

Abstract
Critical events in the life cycle of malaria parasites are controlled by calcium-dependent signalling cascades, yet the molecular mechanisms of calcium release remain poorly understood. The synchronized development of Plasmodium berghei gametocytes relies on rapid calcium release from internal stores within 10 s of gametocytes being exposed to mosquito-derived xanthurenic acid (XA). Here we addressed the function of phosphoinositide-specific phospholipase C (PI-PLC) for regulating gametocyte activation. XA triggered the hydrolysis of PIP2 and the production of the secondary messenger IP3 in gametocytes. Both processes were selectively blocked by a PI-PLC inhibitor, which also reduced the early Ca2+ signal. However, microgametocyte differentiation into microgametes was blocked even when the inhibitor was added up to 5 min after activation, suggesting a requirement for PI-PLC beyond the early mobilization of calcium. In contrast, inhibitors of calcium release through ryanodine receptor channels were active only during the first minute of gametocyte activation. Biochemical determination of PI-PLC activity was confirmed using transgenic parasites expressing a fluorescent PIP2/IP3 probe that translocates from the parasite plasmalemma to the cytosol upon cell activation. Our study revealed a complex interdependency of Ca2+ and PI-PLC activity, with PI-PLC being essential throughout gamete formation, possibly explaining the irreversibility of this process.
Introduction
To be transmitted from the blood stream to a mosquito, malaria parasites rely entirely on highly specialized sexual precursor stages, the gametocytes. While circulating in the blood, mature gametocytes remain in a resting state within erythrocytes, but upon ingestion by a mosquito they rapidly resume development. In response to converging physical and chemical cues from the mosquito midgut environment gametocytes differentiate rapidly into gametes. Activated gametocytes of both sexes emerge from their host erythrocytes and female (macro-) gametocytes are thought to be available for fertilization immediately. Emerged male (micro-) gametocytes, in contrast, require another 10–15 min, during which they enter the cell cycle, complete three cycles of DNA replication and mitosis, assemble axonemes, and then give rise to eight flagellated microgametes in a process termed exflagellation. Gametes fertilize and each zygote then transforms into a motile stage, the ookinete, which from about 20 h post feeding penetrates the mosquito peritrophic matrix and midgut epithelium to establish the infection in the mosquito (Sinden et al., 1996; Alano and Billker, 2005). Triggers of gametocyte activation include a drop in temperature, a rise in pH and the small mosquito-derived molecule, xanthurenic acid (Carter and Nijhout, 1977; Nijhout, 1979; Billker et al., 1997; 1998; Garcia et al., 1997). At a permissive temperature either a rise in pH or xanthurenic acid are sufficient to activate gametocytes (Billker et al., 2000). In search of second messengers regulating activation, pharmacological studies identified roles for cyclic guanosine 3′,5′-monophosphate (cGMP) and Ca2+ in P. berghei and P. falciparum (Kawamoto et al., 1990). Both pathways were recently confirmed in genetic studies. The only known cGMP effector in Plasmodium, protein kinase G (PKG), is essential at an early stage in P. falciparum gametocyte activation (McRobert et al., 2008).Negative regulation of cGMP in P. falciparum gametocytes requires a parasite phosphodiesterase, PDEδ (Taylor et al., 2008). In P. berghei gametocytes cytosolic Ca2+ was measured in a transgenic reporter line expressing a Ca2+ sensitive luciferase, which revealed a rapid release of Ca2+ from intracellular stores within less than 10 s of exposing gametocytes to xanthurenic acid (Billker et al., 2004). In P. berghei Ca2+ controls all constituent events of gametogenesis, including egress from the host cell, male cell cycle progression and exflagellation. Differentiation of the male gametocyte is regulated through a male-specific Ca2+-dependent protein kinase, CDPK4, which is required for the initiation of DNA replication (Billker et al., 2004). After replication and mitosis an atypical mitogen-activated kinase-like protein, MAP-2, that serves as substrate for CDPK4 in vitro, is then needed at the stage of exflagellation for motile microgametes to emerge (Khan et al., 2005; Rangarajan et al., 2005; Tewari et al., 2005). Both kinases are dispensable for macrogametocyte activation and for gametocyte egress from the host cell in either sex, suggesting other Ca2+-dependent events are mediated through different effector pathways.
A parasite receptor for xanthurenic acid has remained elusive and how physical and chemical triggers from the mosquito activate second messenger pathways in gametocytes is largely unknown. In eukaryotes different upstream messengers and channels control Ca2+ release from intracellular compartments. One pathway involves ryanodine receptor (RyR) channels on the endoplasmic reticulum (ER), which are bound tightly by the plant alkaloid ryanodine, but which are controlled in vivo by the intracellular messenger cyclic ADP ribose (cADPR), the product of a specific cyclase (Galione and Churchill, 2002). Toxoplasma gondii can produce cADPR and possesses RyR Ca2+ release channels, which regulate intracellular Ca2+ in a way that is important for microneme secretion, Ca2+-dependent egress and parasite motility (Chini et al., 2005; Nagamune et al., 2008). Although enzymes and channels involved in cADPR signalling have so far only been identified from animals, at least parts of this pathway seem conserved in Apicomplexa.
Another pathway to Ca2+ mobilization relies on phosphoinositide specific phospholipase C (PI-PLC), which hydrolyses the minor membrane lipid phosphatidylinositol-(4,5)-bisphosphate (PIP2), producing the secondary messengers inositol-(1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG); IP3 then triggers Ca2+ release into the cytosol by binding to IP3-gated Ca2+ channels localized predominately in the ER membrane (Berridge et al., 2000).
Phosphatidylinositol is the phospholipid that in erythrocytes infected with P. falciparum asexual stages experiences the highest relative increase due to biosynthetic activity of the parasite, indicating important biological functions in Plasmodium (Vial et al., 1990). Parasite-derived PIP2 synthesis and Ca2+-dependent production of inositol polyphosphates is preponderant in mature asexual blood stage P. falciparum parasites (Elabbadi et al., 1994). The parasite's PI synthase has been characterized (Elabbadi et al., 1994; Wengelnik and Vial, 2007), as has been a phosphatidylinositol 4-phosphate 5-kinase that gives rise to PIP2 (Leber et al., 2009).
PI-PLC is a strong candidate for regulating cellular Ca2+ levels in gametocytes, because IP3 and DAG were found previously to increase in response to gametocyte activation in P. falciparum (Martin et al., 1994). In the current study we examine the role of PIP2 hydrolysis during gametogenesis of P. berghei in the context of our recent advances in understanding the timing of signalling events in this parasite species. We combine a kinetic analysis with pharmacological experiments to place agonist induced activation of PI-PLC with respect to Ca2+ mobilization early in gametocyte activation. We also present evidence for additional roles of IP3 production at late stages of gametogenesis.
Results
PI-PLC inhibition abolishes gametocyte activation
In P. berghei gametocyte activation requires a rapid increase of cytosolic Ca2+ released from intracellular stores, which becomes detectable within 8–10 s of exposing gametocytes to xanthurenic acid at a permissive temperature (Billker et al., 2004). Ca2+ mobilization in gametocytes can be conveniently measured using a transgenic reporter strain of P. berghei that constitutively expresses a Ca2+-dependent luciferase, GFP–aequorin. Using this assay we first examined the effect of a widely used inhibitor of PI-PLC dependent signalling, U73122. Between 0.5 and 5 µM U73122 dose-dependently reduced the XA induced Ca2+ signal in populations of enriched gametocytes (Fig. 1A), consistent with a role for PI-PLC upstream of Ca2+ mobilization. However, at 20 µM U73122 we unexpectedly observed an increase in cytosolic Ca2+, albeit with a time-course atypical of an XA-induced response (Fig. 1A, left). In fact, at this concentration, U73122 mobilized intracellular Ca2+ independently of XA (Fig. 1A, right). We next compared U73122 with its inactive structural analogue, U73343. In Fig. 1B the total luciferase activity during the first 50 s after XA activation is plotted against compound concentration, showing that inhibition of the XA-induced Ca2+ response was specific to U73122 and maximal at around 5 µM. The ‘inactive’ analogue did not reduce the Ca2+ signal but instead enhanced the XA-induced Ca2+ response (Fig. 1A lower panels and Fig. 1B). The selective inhibitory effect of U73122 over its structural analogue would be consistent with an early role for PI-PLC during the first few seconds of gametocyte activation, and upstream of Ca2+ release. Consistent with this hypothesis, 20 µM U73122 inhibited exflagellation completely and selectively over U73343 (Fig. 1C). We next asked whether the addition of inhibitor at different time points after the initial Ca2+ burst would still block exflagellation. Exflagellation remained sensitive to U73122 when the inhibitor was added to the gametocyte culture at any time during at least the first 5 min after activation, but thereafter became resistant (Fig. 1D). This indicates that PI-PLC activation is required beyond the first few seconds of gametocyte activation, during which intracellular Ca2+ is mobilized. The resistance of activated gametocytes after 5 min furthermore shows that neither U73122 nor U73343 exhibited non-specific toxicity towards gametocytes. Exflagellation is a highly dynamic process and inherently difficult to quantify. A more robust measure of male gametocyte activation can be obtained from a [3H]hypoxanthine incorporation assay, which determines DNA synthesis during the rapid threefold genome replication that precedes microgamete release (Raabe et al., 2009). We used this assay to determine the IC50 of U73122 as being just below 3 µM (Fig. 1E). This inhibitor thus blocks gametocyte activation selectively over U73343 at the same concentration, at which rapid Ca2+ mobilization within the first 10 s is also inhibited (Fig. 1A).
Fig. 1.
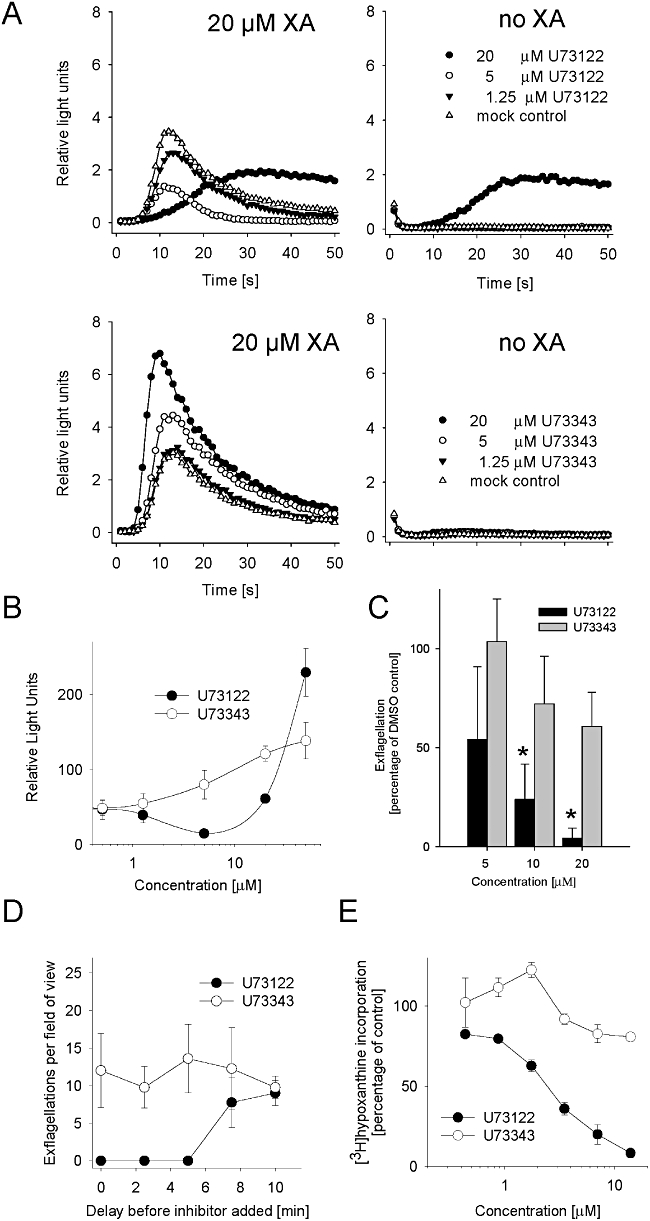
The PI-PLC inhibitor U73122 inhibits Ca2+ mobilization, DNA synthesis and exflagellation in P. berghei gametocytes.
A. Effect of U73122 on light emission over time in gametocytes expressing the Ca2+-dependent luciferase GFP–aequorin. Representative time-courses show effects of U73122 (upper panels) and U73433 (lower panels) on XA induced Ca2+ mobilization (left) compared with effects of compounds alone (right). XA and compounds were added at time point 0 s.
B. Dose-dependent effects of U73122 and U73343 on Ca2+-dependent luciferase activity in the presence of 20 µM XA. Relative light units were integrated over the first 50 s after addition of XA + inhibitor.
C. Dose-dependent effects on exflagellation of inhibitors added at the moment of microgametocyte activation, expressed as a percentage of a DMSO control. Asterisks indicate significant differences from solvent controls (*P < 0.01, Student's t-test).
D. Effects on exflagellation of adding 20 µM U73122 or U73343 at different time points after gametocyte activation by XA + pH 8.0. Exflagellation was counted after 12–15 min. Error bars indicate standard deviations among 10 slides from three different experiments.
E. Dose-response of U73122 and U73343 for [3H]hypoxanthine incorporation as a measure of DNA synthesis during microgametogenesis. Compounds and [3H]hypoxanthine were added simultaneously with the activation medium. Error bars in (B), (C) and (E) show standard deviations from 3–4 samples.
Changes of PI-PLC substrate levels upon gametocyte activation
We next sought to measure the cellular PI-PLC activity in intact cells directly by monitoring the level of radiolabelled cellular PIP2, the substrate of PI-PLC. Incubating preparations of highly enriched gametocytes with [32P]orthophosphate resulted in efficient incorporation of radiolabel into PIP, PIP2 and other phospholipids, as revealed by thin layer chromatography (TLC) of extracted cellular lipids in parallel with lipid standards (Fig. 2A). Label incorporation into phosphoinositides was linear over a 6 h incubation period (Fig. 2B), and male gametocytes retained their ability to differentiate into gametes for up to 3 h of culture in vitro (data not shown). We therefore routinely assayed PI-PLC activity after 2 h of labelling, when gametocytes were still unaffected in their ability to differentiate. When gametocytes were activated by XA, PIP2 levels decreased within the first minute (Fig. 2C) and then remained depressed if compared with time-matched, mock treated control cells. The Ca2+ ionophore A23187 produced a similar drop in cellular PIP2 levels, consistent with the ability of Ca2+ to activate PI-PLC in P. falciparum infected erythrocytes (Elabbadi et al., 1994). We wondered if the PIP2 hydrolysis we observed could be attributed entirely to the parasite, or if some occurred in the host cell compartment. However, at room temperature [32P]orthophosphate incorporation into uninfected erythrocytes was only 4% of gametocyte infected cells (Fig. 2D). Host cell phosphoinositides are thus unlikely the make a significant contribution to the PIP2 hydrolysis shown in Fig. 2C.
Fig. 2.
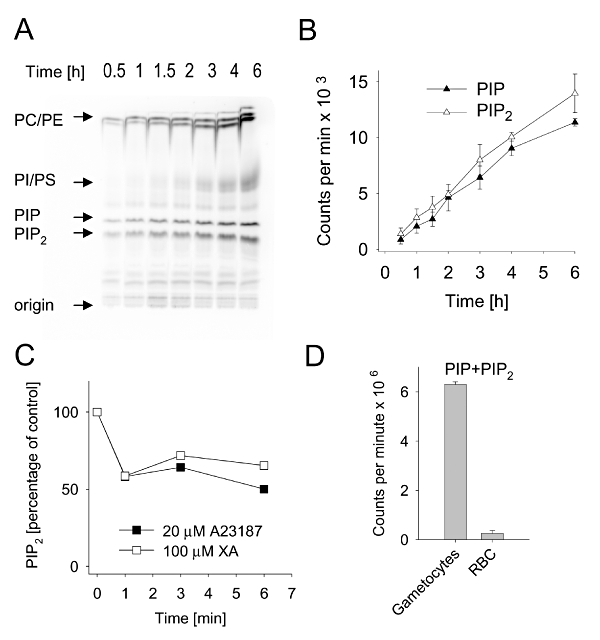
PIP2 hydrolysis during gametogenesis.
A. Phosphoimager scan of a TLC plate showing separation of phospholipids from 9.2 × 107 purified gametocyte-infected erythrocytes labelled with [32P] orthophosphate for 0.5–6 h. The position of lipid standards is indicated. PI, phosphatidylinositol; PS, phosphatidylserine; PC, phosphatidylcholine; PE, phosphatidylethanolamine.
B. PIP and PIP2 bands were quantified by liquid scintillation counting and plotted against labelling period. Error bars show SE of two experiments.
C. PIP2 levels in gametocytes treated with XA or A23187 after previous labelling with 32P for 90 min, expressed as a percentage of time-matched mock treated controls. Shown is one representative experiment of three.
D. 32P incorporation into PIP + PIP2 by gametocyte-infected and uninfected erythrocytes labelled for 6 h. Error bars show standard deviations in duplicate measurements from two independent experiments.
Analysis of PI-PLC product levels upon gametocyte activation
In complementary experiments we also determined the level of IP3, the product of PIP2 hydrolysis, using a Biotrak assay system. XA-independent PI-PLC activation by Ca2+ ionophore A23187 resulted in a rapid and sustained increase of total IP3 levels in gametocyte cultures (Fig. 3A). In contrast, gametocyte activation by XA produced a marked but weaker and more delayed response, in which a rise in IP3 did not become apparent until later than one minute of activation (Fig. 3A, inset). Importantly, XA-induced IP3 production continued throughout gametocyte differentiation (Fig. 3B). The XA-induced rise in cellular IP3 was totally abolished by U73122, but not U73343 (Fig. 3B and C), consistent with PI-PLC being involved. XA-induced IP3 production was completely inhibited by the membrane permeable Ca2+ chelator, BAPTA-AM (Fig. 3C). PI-PLC thus appears to require cellular Ca2+. Surprisingly, however, PI-PLC activity was not sensitive to U73122 when activated by the Ca2+ ionophore A23187 (Fig. 3C). We hypothesized that unphysiologically high Ca2+ levels could overcome PI-PLC inhibition by U73122. However, when we varied extracellular Ca2+ over a wide range of concentrations before adding the ionophore, we failed to find a condition at which Ca2+-induced IP3 production was selectively inhibited by U73122 over U73343 (Fig. 3D). We conclude that direct activation of PI-PLC though Ca2+ may bypass inhibition by U73122, which has an unknown mechanism of action.
Fig. 3.
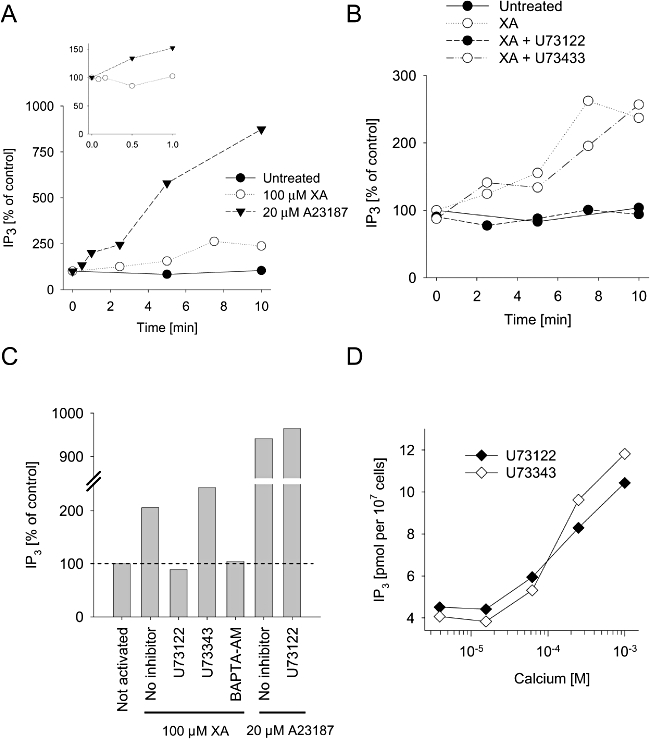
IP3 production during gametogenesis.
A. IP3 content of purified gametocyte-infected erythrocytes at different times after treatment with XA, A23187 or solvent control, expressed as a percentage of the resting level (around 2 pmol per 107 gametocytes). Inset shows immediate onset of IP3 production only in A23187 treated cells. Shown is a representative result from two experiments.
B. Effect of U73122 and U73343 (both 10 µM) on IP3 following activation by 100 µM XA.
C. Effect of inhibitors on cellular IP3 content 10 min after treatment with either XA or A23187.
D. Effect of various Ca2+ concentrations in the culture medium on IP3 content 10 min after ionophore activation (20 µM A23187) in the presence of either 10 µM U73122 or 10 µM U73343.
Single cell imaging using a PIP2/IP3 binding fluorescent reporter protein
We next sought to observe PI-PLC activation at the level of the individual gametocyte. Dynamic changes in cellular PIP2 have been monitored successfully in cultured mammalian cells by single cell imaging of a fluorescent reporter protein fused to the PH domain of human phospholipase Cδ1 (hPLCδ1) (Violin et al., 2003). PH domains can bind both, PIP2 and IP3. Resting cells contain low IP3 levels and a PH domain-containing reporter protein is targeted mostly to the plasma membrane where PIP2 resides. PI-PLC activation and IP3 production then leads to translocation of the probe to the cytoplasm (Fig. 4A). We generated a P. berghei expression cassette, in which the strong constitutive ef1α promoter controls expression of a fusion protein consisting of the PH domain of hPLCδ1 fused to yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP). A vector containing this reporter cassette, together with a Tgdhfr/ts selection marker for antimalarial drug resistance, was then introduced into P. berghei schizonts by electroporation, and maintained as episome by selecting for the resistance marker. In most resting gametocytes of either sex the CFP–PH–YFP protein was clearly detectable in the periphery of the cells, consistent with a localization at the plasma membrane (Fig. 4B). By time lapse microscopy we observed that within two minutes of activation by XA, the CFP–PH–YFP protein began to redistribute to the cytosol, a process that was typically complete 5 min after gametocyte activation (Fig. 4B). A quantitative analysis in randomly selected macrogametocytes found that CFP–PH–YFP redistributed to the cytosol in about half of the cells (red lines in Fig. 4C). A few cells showed a high proportion of peripherally located CFP–PH–YFP that did not change upon addition of XA (blue lines in Fig. 4C); these cells may have been immature gametocytes still unable to respond to XA. Other gametocytes had a relatively high level of cytosolic fluorescence that remained unchanged (black lines in Fig. 4C). The latter response was typical of the cytosolic localization in the control cell line expressing GFP without a PH domain (Fig. 4D and E). CFP–PH–YFP expressing cells with cytosolic localization of the marker may have responded already during the minute that typically elapsed between gametocyte activation and recording of the first image. Male and female gametocytes both showed redistribution of the CFP–PH–YFP reporter constructs, but due to the choice of promoter the reporter protein was more strongly expressed and easier to detect in macrogametocytes (not shown). In the vast majority of gametocytes CFP–PH–YFP accumulated transiently in a disc-like structure in the cell periphery (Fig. 4F). We have no explanation for this structure, but believe it may indicate a transient heterogeneity in membrane lipid composition of differentiating gametocytes that could be linked to the marked changes in cell shape and volume during gametogenesis (Sinden and Croll, 1975).
Fig. 4.
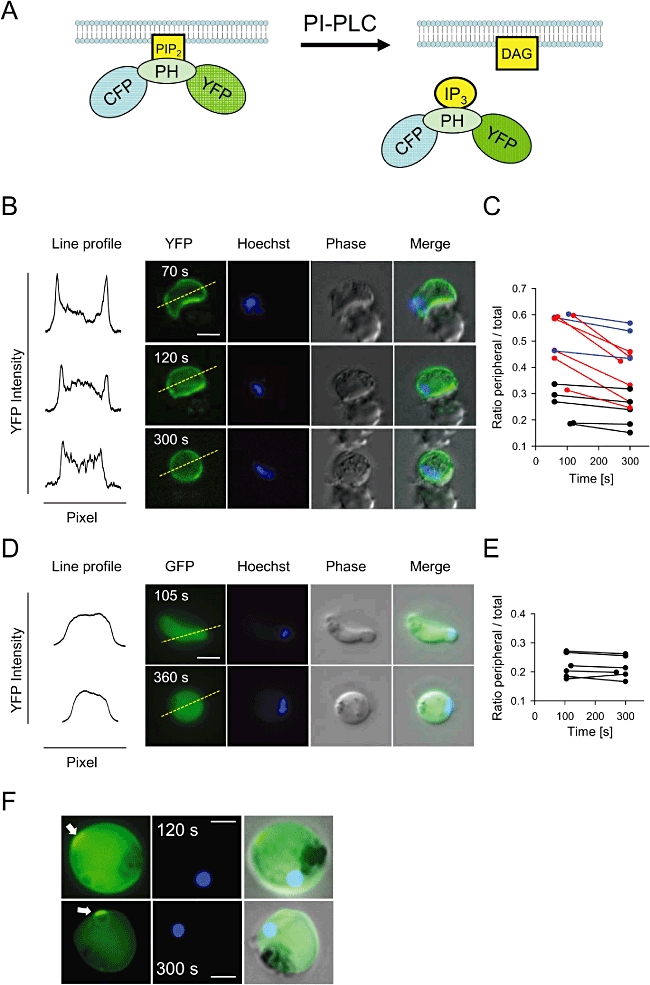
A fluorescent reporter protein to monitor PI-PLC activation in single live P. berghei gametocytes.
A. Scheme illustrating translocation from the plasma membrane to the cytosol of the CFP–PH–YFP protein upon PI-PLC activation.
B. Light microscopic images of a CFP–PH–YFP expressing macrogametocyte recorded at different time points after activation with XA. The first column shows the line profiles of the intensity of YFP fluorescence along the dashed lines in the corresponding YFP images.
C. CFP–PH–YFP redistribution in 14 randomly selected macrogametocytes. See text for an explanation of line colours.
D. Time lapse microscopy and intensity profiles after activation of macrogametocytes expressing GFP without a PH domain as a control probe.
E. Absence of peripheral localization and redistribution of GFP lacking a PH domain.
F. Fluorescence images of typical CFP–PH–YFP expressing macrogametocytes showing a disc-like structure (arrows) seen in the majority of gametocytes after rounding up. All indicated times are seconds post activation. The scale bar is 2 µm in all panels.
Analysis of RyR channels in Ca2+ release
In many mammalian tissues IP3 receptor channels coexist and interact with Ca2+ release through RyR channels, for instance during Ca2+-induced Ca2+ release (Berridge et al., 2000; 2003;). In view of the importance of RyR in agonist-induced signalling in the closely related parasite Toxoplasma gondii, we examined two inhibitors that have been validated for blocking RyR in this species, dantrolene and ruthenium red (Chini et al., 2005; Nagamune et al., 2008), in P. berghei gametocytes. Both compounds significantly inhibited the rapid Ca2+ response to XA (Fig. 5A) and also inhibited exflagellation (data not shown), an effect we quantified in the [3H]hypoxanthine incorporation assay for dantrolene (Fig. 5B). Reduced IP3 production in gametocytes treated with ruthenium red (Fig. 5C) indicated that RyR mediated Ca2+ mobilization may be required for sustained PI-PLC activity during gametogenesis, not only for the early agonist mediated Ca2+ burst. Having shown that exflagellation remained sensitive to PI-PLC inhibition by U73122 even beyond the initial XA-induced Ca2+ burst (Fig. 1D), we wondered whether a RyR channel antagonist would be more selective for early events leading to rapid Ca2+ mobilization. To examine this possibility we added either dantrolene or U73122 at different time points after activation, and asked when [3H]hypoxanthine incorporation became insensitive to the inhibitors (Fig. 5D). The window of sensitivity differed markedly between both compounds. Dantrolene only exerted its full inhibitory effect when added simultaneously with XA. In contrast, gametocyte differentiation remained sensitive to addition of U73122 for an extended period of 4–6 min after activation. Taken together, these data suggest that ryanodine receptors and IP3-mediated mechanisms are likely to be responsible for the rapid early Ca2+ mobilization in activated gametocytes. In contrast, in order for gametogenesis to be completed sustained activation of PI-PLC for a more extended period, resulting in increasing IP3 levels, is required.
Fig. 5.
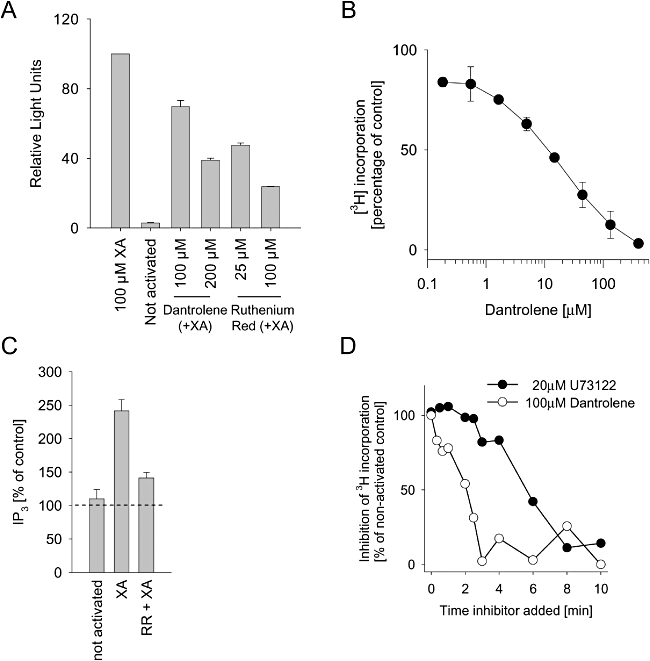
RyR receptor channel antagonists inhibit Ca2+ mobilization and IP3 production upon gametocyte activation.
A. Effect of dantrolene and ruthenium red on induction of Ca2+ mobilization by 100 µM XA, measured as light emitted over 50 s in GFP–aequorin expressing gametocytes. Error bars show standard deviations (n = 4).
B. Effect of dantrolene on microgametogenesis, measured as [3H]hypoxanthine incorporation into DNA. Error bars show standard deviations (n = 3).
C. Effect of 100 µM ruthenium red on IP3 produced by gametocyte-infected erythrocytes activated by 100 µM XA or mock treatment (pipetting). IP3 is given as a percentage of untreated control cells. Error bars show standard deviations (n = 4). Inhibitors in panels A-C were added at the time of gametocyte activation by XA.
D. Effect of delayed addition of agents on inhibition of microgametogenesis, as measured by DNA synthesis ([3H]hypoxanthine incorporation) during the first 20 min of activation. Dantrolene was used at 100 µM, U73122 at 20 µM. Inhibitors were added after 100 µM XA-induced onset of gametogenesis at indicated time points. The inhibition of label incorporation is expressed relative to non-activated control cells. Shown is one representative experiment of three.
Discussion
Ca2+ is an important second messenger regulating key events throughout the life cycle of apicomplexan parasites (Billker et al., 2009). In Plasmodium the activation of gametocytes currently provides the best documented example of a signal transduction pathway leading from extracellular signals, via the rapid release of Ca2+ from intracellular stores, to a stage-specific Ca2+ effector pathway for cellular differentiation (Billker et al., 2004). How extracellular signals are linked to Ca2+ release in malaria parasites is an important question that is difficult to address experimentally, since candidate genes for signalling receptors have not been identified in Plasmodium. To help close this gap we have here investigated the role for PI-PLC in XA induced activation of gametocytes in P. berghei.
All malaria species encode in their genome a single candidate gene for PI-PLC (e.g. P. falciparum PF10_0132 and, P. berghei PBANKA_121190), which is characterized by a predicted domain organization largely conserved from human PLCδ isoforms to yeast PI-PLC (Williams and Katan, 1996). Like its T. gondii orthologue (Fang et al., 2006), Plasmodium PI-PLC has a predicted N-terminal Pleckstrin homology (PH) domain presumably required for targeting PI-PLC to the plasma membrane, and a bipartite catalytic domain flanked by Ca2+-binding EF hands and a C2 domain that could be involved in binding to membrane phospholipids in a Ca2+-dependent or independent manner. Consistent with this conserved domain organization, recombinant TgPI-PLC, like PI-PLC isozymes from other organisms, is strictly Ca2+-dependent in vitro (Fang et al., 2006).
Measuring different inositol phosphates after metabolic labelling with [3H]myo-inositol, Martin et al. (1994) demonstrated that IP3 is produced in activated P. falciparum gametocytes and showed that it can be degraded via two routes: dephosphorylation to yield Ins(1,4)P2 or phosphorylation to yield Ins(1,3,4,5)P4. All our biochemical and functional assays clearly establish that PI-PLC activity is stimulated upon gametocyte activation by XA in P. berghei. We find that levels of PIP2 drop within a minute of activation and thereafter remain below resting level (Fig. 2C). An increase in IP3 becomes measurable with some delay, but then high levels of IP3 persist for up to 20 min (Fig. 3B and data not shown). A continuous rise in IP3 levels reflects well the persistent reduction of PIP2 levels below baseline, suggesting continuous activity of PI-PLC, as in P. falciparum. The kind of CFP–PH–YFP probe that we used here binds both PIP2 and IP3 and has been described to translocate to the cytoplasm more as a consequence of rising IP3 levels than of decreasing PIP2 levels at the plasma membrane (Hirose et al., 1999). The time-course we report here for the cytosolic translocation of the PH-YFP probe 3–5 min after activation (Fig. 4B) also seems to mirror the increase in IP3 (Fig. 3A) better than the decrease in PIP2 levels (Fig. 2C).
Gametocyte PIP2 is clearly not depleted entirely upon activation (Fig. 2C), as it would be in some mammalian model systems (Suh and Hille, 2007). Why is a large proportion of PIP2 not hydrolysed? We show that erythrocytes hardly incorporate [32P]orthophosphate into phosphoinositides at room temperature, and thus a major contribution of host lipids to our PIP2 measurements is unlikely. We have used a fluorescent probe that reports specifically on PI-PLC activity in the parasite cytosol and the data are consistent with overall changes in lipid composition upon gametocyte activation being primarily due to changes in the parasite. It remains a theoretical possibility, however, that a Plasmodium infection activates host cell PI-kinases in the red blood cell cytoplasm resulting in elevated PIP2 levels that would be inaccessible to the parasite's PI-PLC enzyme. It has been published that in mature human erythrocytes a calcium-dependent ‘phosphoinositidase C’ activity can be induced upon treatment with an ionophore (Gascard et al., 1989). A physiological trigger, however, and the function of the reaction products DAG and IP3 have, to our knowledge, not been identified, and common downstream effectors like the IP3-receptor and protein kinase C are thought to be absent from erythrocytes. Alternatively, if a large proportion of gametocytes were non-responsive to XA, this could explain why more than half of the labelled PIP2 appears unhydrolysed upon gametocyte activation. However, this is also unlikely since PI-PLC in such non-responsive gametocytes would presumably still be activated by the Ca2+ ionophore. We find it most likely that PIP2 is re-synthesized at a rate similar to its hydrolysis. Consistent with this hypothesis, we find the IP3 levels keep rising throughout gametogenesis, which requires PIP2 to be replenished. The last step in PIP2 biosynthesis is catalysed by a phosphatidylinositol 4-phosphate 5-kinase (PIP5K). This enzyme has been characterized in P. falciparum (Leber et al., 2009) as part of a putatively bifunctional protein contain N-terminally EF-hand-like motifs found in a family of neuronal Ca2+ sensor (NCS) proteins. It has been suggested that Ca2+ sensor domains could regulate PfPIP5K activity, linking directly cytosolic Ca2+ to PIP2 synthesis (Leber et al., 2009). This might lead to enhanced PIP2 synthesis and could be crucial for the sustained IP3 production we observe following the initial Ca2+ release after gametocyte activation. Alternatively, incomplete PIP2 hydrolysis could result from a partly inaccessible PIP2 pool. It is intriguing to speculate that the disk-like peripheral structure, in which the PIP2 binding PH-YFP reporter protein accumulates transiently after gametocyte activation (Fig. 4B and F), could be a specialized membrane domain or compartment, in which PIP2 is protected from hydrolysis.
The pharmacology of PI-PLC is still poorly understood. Our attempts to produce recombinant Plasmodium PI-PLC protein have been unsuccessful, preventing biochemical characterization of purified enzyme. We have indications that deletion or overexpression of the PI-PLC gene is deleterious for P. berghei blood stage development (A.C. Raabe, O. Billker, K. Wengelnik, unpublished) excluding genetic approaches. Thus, we rely on pharmacology to place PI-PLC activity with respect to agonist-induced Ca2+ mobilization during gametocyte activation. We find that Ca2+ release in gametocytes is selectively inhibited by U73122 over its structural analogue, U73343, placing PI-PLC upstream of the rapid calcium release. The aminosteroid U73122 and its control compound are used widely to infer PI-PLC in signalling processes, but some studies have also reported significant off-target effects (Horowitz et al., 2005 and references therein). Consistent with this we find that U73122 and its ‘inactive’ analogue, U73343, can both non-selectively facilitate the mobilization of Ca2+ release in gametocytes at concentrations only just above those at which U73122 selectively inhibits XA-induced calcium release (Fig. 1B). The molecular mechanism of inhibition of PI-PLC by U73122 remains controversial. Some studies report direct inhibition of catalytic activity by U73122 of recombinantly expressed PLC isozymes (Staxen et al., 1999). However, other PI-PLC enzymes, including that from T. gondii, are not inhibited in vitro (Fang et al., 2006). We find that Ca2+/ionophore-activated PI-PLC is resistant to U73122, suggesting the compound does not target the catalytic site but interferes in some other way with enzyme activation in intact cells. Nevertheless, U73122 in Plasmodium is clearly able to uncouple PI-PLC from its natural upstream activators.
The additional ability of U73122 to mobilize gametocyte Ca2+ (Fig. 1A) at 20 µM probably relies on a different mechanism that is independent of PI-PLC, since at the same high concentration IP3 production is effectively inhibited (Fig. 3B). The highly lipid-soluble and chemically reactive U73122 cation may exert its non-specific effects by sequestering membrane lipids, or by covalently modifying membrane proteins (Horowitz et al., 2005), which could explain the complex results some investigators have obtained with this compounds (Mogami et al., 1997). We have therefore used one functional and two biochemical assays to demonstrate that U73122 inhibits IP3 production selectively over its control compound, U73343, and that at the appropriate concentration this inhibition is strictly correlated with a block in Ca2+ release, and gametocyte differentiation.
That PI-PLC plays a key role early in gametocyte activation, at the time of rapid Ca2+ release, is supported by the selective inhibitory effect of U73122 (Fig. 3B and C), by our observation that PIP2 hydrolysis is initiated during the first minute of activation (Fig. 2C) and by previous evidence that in P. falciparum gametocytes IP3 levels shoot up within 30 s of activation (Martin et al., 1994). In P. berghei accumulation of IP3 appeared to trail PIP2 hydrolysis, becoming measurable only from 2 min after activation (Fig. 3A and B), although both might be expected to reflect PI-PLC activity. We can only speculate that an initial rapid increase in IP3 may be too small to become detectable by the assays we used, or that the first burst of IP3 may be rapidly degraded or metabolized to more highly phosphorylated inositol phosphates.
Which function PI-PLC might have during late stages of gametogenesis is unclear. Is it required to keep Ca2+ levels elevated? Our GFP–aequorin reporter assay is optimized to detect the initial Ca2+ release with exquisite sensitivity (Billker et al., 2004). However, it is unable to measure absolute Ca2+ levels reliably, due to the rapid depletion of the luciferase-substrate complex. The use of membrane permeable fluorescent Ca2+ sensor dyes (Garcia et al., 1996) could overcome this limitation. However, in our hands these dyes proved impractical in gametocytes since they were either hardly incorporated, showed high bleaching rates, or lead to exflagellation without XA stimulation (A.C. Raabe, K. Wengelnik, unpublished). We therefore do not know whether sustained IP3 production results in constantly elevated levels of cellular free Ca2+. It is also unknown whether diacylglycerol, the other product of PIP2 hydrolysis, has a signalling role in Plasmodium. A major target for DAG in other eukaryotes is protein kinase C, which has no obvious orthologue in Apicomplexa.
While on the one hand IP3 thus appears to be required for Ca2+ release, Ca2+ may in turn enhance PI-PLC activity. In support of this we found that a Ca2+ ionophore is sufficient to trigger activation of PI-PLC in resting P. berghei gametocytes in an XA-independent manner (Figs 2C and 3A), and that XA-mediated activation of PI-PLC is prevented when cytosolic Ca2+ is chelated by BAPTA-AM (Fig. 3C). As is typical of PLCδ isoforms, recombinant T. gondii PI-PLC was shown to be Ca2+-dependent (Fang et al., 2006). We were unable to express recombinant Plasmodium PI-PLC to confirm this, but Ca2+ binding C2 and EF hand domains appear to be intact in its conserved sequence.
We were intrigued to find that the RyR antagonists dantrolene and ruthenium red also inhibit early Ca2+ release and PI-PLC activation. The natural RyR agonist cADPR may thus be involved in the initial mobilizationof Ca2+, which could then trigger or support the more sustained activation of PI-PLC, resulting in Ca2+ release through IP3 receptor channels and irreversible activation of the gametocyte. Consistent with this model we find that male gametogenesis started to become insensitive to dantrolene within seconds of activation, but remained sensitive to U73122 for a much longer period (Fig. 5D). It is tempting to speculate that, following RyR activation during the initial rise of Ca2+ levels, positive feedback regulation of PI-PLC and Ca2+ could become important for gametocyte activation and for the continuous production of IP3 throughout gametogenesis. The likely irreversible nature of this process may be one reason why the initiation of gametogenesis needs to be so well controlled by multiple converging environmental factors.
Work in T. gondii has demonstrated that both IP3 and cADPR can trigger the release of Ca2+ from ER-derived membrane microsomes in vitro, and has validated in an apicomplexan parasite the use of ruthenium red and dantrolene as compounds that selectively block RyR (Chini et al., 2005). Both pathways may be involved in regulating gliding motility (Chini et al., 2005). More recently abscisic acid, previously known only as a plant hormone, was discovered to be an endogenously produced inducer of cADPR production, leading to tachyzoite egress (Nagamune et al., 2008). It will be interesting to investigate whether Plasmodium gametocytes posses an ADP-ribosyl cyclase activity that is stimulated by XA. Genes encoding IP3 receptors, RyR receptors or a ADP-ribosyl cyclase to produce cADPR have so far only been identified in animals and no obvious homologues are present in apicomplexan genomes (Billker et al., 2009). In mammalian cells activation of PI-PLC relies on heterotrimeric G-proteins or phosphorylating receptors (Rebecchi and Pentyala, 2000), which also appear to be absent from Apicomplexa. The identification of novel receptor mechanisms that link extracellular signals to Ca2+ release in Plasmodium will therefore be a major challenge for future research.
Experimental procedures
Solutions and chemicals
All chemicals were purchased from Sigma (France) unless otherwise stated. Stock solutions for dantrolene (10 mM), U73122 (2 mM), U73343 (2 mM), A23187 (4 mM), 8-Br-A23187 (4 mM) were made up in DMSO. As dantrolene is reportedly light sensitive, it was prepared fresh for each experiment. The final concentration of DMSO in all assays did not exceed 1%, a concentration that does not inhibit exflagellation or Ca2+ mobilization in gametocytes. Stock solutions for ruthenium red (10 mM) and xanthurenic acid (10 mM) were made up in water. Radioactive hypoxanthine was purchased from GE Healthcare, France ([3H]hypoxanthine stock solution: 52 µM hypoxanthine in water/ethanol 1:1; specific activity 1 mCi ml−1). Radioactive phosphate was purchased from Perkin Elmer ([32P] phosphorus as H3[32P]O4, 5 mCi ml−1 in water with specific activity of 285.6 Ci mg−1 at calibration). TLC plates used were 20 × 20 cm silica-coated glass plates (Silica 60) with a concentration zone (Merck, Germany).
Parasite maintenance and gametocyte purification
All parasites used in this study were derived from the P. berghei ANKA clone 2.34. For Ca2+ measurements, the clone 1.7.8 was used as previously described (Billker et al., 2004). Parasites were maintained in female NMRI mice (Charles River). This research adhered to the Principles of Laboratory Animal Care. The animal study was approved by the local animal use committees in compliance with European regulations and national legislation. Gametocytes were purified as described previously (Billker et al., 2004) with minor modifications. Mice were pre-treated with 0.1 ml phenylhydrazine (25 mg ml−1 in PBS) and infected 2 days later with 0.5–2 × 107 parasites from frozen blood stocks. On day 4 p.i. 20 mg l−1 sulfadiazine in drinking water was applied to kill asexual stages. On day 6 p.i., mice were bled by cardiac puncture, the blood washed in gametocyte maintenance buffer (GMB: 137 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 20 mM glucose, 20 mM Hepes, 4 mM sodium bicarbonate, 0.1% BSA, [pH 7.24–7.29]) and white blood cells were removed on CF11 cellulose (Whatman) columns. Gametocytes were purified on a 48% Nycodenz/GMB cushion [Nycodenz stock solution: 27.6% w/v Nycodenz in 5 mM Tris-HCl (pH 7.2), 3 mM KCl, 0.3 mM EDTA]. After purification gametocytes were resuspended in GMB and kept at 20°C and their purity examined on Giemsa stained blood films. On average, gametocytes were enriched to approximately 95% with contaminants mainly being late stage trophozoites (∼4%), few red blood cells and occasionally very few white blood cells.
Gametocyte activation, exflagellation and Ca2+ measurements
All experiments were carried out at room temperature (20–26°C) unless otherwise indicated. For phospholipid measurements gametocytes were activated by transferring them to gametocyte activation medium (GAM), RPMI 1640 with 20 mM Hepes, 4 mM sodium bicarbonate, pH 8.0 containing 100 µM XA, unless otherwise stated. To count exflagellation events either 10 µl purified gametocytes were resuspended in 50 µl GAM or 3 µl of tail blood from an infected mouse were washed rapidly in 1 ml GMB, pelleted and resuspended in 30 µl GAM. A drop was then placed on a microscope slide and covered with a Vaseline rimmed coverslip. Exflagellation events were then counted at 400× magnification after 12–15 min. DNA synthesis during microgametogenesis was measured through incorporation of radioactive [3H]hypoxanthine as described elsewhere (Raabe et al., 2009). The luminometric Ca2+ assay was performed on enriched gametocytes exactly as described previously (Billker et al., 2004). Briefly, gametocytes were loaded with the luciferin, coelenterazine-fcp (Biotrend, Germany), for 30 min at 20°C, in loading buffer containing 1 mM EGTA, pH 7.25. Washed gametocytes were then auto-injected, one well at a time into 96-well plates containing test compounds and 100 µM XA or control solutions. Bioluminescence was counted in a Berthold Orion II luminometer. The presence of either 20 µM or 100 µM XA in the different activation media had no influence on the results and both concentrations result in maximal activation of gametocytes.
Measurement of IP3 and PIP2
The Biotrak radioreceptor assay (GE Healthcare, product code TRK1000) was used, which determines IP3 in a sample by its ability to displace a [3H]IP3 radiotracer from a high affinity IP3 receptor protein (Palmer et al., 1989). Fifty microlitres of enriched gametocytes in GMB at 20°C (0.5–2 × 107 cells) was activated by transfer into 150 µl GAM. Parasite development was stopped by addition of 200 µl ice cold 10% (v/v) 1 M perchloric acid. IP3 was extracted using freon/octylamide as described in the manufacturer's manual.
32P labelling and analysis of phosphoinositides by thin layer chromatography
A total of 3 × 108 purified gametocytes were resuspended in 800 µl GMB containing the radioactive label 32P (final concentration depending on specific activity, usually 1 mCi) and incubated for 1.5–2 h at 20°C under agitation in an Eppendorf Thermomixer at 1000 r.p.m. Following three GMB washes in the presence of 10 mM cold phosphate, cells were resuspended and incubated at 20°C, 1000 r.p.m. for another 3 min to purge the cells of radioactive ATP. Gametocytes were then activated by addition of XA or A23187 and the reactions stopped by transferring aliquots (200 µl) into screw cap glass tubes containing 1.2 ml ice cold methanol. The following solvents were added with intermittent vortexing steps: 600 µl chloroform, 20 µl HCl (12 M), 600 µl chloroform, 600 µl KCl (2 M). Following centrifugation (3000 r.p.m., 20°C, 5 min) the lower phase was transferred to a new glass tube. The upper phase was washed by adding 2 ml chloroform and lower phases pooled. The solvent of the pooled fractions was evaporated under N2 flow at room temperature. For loading on a TLC plate, lipids were resuspended in 100 µl chloroform/methanol (2:1, v/v) and 10–20 µl applied onto the concentration zone of a TLC plate, which had previously been incubated for 15 min in oxalate solution [1% Potassium-Oxalate, 2 mM EDTA)/(Methanol) 1/1 (v/v)] and heat activated at 100°C for 1 h. TLC was performed with [CHCl3/CH3COCH3/CH3OH/ CH3COOH/H2O, 80/30/26/24/14 (v/v)]. TLC plates were revealed using a phosphoimaging scanner. The quantification of PIP and PIP2 was done by either scraping off the silica of bands of interest and analysis in a Beckman Coulter Multi-Purpose Scintillation Counter, or by using the Image Quant v5.2 software to obtain relative intensity levels.
Cloning of CFP–PH–YFP
The PH domain of human phospholipase Cδ1 (hPLCδ1) fused to CFP and YFP was isolated from a pcDNA3.1(+)-based plasmid containing the CYPHR fusion protein (Violin et al., 2003) as a HindIII/XbaI fragment and subsequently blunted. The P. berghei expression vector pDEFGFPM3A encodes the green fluorescent protein (GFP) and is equivalent to MR4 reagent MRA-786 (pL0017) differing from the pPbGFPCON (Franke-Fayard et al., 2004) only by the presence of an additional XbaI site immediately following the stop codon of gfp. The gfp coding sequence was removed by a BamHI/XbaI digest, the vector blunted and the CFP–PH–YFP sequence inserted. Correct insertion and the sequence of CFP–PH–YFP were confirmed by sequencing. Plasmid pDEFGFPM3A was used as control construct without a PH domain. Parasites harbouring the plasmids as episomes were generated as described (Janse et al., 2006) by electroporation of enriched schizonts followed by selection with pyrimethamine in the drinking water of infected mice.
Acknowledgments
We are grateful to Alexandra C. Newton, and Chris Janse for providing plasmids. This work is part of the activities of the BioMalPar and EVIMalaR European Networks of Excellence (LSHP-CT-2004–503578 and No. 242095). KW and HJV are INSERM investigators. OB was supported by the Medical Research Council (grant numbers G0501670) and the Wellcome Trust (WT089085/Z/09/Z).
References
- Alano P, Billker O. Gametocytes and gametes. In: Sherman IW, editor. Molecular Approaches to Malaria. Washington, DC: ASM Press; 2005. pp. 191–219. Chapter 10. [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Billker O, Shaw MK, Margos G, Sinden RE. The roles of temperature, pH and mosquito factors as triggers of male and female gametogenesis of Plasmodium berghei in vitro. Parasitology. 1997;115:1–7. doi: 10.1017/s0031182097008895. [DOI] [PubMed] [Google Scholar]
- Billker O, Lindo V, Panico M, Etienne AE, Paxton T, Dell A, et al. Identification of xanthurenic acid as the putative inducer of malaria development in the mosquito. Nature. 1998;392:289–292. doi: 10.1038/32667. [DOI] [PubMed] [Google Scholar]
- Billker O, Miller AJ, Sinden RE. Determination of mosquito bloodmeal pH in situ by ion-selective microelectrode measurement: implications for the regulation of malarial gametogenesis. Parasitology. 2000;120:547–551. doi: 10.1017/s0031182099005946. [DOI] [PubMed] [Google Scholar]
- Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell. 2004;117:503–514. doi: 10.1016/s0092-8674(04)00449-0. [DOI] [PubMed] [Google Scholar]
- Billker O, Lourido S, Sibley LD. Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe. 2009;5:612–622. doi: 10.1016/j.chom.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter R, Nijhout MM. Control of gamete formation (exflagellation) in malaria parasites. Science. 1977;195:407–409. doi: 10.1126/science.12566. [DOI] [PubMed] [Google Scholar]
- Chini EN, Nagamune K, Wetzel DM, Sibley LD. Evidence that the cADPR signalling pathway controls calcium-mediated microneme secretion in Toxoplasma gondii. Biochem J. 2005;389:269–277. doi: 10.1042/BJ20041971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elabbadi N, Ancelin ML, Vial HJ. Characterization of phosphatidylinositol synthase and evidence of a polyphosphoinositide cycle in Plasmodium-infected erythrocytes. Mol Biochem Parasitol. 1994;63:179–192. doi: 10.1016/0166-6851(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Fang J, Marchesini N, Moreno SN. A Toxoplasma gondii phosphoinositide phospholipase C (TgPI-PLC) with high affinity for phosphatidylinositol. Biochem J. 2006;394:417–425. doi: 10.1042/BJ20051393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke-Fayard B, Trueman H, Ramesar J, Mendoza J, van der KM, van der LR, et al. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Galione A, Churchill GC. Interactions between calcium release pathways: multiple messengers and multiple stores. Cell Calcium. 2002;32:343–354. doi: 10.1016/s0143416002001902. [DOI] [PubMed] [Google Scholar]
- Garcia CR, Dluzewski AR, Catalani LH, Burting R, Hoyland J, Mason WT. Calcium homeostasis in intraerythrocytic malaria parasites. Eur J Cell Biol. 1996;71:409–413. [PubMed] [Google Scholar]
- Garcia GE, Wirtz RA, Rosenberg R. Isolation of a substance from the mosquito that activates Plasmodium fertilization. Mol Biochem Parasitol. 1997;88:127–135. doi: 10.1016/s0166-6851(97)00086-8. [DOI] [PubMed] [Google Scholar]
- Gascard P, Journet E, Sulpice JC, Giraud F. Functional heterogeneity of polyphosphoinositides in human erythrocytes. Biochem J. 1989;264:547–553. doi: 10.1042/bj2640547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 1999;284:1527–1530. doi: 10.1126/science.284.5419.1527. [DOI] [PubMed] [Google Scholar]
- Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse CJ, Franke-Fayard B, Mair GR, Ramesar J, Thiel C, Engelmann S, et al. High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol. 2006;145:60–70. doi: 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kawamoto F, Alejo-Blanco R, Fleck SL, Kawamoto Y, Sinden RE. Possible roles of Ca2+ and cGMP as mediators of the exflagellation of Plasmodium berghei and Plasmodium falciparum. Mol Biochem Parasitol. 1990;42:101–108. doi: 10.1016/0166-6851(90)90117-5. [DOI] [PubMed] [Google Scholar]
- Khan SM, Franke-Fayard B, Mair GR, Lasonder E, Janse CJ, Mann M, Waters AP. Proteome analysis of separated male and female gametocytes reveals novel sex-specific Plasmodium biology. Cell. 2005;121:675–687. doi: 10.1016/j.cell.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Leber W, Skippen A, Fivelman QL, Bowyer PW, Cockcroft S, Baker DA. A unique phosphatidylinositol 4-phosphate 5-kinase is activated by ADP-ribosylation factor in Plasmodium falciparum. Int J Parasitol. 2009;39:645–653. doi: 10.1016/j.ijpara.2008.11.015. [DOI] [PubMed] [Google Scholar]
- McRobert L, Taylor CJ, Deng W, Fivelman QL, Cummings RM, Polley SD, et al. Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol. 2008;6:e139. doi: 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SK, Jett M, Schneider I. Correlation of phosphoinositide hydrolysis with exflagellation in the malaria microgametocyte. J Parasitol. 1994;80:371–378. [PubMed] [Google Scholar]
- Mogami H, Lloyd MC, Gallacher DV. Phospholipase C inhibitor, U73122, releases intracellular Ca2+, potentiates Ins(1,4,5)P3-mediated Ca2+ release and directly activates ion channels in mouse pancreatic acinar cells. Biochem J. 1997;324:645–651. doi: 10.1042/bj3240645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamune K, Hicks LM, Fux B, Brossier F, Chini EN, Sibley LD. Abscisic acid controls calcium-dependent egress and development in Toxoplasma gondii. Nature. 2008;451:207–210. doi: 10.1038/nature06478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout MM. Plasmodium gallinaceum: exflagellation stimulated by a mosquito factor. Exp Parasitol. 1979;48:75–80. doi: 10.1016/0014-4894(79)90056-0. [DOI] [PubMed] [Google Scholar]
- Palmer S, Hughes KT, Lee DY, Wakelam MJ. Development of a novel, Ins(1,4,5)P3-specific binding assay. Its use to determine the intracellular concentration of Ins(1,4,5)P3 in unstimulated and vasopressin-stimulated rat hepatocytes. Cell Signal. 1989;1:147–156. doi: 10.1016/0898-6568(89)90004-1. [DOI] [PubMed] [Google Scholar]
- Raabe AC, Billker O, Vial HJ, Wengelnik K. Quantitative assessment of DNA replication to monitor microgametogenesis in Plasmodium berghei. Mol Biochem Parasitol. 2009;168:172–176. doi: 10.1016/j.molbiopara.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan R, Bei AK, Jethwaney D, Maldonado P, Dorin D, Sultan AA, Doerig C. A mitogen-activated protein kinase regulates male gametogenesis and transmission of the malaria parasite Plasmodium berghei. EMBO Rep. 2005;6:464–469. doi: 10.1038/sj.embor.7400404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Sinden RE, Croll NA. Cytology and kinetics of microgametogenesis and fertilization in Plasmodium yoelii nigeriensis. Parasitology. 1975;70:53–65. doi: 10.1017/s0031182000048861. [DOI] [PubMed] [Google Scholar]
- Sinden RE, Butcher GA, Billker O, Fleck SL. Regulation of infectivity of Plasmodium to the mosquito vector. Adv Parasitol. 1996;38:53–117. doi: 10.1016/s0065-308x(08)60033-0. [DOI] [PubMed] [Google Scholar]
- Staxen I, Pical C, Montgomery LT, Gray JE, Hetherington AM, McAinsh MR. Abscisic acid induces oscillations in guard-cell cytosolic free calcium that involve phosphoinositide-specific phospholipase C. Proc Natl Acad Sci USA. 1999;96:1779–1784. doi: 10.1073/pnas.96.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of KCNQ channels by manipulation of phosphoinositides. J Physiol. 2007;582:911–916. doi: 10.1113/jphysiol.2007.132647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CJ, McRobert L, Baker DA. Disruption of a Plasmodium falciparum cyclic nucleotide phosphodiesterase gene causes aberrant gametogenesis. Mol Microbiol. 2008;69:110–118. doi: 10.1111/j.1365-2958.2008.06267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R, Dorin D, Moon R, Doerig C, Billker O. An atypical mitogen-activated protein kinase controls cytokinesis and flagellar motility during male gamete formation in a malaria parasite. Mol Microbiol. 2005;58:1253–1263. doi: 10.1111/j.1365-2958.2005.04793.x. [DOI] [PubMed] [Google Scholar]
- Vial HJ, Ancelin ML, Philippot JR, Thuet MJ. Biosynthesis and dynamics of lipids in Plasmodium-infected mature mammalian erythrocytes. Blood Cells. 1990;16:531–555. [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengelnik K, Vial HJ. Characterisation of the phosphatidylinositol synthase gene of Plasmodium species. Res Microbiol. 2007;158:51–59. doi: 10.1016/j.resmic.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Williams RL, Katan M. Structural views of phosphoinositide-specific phospholipase C: signalling the way ahead. Structure. 1996;4:1387–1394. doi: 10.1016/s0969-2126(96)00146-3. [DOI] [PubMed] [Google Scholar]