda RK, Mullen MJ, Bhagat K, Taylor M, et al. Acute systemic inflammation impairs endothelium-dependent dilation in humans. Circulation. 2000;102:994–99. doi: 10.1161/01.cir.102.9.994. [DOI] [PubMed] [Google Scholar]
- 45.Motterlini R, Kerger H, Green CJ, Winslow RM, Intaglietta M. Depression of endothelial and smooth muscle cell oxygen consumption by endotoxin. Am J Physiol. 1998;275:H776–H782. doi: 10.1152/ajpheart.1998.275.3.H776. [DOI] [PubMed] [Google Scholar]
- 46.Karimova A, Pinsky DJ. The endothelial response to oxygen deprivation: biology and clinical implications. Intensive Care Med. 2001;27:19–31. doi: 10.1007/s001340000790. [DOI] [PubMed] [Google Scholar]
- 47.Doerschug KC, Delsing AS, Schmidt GA, Haynes WG. Impairment in microvascular reactivity is related to organ failure in human sepsis. Am J Physiol Heart Circ Physiology. 2007;293:H1065–H1071. doi: 10.1152/ajpheart.01237.2006. [DOI] [PubMed] [Google Scholar]
- 48.Engelberger RP, Pittet YK, Henry H, Delodder F, Hayoz D, Chioléro RL, et al. Acute endotoxemia inhibits microvascular nitric oxide-dependent vasodilation in humans. Shock. 2011;35:28–34. doi: 10.1097/SHK.0b013e3181ec71ab. [DOI] [PubMed] [Google Scholar]
- 49.Bonanno F. Extending damage control philosophy to non-haemorrhagic situations: implications for a reclassification of shock states. ANZJS. 2008;78:634–7. doi: 10.1111/j.1445-2197.2008.04601.x. [DOI] [PubMed] [Google Scholar]
- 50.Boffard KD. Resuscitative physiology. In: Boffard KD, editor. Manual of Definitive Trauma Care. London: Arnold publisher; 2003. pp. 7–38. [Google Scholar]
- 51.Donnino MW, Nguyen B, Jacobsen G, Tomlanovich M, Rivers E. Cryptic septic shock: a subanalysis of early, goal directed therapy. Chest. 2003;124:905. [Google Scholar]
- 52.Lima A, Jansen TC, Van Bommel J, Ince C, Bakker J. The prognostic value of the subjective assessment of peripheral perfusion in critically ill patients. Crit Care Med. 2009;37:934–8. doi: 10.1097/CCM.0b013e31819869db. [DOI] [PubMed] [Google Scholar]
- 53.Cryer HG, Leong K, McArthur DL, Demetriades D, Bongard FS, Fleming AW, et al. Multiple organ failure: by the time you predict it, it is already there. J Trauma. 1999;46:597–604. doi: 10.1097/00005373-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 54.Douzinas EE, Andrianakis I, Livaditi O, Paneris P, Tasoulis M, Pelekanou A, Betrosian A, Giamarellos-Bourboulis EJ. The level of hypotension during hemorrhagic shock is a major determinant of the post-resuscitation systemic inflammatory response: an experimental study. BMC Physiol. 2008;18;8:15. doi: 10.1186/1472-6793-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cohen ES, Hollenberg SM. Tissue oxygenation and sepsis. Crit Care Med. 2001;29:1479–80. doi: 10.1097/00003246-200107000-00032. [DOI] [PubMed] [Google Scholar]
- 56.Otero RM, Nguyen HB, Huang DT, Gaieski DF, Goyal M, Gunnerson KJ, et al. Early goal-directed therapy in severe sepsis and septic shock revisited: concepts, controversies and contemporary findings. Chest. 2006;130:1579–85. doi: 10.1378/chest.130.5.1579. [DOI] [PubMed] [Google Scholar]
- 57.Rixen D, Siegel JH. Bench-to-bedside review: oxygen debt and its metabolic correlates as quantifiers of the severity of hemorrhagic and post-traumatic shock. Crit Care. 2005;9:441–53. doi: 10.1186/cc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann Surg. 2007;245:812–8. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsumoto H, Mashiko K, Sakamoto Y, Kutsukata N, Hara Y, Yokota H. A new look at criteria for damage control surgery. J Nippon Med Sch. 2010;77:13–20. doi: 10.1272/jnms.77.13. [DOI] [PubMed] [Google Scholar]
- 60.Ince C, Sinaappel M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med. 1999;27:1369–77. doi: 10.1097/00003246-199907000-00031. [DOI] [PubMed] [Google Scholar]
- 61.Fink MP. Bench-to-bedside review: Cytopathic hypoxia. Crit Care. 2002;6:491–9. doi: 10.1186/cc1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deitch EA. The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch Surg. 1990;125:403–4. doi: 10.1001/archsurg.1990.01410150125024. [DOI] [PubMed] [Google Scholar]
- 63.Deeb GM, Grum CM, Lynch MJ, Guynn TP, Gallagher KP, Ljungman AG, et al. Neutrophils are not necessary for induction of ischemia-reperfusion lung injury. J Appl Physiol. 1990;68:374–81. doi: 10.1152/jappl.1990.68.1.374. [DOI] [PubMed] [Google Scholar]
- 64.Saadia R, Schein M, McFarlane J, Boffard K. Gut barrier and the surgeon. Br J Surg. 1990;77:487–92. doi: 10.1002/bjs.1800770505. [DOI] [PubMed] [Google Scholar]
- 65.Steimle CN, Guynn TP, Morganroth ML, Bolling SF, Carr K, Deeb GM. Neutrophils are not necessary for ischemia-reperfusion lung injury. Ann Thorac Surg. 1992;53:64–72. doi: 10.1016/0003-4975(92)90758-v. [DOI] [PubMed] [Google Scholar]
- 66.Deitch EA, Xu D, Franco L, Ayala A, Chaudry IH. Evidence favouring the role of the gut as cytokine-generating organ in rats subjected to haemorrhagic shock. Shock. 1994;1:141–5. doi: 10.1097/00024382-199402000-00010. [DOI] [PubMed] [Google Scholar]
- 67.Koike K, Moore EE, Moore FA, Read RA, Carl VS, Banerjee A. Gut ischemia/reperfusion produces lung injury independent of endotoxin. Crit Care Med. 1994;9:1438–44. doi: 10.1097/00003246-199409000-00014. [DOI] [PubMed] [Google Scholar]
- 68.Peitzman AB, Billiar TR, Harbrecht BG, Kelly E, Udekwu AO, Simmons RL. Hemorrhagic shock. Curr Probl Surg. 1995;32:925–1002. doi: 10.1016/s0011-3840(05)80008-5. [DOI] [PubMed] [Google Scholar]
- 69.Schein M, Witmann DH, Aprahamian CC, Condor ER. The abdominal compartment syndrome: the physiological and clinical consequences of elevated intra-abdominal pressure. J Am Coll Surg. 1995;180:745–53. [PubMed] [Google Scholar]
- 70.Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma. 1996;40:501–12. doi: 10.1097/00005373-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 71.Tamion F, Richard V, Lyoumi S, Daveau M, Bonmarchand G, Leroy J, et al. Gut ischemia and mesenteric synthesis of inflammatory cytokines after hemorrhagic or endotoxic shock. Am J Physiol - Gastrointestinal and Liver Physiology. 1997;273:314–21. doi: 10.1152/ajpgi.1997.273.2.G314. [DOI] [PubMed] [Google Scholar]
- 72.Yassin MM, Barros D’Sa AA, Parks TG, McCaigue MD, Leggett P, Halliday MI, et al. Lower limb ischaemia-reperfusion injury alters gastrointestinal structure and function. Br J Surg. 1997;84:1425–29. [PubMed] [Google Scholar]
- 73.Yassin MM, Barros D’Sa AA, Parks TG, Soong CV, Halliday MI, McCaigue MD, et al. Lower limb ischaemia-reperfusion injury causes endotoxaemia and endogenous antiendotoxin antibody consumption but not bacterial translocation. Br J Surg. 1998;85:785–9. doi: 10.1046/j.1365-2168.1998.00717.x. [DOI] [PubMed] [Google Scholar]
- 74.Garrison RN, Spain DA, Wilson MA, Keelen PA, Harris PD. Microvascular changes explain the “two-hit” theory of multiple organ failure. Ann Surg. 1998;227:851–60. doi: 10.1097/00000658-199806000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rowlands BJ, Soong CV, Gardiner KR. The gastrointestinal tract as a barrier in sepsis. Br Med Bull. 1999;55:196–211. doi: 10.1258/0007142991902213. [DOI] [PubMed] [Google Scholar]
- 76.Reilly PM, Wilkins KB, Fuh KC, Haglund U, Bulkley GB. The mesenteric hemodynamic response to circulatory shock: an overview. Shock. 2001;15:329–43. doi: 10.1097/00024382-200115050-00001. [DOI] [PubMed] [Google Scholar]
- 77.Saini MS, Liberati DM, Diebel LN. Sequential changes in mucosal immunity after haemorrhagic shock. Am Surg. 2001;67:797–801. [PubMed] [Google Scholar]
- 78.Baylor AE, Diebel LN, Liberati DM, Dulchawski SA, Brown WJ, Diglio CA. The synergistic effects of hypoxia/reoxygenation or tissue acidosis and bacteria on intestinal apoptosis. J Trauma. 2003;55:241–8. doi: 10.1097/01.TA.0000079249.50967.C5. [DOI] [PubMed] [Google Scholar]
- 79.Niu CY, Hou YL, Zhao ZG, Zhang YF, Ji JJ, Qiao HX, et al. Role of intestinal lymphatic pathway in pathogenesis of intestine derived bacteria/endotoxin translocation in rats in shock. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2007;19:266–9. [PubMed] [Google Scholar]
- 80.Perl M, Chung CS, Perl U, Lomas-Neira J, de Paepe M, Cioffi WG, et al. Fas induced pulmonary apoptosis and inflammation during indirect lung injury. Am J Respir Crit Care Med. 2007;176:591–601. doi: 10.1164/rccm.200611-1743OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Senthil M, Watkins A, Barlos D, Xu DZ, Lu Q, Abungu B, et al. Intravenous injection of trauma-hemorrhagic shock mesenteric lymph causes lung injury that is dependent upon activation of the inducible nitric oxide synthase pathway. Ann Surg. 2007;246:822–30. doi: 10.1097/SLA.0b013e3180caa3af. [DOI] [PubMed] [Google Scholar]
- 82.Watkins AC, Caputo FJ, Badami C, Barlos D, Xu da Z, Lu Q, et al. Mesenteric lymph duct ligation attenuates lung injury and neutrophil activation after intraperitoneal injection of endotoxin in rats. J Trauma. 2008;64:126–30. doi: 10.1097/TA.0b013e3181574a8a. [DOI] [PubMed] [Google Scholar]
- 83.Zhao ZG, Niu CY, Zhang J, Chen RH, Zhang YP, Jiang H, et al. Effect of ligation of mesenteric lymph duct on inflammation response of liver hemorrhagic shock in rats. Zhongguo Wei Zhong Bing Ji Jiu Xue. 2008;20:385–9. [PubMed] [Google Scholar]
- 84.Feinman R, Deitch EA, Watkins AC, Abungu B, Colorado I, Kannan KB, et al. Hypoxia Inducible Factor (HIF-1) mediates pathogenic inflammatory responses to intestinal ischemia reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2010;299:G833–43. doi: 10.1152/ajpgi.00065.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rutherford RB, Arora S, Fleming PW, Monaghan T, Lowenstein DH. Delayed onset pulmonary insufficiency in primates resuscitated from hemorrhagic shock. J Trauma. 1979;19:422–31. doi: 10.1097/00005373-197906000-00006. [DOI] [PubMed] [Google Scholar]
- 86.Regel G, Grotz M, Weltner T, Sturm JM, Tscherne H. Pattern of organ failure following severe trauma. World J Surg. 1996;20:422–9. doi: 10.1007/s002689900067. [DOI] [PubMed] [Google Scholar]
- 87.Yassin MM, Harkin DW, Barros D’Sa AA, Halliday MI, Rowlands BJ. Lower limb ischemia-reperfusion injury triggers a systemic inflammatory response and multiple organ dysfunction. World J Surg. 2002;26:115–21. doi: 10.1007/s00268-001-0169-2. [DOI] [PubMed] [Google Scholar]
- 88.Yassin MM, Barros D’Sa AA, Parks G, Abdulkadir AS, Halliday I, Rowlands BJ. Mortality following lower limb ischemia-reperfusion: a systemic inflammatory response? World J Surg. 1996;20:961–7. doi: 10.1007/s002689900144. [DOI] [PubMed] [Google Scholar]
- 89.Kologlu M, Sayek I, Kologlu B, Eng C, Onat D. Effect of persistently elevated intra-abdominal pressure on healing of colonic anastomoses. Am J Surg. 1999;178:293–7. doi: 10.1016/s0002-9610(99)00175-0. [DOI] [PubMed] [Google Scholar]
- 90.Kologlu M, Yorganci K, Renda N, Sayek I. Effect of local and remote ischemia-reperfusion injury on healing of colonic anastomoses. Surgery. 2000;128:99–104. doi: 10.1067/msy.2000.107414. [DOI] [PubMed] [Google Scholar]
- 91.Lefer AM, Lefer DJ. Pharmacology of the endothelium in ischemia-reperfusion and circulatory shock. Annu Rev Pharmacol Toxicol. 1993;33:71–90. doi: 10.1146/annurev.pa.33.040193.000443. [DOI] [PubMed] [Google Scholar]
- 92.Nathan C. Oxygen and the inflammatory cell. Nature. 2003;17:675–6. doi: 10.1038/422675a. [DOI] [PubMed] [Google Scholar]
- 93.Assalia A, Bitterman H, Hirsh TM, Krausz MM. Influence of hypertonic saline on bacterial translocation in controlled hemorrhagic shock. Intensive Care Med. 2004;30:1011–13. doi: 10.1097/00024382-200115040-00010. [DOI] [PubMed] [Google Scholar]
- 94.Sessler C, Windsor A, Schwartz M. Circulating ICAM-1 is increased in septic shock. Am J Respir Crit Care Med. 1995;151:1420–7. doi: 10.1164/ajrccm.151.5.7735595. [DOI] [PubMed] [Google Scholar]
- 95.Muller AM, Cronen C, Muller KM, Kirkpatrick CJ. Heterogeneous expression of cell adhesion molecules by endothelial cells in ARDS. J Pathol. 2002;198:270–5. doi: 10.1002/path.1186. [DOI] [PubMed] [Google Scholar]
- 96.Vallet B. Bench-to-bedside review: Endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Critical Care. 2003;7:130–8. doi: 10.1186/cc1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Levi M, De Jonge E, Van Der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. 2003;16:43–7. doi: 10.1023/B:THRO.0000014592.27892.11. [DOI] [PubMed] [Google Scholar]
- 98.Bonanno GF. Physiopatholgy of shock. J Emergencies, Trauma, and Shock. 2011;4:222–32. doi: 10.4103/0974-2700.82210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rutherford RB, Balis JV, Trows RS, Graves GM. Comparison of hemodynamic and regional blood flow at equivalent stages of endotoxin and hemorrhaghic shock. J Trauma. 1976;16:886–97. doi: 10.1097/00005373-197611000-00007. [DOI] [PubMed] [Google Scholar]
- 100.McMillen MA, Huribal M, Sumpio B. Common pathway of endothelial-leukocyte interaction in shock, ischemia, and reperfusion. Am J Surg. 1993;166:557–62. doi: 10.1016/s0002-9610(05)81153-5. [DOI] [PubMed] [Google Scholar]
- 101.Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res. 1999;41:55–64. doi: 10.1016/s0008-6363(98)00230-2. [DOI] [PubMed] [Google Scholar]