

Abstract
Nonceruloplasmin-bound copper (“free”) is reported to be elevated in Alzheimer's disease (AD). In Wilson's disease (WD) Cu-ATPase 7B protein tightly controls free copper body levels. To explore whether the ATP7B gene harbours susceptibility loci for AD, we screened 180 AD chromosomes for sequence changes in exons 2, 5, 8, 10, 14, and 16, where most of the Mediterranean WD-causing mutations lie. No WD mutation, but sequence changes corresponding to c.1216 T>G Single-Nucleotide Polymorphism (SNP) and c.2495 A>G SNP were found. Thereafter, we genotyped 190 AD patients and 164 controls for these SNPs frequencies estimation. Logistic regression analyses revealed either a trend for the c.1216 SNP (P = .074) or a higher frequency for c.2495 SNP of the GG genotype in patients, increasing the probability of AD by 74% (P = .028). Presence of the GG genotype in ATP7B c.2495 could account for copper dysfunction in AD which has been shown to raise the probability of the disease.
1. Introduction
There is a general agreement on the existence of a link between Alzheimer's disease (AD) and oxidative stress phenomena triggered by transition metals [1, 2]. The existence of systemic copper dysfunctions in AD has been a controversial issue for many years. In fact, many studies have reported an increase of circulating copper in AD patients with respect to healthy controls [3–13], many others no variation [14–23], and two very recent studies even a decrease of plasma [24] and serum [25] copper in AD patients. Recently, to gain an objective evaluation to the question whether systemic copper variations are associated to AD a meta-analysis of all the studies carried out on serum/plasma copper in AD and healthy cohorts between 1983 and 2010 was run [26]. This analysis demonstrated that AD patients have higher levels of serum Cu than healthy controls. Even though moderate, the assessed copper increase was sufficient to unambiguously distinguish AD patients from healthy controls.
Abnormalities in serum copper not bound to ceruloplasmin (“free” copper [27]) can be advocated as an explanatory variable of copper disturbances in AD [26, 28], as several research groups recently confirmed [3, 25, 29, 30]. Normally, most human serum copper binds tightly to ceruloplasmin [27]. The remaining copper, that is free copper, is distributed and exchanged among albumin, alpha 2 macroglobulin, and low-molecular-weight compounds such as peptides and amino acids (e.g., histidine [31]. A key difference between the two pools lies in the fact that the low-molecular-weight compounds allow free copper to easily cross the Blood-Brain Barrier (BBB) [32, 33]. A recent study confirmed the evidence that the copper transport into the brain is mainly achieved through the BBB as free copper ion, and the blood-cerebrospinal fluid barrier may serve as a main regulatory site of copper in the cerebrospinal fluid (CSF) [34].
Wilson's disease is the paradigmatic example of copper toxicosis or accumulation, in which large amounts of free copper enter the brain and cause cognitive impairment [35, 36], abnormal glial cells and degenerated ganglion cells in cerebral cortex, putamen, and dentate nucleuses [35, 36]. In WD, free copper levels are disproportionately high due to defects in the ATPase 7B (WD protein) and represent most of the circulating copper. Systemic copper abnormalities in AD resemble those observed in WD, though they are very much less severe [10, 37]. Moreover, free copper correlates with the typical deficits [9, 38–40] and markers of AD, namely, CSF Amyloid Beta (Aβ) and Tau proteins [9], as well as an unfavourable prognosis of the disease [40], and tend to predict the annual worsening in Minimental State Examination (MMSE) [10, 41]. WD is an autosomal recessive genetic disorder due to mutations in the ATP7B gene (WD gene), and the rate of occurrence of a single abnormal copy is 1 in 90 people [36]. Based on this, we initiated a hypothesis-driven candidate gene project to determine whether the WD ATP7B gene harbours susceptibility loci for late-onset AD [42, 43]. In particular, in the study presented, we explored the hypothesis that ATP7B sequence changes in exon 2, 5, 8, 10, 14, and 16—where most of the Mediterranean WD-causing mutations lie—have a higher frequency in a group of patients affected by mild or moderate AD compared to a group of healthy individuals.
2. Materials and Methods
190 patients with AD and 164 elderly controls were recruited by two specialized dementia care centres: the Department of Neuroscience, Fatebenefratelli Hospital, Isola Tiberina, in Rome, and the Department of Neurology, Campus Bio-Medico University, Rome, Italy, using a common standardized clinical protocol [10].
The AD patients sample consisted of individuals with a diagnosis of probable AD according to NINCDS-ADRDA criteria [44, 45] and an MMSE score of 25 or less [41]. All AD patients underwent general medical, neurologic, and psychiatric assessments. Neuroimaging diagnostic procedures (magnetic resonance imaging or computed tomography) and complete laboratory analyses were performed to exclude other causes of progressive or reversible dementia. The control sample consisted of healthy volunteers with no clinical evidence of neurological and psychiatric disease. Criteria for exclusion of both patients and controls were conditions known to affect copper metabolism and biological variables of oxidative stress (e.g., diabetes mellitus, inflammatory diseases, recent history of heart or respiratory failure, chronic liver or renal failure, malignant tumors, and a recent history of alcohol abuse).
Among the study populations, 28 AD cases and 41 controls were not analyzed for c.1216 T>G and 10 cases and 13 controls for c.2495 A>G because during the analyses it was not possible to assess the genotype (insufficient DNA/blood sample, sequence analysis failure).
The study was approved by the local IRB, and all participants or legal guardians signed an informed consent.
2.1. SNPs Genotyping
We collected approximately 10 mL of peripheral blood samples from study participants. Genomic DNA from fresh whole blood was prepared using the conventional method for DNA isolation (QLAamp DNA Blood Midi kit).
Polymerase chain reaction (PCR) was performed to amplify the exons and flanking regions of the ATP7B gene. DNA amplification was carried out in a total volume of 25 μL containing 50–100 ng of genomic DNA, 10 pmol of each primer, 0.4 mM of dNTPs, 3 mM MgCl2, and 1 unit of Taq polymerase (Taq Gold, Applied Biosystems) in a thermocycler (2720 Thermal Cycler Applied Biosystem). The conditions were denaturation at 95°C for 30 s, 30 s of appropriate annealing temperature (varying between 53°C–58°C), and 30 s of extension temperature at 72°C for 30 cycles with 5 min at 72°C final extension. Primers, sequences and annealing temperatures are reported in Table 1.
Table 1.
Oligonucleotides sequences.
| Primer forward for sequencing | Primer reverse for sequencing | Annealing temperature | PCR product | |
|---|---|---|---|---|
| Exon 2a | 5'AGAGGCCGTCATCACTTATC 3′ | 5'CAATGGCAATCAGAGTGGTA 3' | 57°C | 255 bp |
| Exon 2b | 5'AGCTCCTAGGGGTTCAAAGT 3′ | 5' CAAGGAAAGTTTGCAGGATT 3′ | 57°C | 584 bp |
| Exon 5 | 5'TTTCACAGGCTTTCCTTGAT 3′ | 5' ATTTCCATGGGAAAAGTTGA 3' | 53°C | 336 bp |
| Exon 8 | 5'CGACTGTGCACAAAGCTAGA 3′ | 5'CATGGTGTTCAGAGGAAGTG 3' | 54°C | 386 bp |
| Exon 10 | 5′CAGCTGGCCTAGAACCTGAC 3′ | 5'TATCCTCCTGAGGGAACAT 3' | 53°C | 234 bp |
| Exon 14 | 5'CTGTGCAGGTGTCTTGTTTC 3′ | 5'TTTTCCAGACCACACAGAGA 3' | 57°C | 407 bp |
| Exon 16 | 5'TGTCCTAAAGGATGCTGTCA 3′ | 5'GGAAAACAGGCCTGAAATTA 3' | 55°C | 451 bp |
| Primer forward for allele-specific PCR | Primer reverse for allele-specific PCR | |||
| Exon 10 | 5′CAGCTGGCCTAGAACCTGACCC 3′ | 5′GAAACTTTCCCCCAGGGACCACCT 3′ 5′ACTTTCCCCCAGGGACCACCC 3′ |
63°C | 141 bp |
| Beta actin | 5'GTCACATCCAGGGTCCTCAC 3' | 5′CACCTTCACCGTTCCAGTTT 3' | 65°C | 350 bp |
The PCR products that were free of contaminating bands due to nonspecific amplification were column-purified using Nucleo Spin Extract II (Macherey-Nagel). Sequencing PCR reaction was performed in a total volume of 20 μL containing 2 μL Terminator Ready Reaction mix (Applied Biosystems), 3.2 pmol primers, 3 μL Dilution Buffer, 6 ng purified PCR product.
Bidirectional sequencing of exons 2, 5, 8, 10, 14, and 16 of the ATP7B gene was performed using an ABI prism 310 DNA analyzer (Applied Biosystems) with dye-termination chemistry.
Nucleotide changes were detected by comparing the sequence obtained in the chromatogram with the normal gene sequence [NG_008806.1; Homo sapiens ATPase, Cu++ transporting, beta polypeptide (ATP7B) on chromosome 13] using SeqScape software version 2.5 (Applied Biosystems).
PCR-restriction fragment length polymorphism (RFLP) assay was applied for detection of c.1216 T>G (rs1801243) ATP7B SNP in AD and healthy controls (Figure 1). PCR- RFLP reaction was the same as the one reported above using specific oligonucleotide primers (Table 1). The T>G transition at the exon 2 creates an MspA1I (Promega) restriction-endonuclease recognition site. The 584 bp PCR product was digested with MspA1I only if the substitution was present. MspA1I reactions were performed at 37°C for 2 h 30 min. All restriction products were analyzed on a 1.5% agarose gel by electrophoresis and visualized by staining the gel using ethidium bromide. Homozygous alleles of the TT genotype appeared as a 584 bp DNA band on the gel, and homozygous alleles of the GG genotype appeared as a 375 bp and an 209 bp DNA band. Heterozygote alleles displayed a combination of the bands (584 bp, 375 bp and 209 bp). Direct DNA bidirectional sequencing was performed for 15% of the PCR products, which were randomly selected and analyzed to confirm the genotypes.
Figure 1.
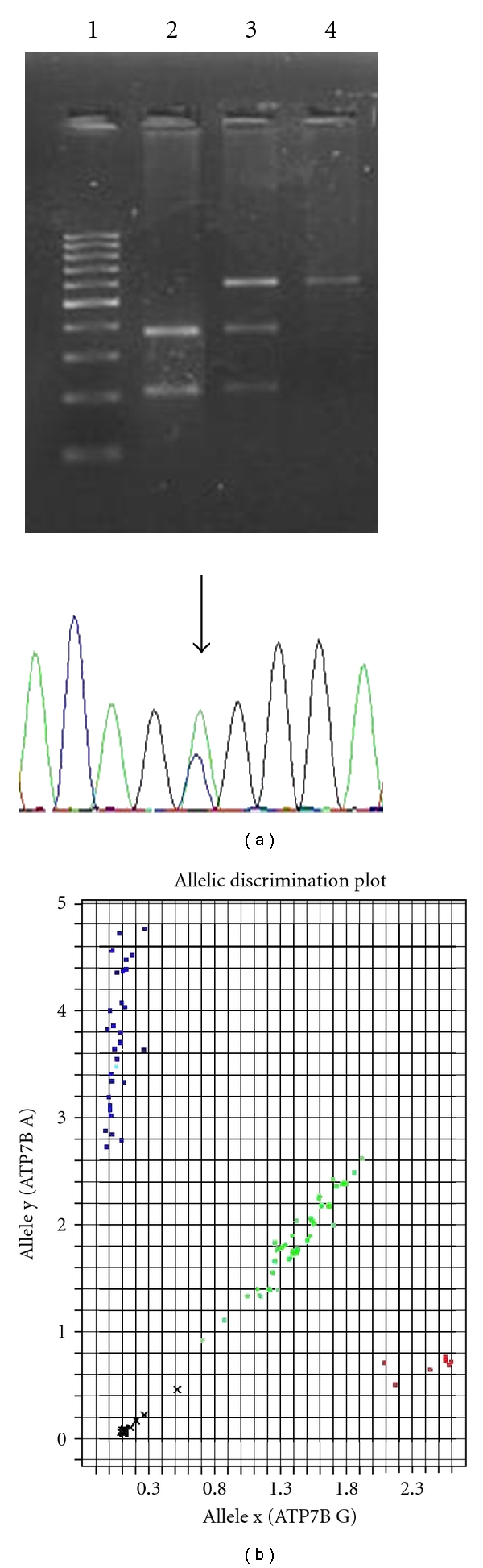
Analytical procedures for ATP7B SNPs detection. (a) PCR-restriction fragment length polymorphism (RFLP) assay for detection of c.1216 T>G SNP. Lane 1 : PCR 100 bp low ladder. Lane 2 : GG genotype (375 bp and 209 bp RFLP). Lane 3 : TG genotype (584 bp, 375 bp and 209 bp RFLP). Lane 4 : TT genotype (584 bp RFLP). In the electropherogram the arrow indicates the TG genotype. (b) TaqMan allelic discrimination assays for detection of c.2495 A>G SNP. Blue: AA genotype. Green: TG genotype. Red: GG genotype; x: undetermined.
Detection of c.2495 A>G (rs1061472) ATP7B polymorphism was performed by direct bidirectional sequencing of exon 10.
Bidirectional sequencing of exon 10 was performed using an ABI prism 310 DNA analyzer (Applied Biosystem, Foster City CA) with dye-termination chemistry. PCR reaction was the same as the one reported above. Oligonucleotides are detailed in Table 1.
Genotyping of SNPs rs1061472 was achieved by the TaqMan allelic discrimination assays from Applied Biosystems Inc. (Foster City CA) (Figure 1). The predesigned SNPs genotyping assay ID is ID C_1919004_30 (Applied Biosystems). The total reaction volume per well was 20 μL, including 5ng genomic DNA, 1 μL TaqMan SNP genotyping assay (containing two PCR primers and two dye (VIC or FAM)-labelled TaqMan MGB probes) and 10 μL TaqMan Universal PCR Master Mix (Applied Biosystems), according to the manufacturer's manual.
PCR was performed at 95°C for 10 min and 40 cycles at 95°C for 15 s and 60°C for 1 min. The samples were amplified, read, and analyzed using the ABI Prism 7900HT Sequence Detection System and ABI Prism SDS 2.4 software. Two blank controls in each 96-well plate were used for the assay quality control.
Apolipoprotein E (APOE) genotyping was performed according to established methods [46].
2.2. Statistical Analyses
Demographic and clinical characteristics in our patient and control samples were described either in terms of mean ± SD if quantitative, or in terms of proportions.
To calculate the power analysis of our study we considered data reported in general population (CEPH) [47, 48] of SNPs allele distribution. As the presence of TG heterozygosis in healthy individuals was about 40% for c.1216 SNP and that of AG heterozygosis was 52% for c.2945 SNP [47, 48], we estimated that, with our sample size, the power was 80% to recognize as significant (at bilateral alpha level of 0.05) a higher prevalence in AD with respect to controls of 12% (or more) for c.1216 SNP and of 11% for c.2945 SNP.
After checking for normality, Student's t-tests were used when appropriate to evaluate differences in quantitative variables. The differences in the overall distribution of the alleles among normal and AD chromosomes were evaluated by χ2 test. The association of the allele with the largest positive deviation between the observed and the expected frequency under null hypothesis was represented with a 2 × 2 table and tested by means of χ2 test. The relative risk of having AD was estimated by Odds Ratios (ORs), and corresponding 95% CIs were also provided. Two-sided χ2 tests were used to verify Hardy-Weinberg equilibrium. Logistic regression analysis with group (cases and controls) as dependent variable and genetic and demographic measures as independent variables allowed identifying the characteristics more able to discriminate the two groups.
Coefficient pairwise Linkage Disequilibrium (LD; D') between ATP 7B SNPs was estimated using Haploview version 4.2 [49].
All analyses were conducted with SPSS software version 16.0 (SPSS Ltd., Surrey, UK). A P value less than .05 was considered significant in all statistical analysis.
3. Results
Main demographic and clinical characteristics of the subjects participating to this study were reported in Table 2. AD patients and controls did not differ for sex, but differed in age, mean MMSE score, and APOE ε4 allele frequency (Table 2). As the age effect was considered a potentially confounding factor, it was taken into account in the statistical analyses. As expected, the mean MMSE score was lower in patients than in controls. Education did not differ between the 2 groups, while the presence of at least one APOE ε4 allele was more frequent in patients than in controls (OR = 3.7; 95% CI = 2.1–6.5; P < .001).
Table 2.
Demographic characteristics of the investigated groups. Data are mean (SD).
| AD patients | Controls | |
|---|---|---|
| Number of subjects | 190 | 164 |
| Sex F (%) | 70 | 68 |
| Education mean (SD) | 9 (5.0) | 9.3 (4.6) |
| Age (years) mean (SD) | 74.4 (7.9)a | 67.7 (11.2) |
| MMSE mean (SD) | 18.6 (5.8)a | 28.3 (1.2) |
| APOE ε4 frequency(%) | 35a | 13 |
*Correlation is significant at the 0.05 level (2-tailed).
aSignificantly different between AD and control group (P < .001).
bSignificantly different between AD and control group (P < .05).
The genetic screening for WD mutations in the sole AD cohort by direct sequencing was restricted to exons 2, 5, 8, 14, and 16 of the ATP7B gene in 180 chromosomes, while it was carried out in 360 AD chromosomes and 302 control chromosomes for exon 10. The study revealed no mutations, but sequence changes corresponding to the c.1216 T>G (Ser406Ala) in exon 2 and 2495 A>G (Lys832Arg) in exon 10 SNPs occurred.
The Hardy-Weinberg equilibrium was checked in each group. No statistically significant differences were found.
3.1. C.1216 T>G SNP (Exon 2) in AD and Healthy Controls
Genotype frequencies of c.1216 T>G SNP in our control panel were as follows: TT 30.9%, TG 49.6%, and GG 19.5%. In AD patients they were not different, being TT 24.1%, TG 50.6%, and GG 25.3% (χ2 = 2.25, P = .325). Also allele frequency did not differ between groups (Table 3). When we merged data of TG and TT genotype carriers together and compared their pooled frequency versus that of GG genotype in a model of logistic regression analysis taking into account the age effect, we observed a higher frequency of GG in patients than in controls, although the difference was only marginally significant (OR = 1.773; 95% CI = 0.947–3.320; P = .074; Table 3).
Table 3.
c.1216 T>G ATP7B (exon 2) SNP allele distribution in AD patients and healthy controls and comparison of GG versus TG + TT.
| c.1216 T>G ATP7B (exon 2) | AD patients (162) | Controls (123) | P value |
|---|---|---|---|
| Allele T frequency n (%) | 160 (49%) | 137 (55.7%) |
χ2 = 2.23; df = 1;
P = .13 |
| Allele G frequency n (%) | 164 (51%) | 109 (44.3%) | |
| GG n (%) | 41 (25.3%) | 24 (19.5%) | P = .074* |
| TG + TT n (%) | 121 (74.7%) | 99 (80.5%) | |
Correlation is significant at the .05 level (2-tailed). *The analyses were corrected for the age effect.
3.2. C.2495 A>G SNP (Exon 10) in AD and Healthy Controls
c.2495 A>G genotype frequencies in our control panel were as follows: AA 14.6%, AG 56.3%, and GG 29.1%. In AD patients they were not different, being AA 9.4%, AG 57.1%, and GG 38.9%. The overall χ2 indicated that the 2 distributions were not clearly different (χ2 = 4.42, P = .110), although the linear component (considering the number of G alleles: 0,1,2) suggested there was an association (χ2 = 4.441, P = .036). G allele frequency was higher in AD than in controls (χ2 = 3.8, P = .05). Furthermore, when we merged data of AG and GG genotype carriers together and compared their pooled frequency versus that of GG genotype, in a model of logistic regression, GG category was significantly more frequent in AD patients than in controls (Table 4). In particular, GG genotype was carried by 39% of AD patients versus 29% of healthy controls and resulted in a significant odds ratio (OR = 1.741; 95% CI = 1.060–2.858; P = .028).
Table 4.
c.2495 A>G ATP7B (exon 10) SNP allele distribution in AD patients and healthy controls and comparison between GG versus AG + AA.
| c.2495 A>G ATP7B (exon 10) SNP | AD patients (180) | Controls (151) | P value |
|---|---|---|---|
| Allele A frequency n (%) | 127 (35%) | 129 (43%) |
χ2 = 3.8; df = 1;
P = .05 |
| Allele G frequency n (%) | 233 (65%) | 173 (57%) | |
| GG n (%) | 70 (39%) | 44 (29%) | P = .028* |
| AG + AA n (%) | 110 (61%) | 107 (71%) | |
Correlation is significant at the .05 level (2-tailed). *The analyses were corrected for the age effect.
Allele frequency of the 2 SNPs in our cohorts (Tables 3 and 4) resembles those reported in HapMap for European origin populations.
To verify whether c.1216 T>G and c.2495 A>G SNPs were in linkage disequilibrium (LD), we constructed plots for our 2 cohorts and compared them with those reported in HapMap database (http://www.hapmap.org/) for European origin population (Figure 1). The analysis revealed that the 2 ATP7B SNPs were not in high LD in our population, either when analysing the subjects' sample separately as an AD (D′ = 0.74; D' confidence bounds-Conf. Bounds-0.60–0.84 ) and a control (D′ value = 0.64; Conf. Bounds 0.5–0.77) cohort or when considering the subjects as a combined population (D′ value = 0.70; Conf. Bounds 0.59–0.79). The degree of LD calculated for the Italian Tuscan population (TSI; D′ = 90; Conf. Bounds 0.77–0.97) and for the Utah residents with Northern and Western European ancestry from CEPH collection (CEU; D′ = 93; 0.85–0.98), on the basis of data reported in HapMap database, was higher than that we identified (Figure 2).
Figure 2.

Pairwise LDs are shown, calculated between the ATP7B gene SNPs in AD, controls, whole sample and in HapMap European origin populations (CEU: Utah residents with Northern and Western European ancestry from the CEPH collection; TSI: Tuscans in Italy). The top panel depicts the location of the SNPs in the AT7PB gene. The intensity of the box shading is proportional to the strength of the LD (D′ confidence bounds) for the marker pair, which is also indicated as a percentage within each box.
APOE ε4 and ATP7B (both c.1216 T>G and c.2495 A>G) SNPs were independent AD risk factors, since there was no difference in the frequency of the ATP7B SNPs between carriers and noncarriers of the APOE ε4 allele (consistently P > .2), in addition to when the analysis was restricted to assessment of only the AD population (consistently P > .2).
4. Discussion
We have focused the current investigation on ATP7B WD gene, which is a tight control balance regulator for free copper levels in the body [42, 43, 50]. ATPase 7B protein is expressed at high levels in the liver and kidney and at lower levels in the lung, placenta, and brain [50]. It is localized to the trans-Golgi membrane where it maintains intracellular copper concentration by transporting copper from the cytosol across the Golgi lumen. In the Golgi lumen, ATPase 7B mediates the incorporation of copper atoms into ceruloplasmin during its biosynthesis [51–53]. Under elevated copper concentrations ATPase 7B undergoes a reversible, copper-mediated translocation from the trans-Golgi to the apical canalicular membrane where it pumps copper directly into the bile [51–56]. In WD, defects of ATPase 7B prevents copper translocation to the secretory pathway as well as the excretion trough the bile, resulting in free copper increased levels and in the secretion of apoceruloplasmin which, being unstable, is rapidly degraded in the blood [54, 57]. In a dedicated study, we have shown that a conspicuous amount of apoceruloplasmin is present in the CSF of AD patients [58]. We have also reported fragmentation of ceruloplasmin, revealed by the presence of low-molecular-weight fragments (<50 KDa) of ceruloplasmin in AD samples from selected patients with higher-than-normal levels of free copper [59].
The most common Mediterranean WD mutations were reported to lie primary in exons 2, 5, 8, 10, 14, and 16 [47, 48, 60–68]. In particular, the Cys271Stop mutation in exon 2 was reported to account for 19% of the total mutations in the European and Turkish population [61]. 1708-1 G>C, 1785 delT, and 1823 del3 have been identified in the Italian population (exon 5) [62–65]. The 2299insC mutation in exon 8 was found in Continental Italians [63]. In the same exon lies the Arg 778 Leu mutation, which is the prevalent mutation of the Mongoloid population [60]. The 2464delC in exon 10 was found in Sardinian and the 2533delA in Sardinian, Continental Italian, Turkish, and Albanian populations [62]. His1069Gln in exon 14, which was reported to account for 17.5% of WD mutations in Mediterraneans, was also found in 20–40% of the WD cases in different Caucasian population groups [47, 63, 65, 66, 68]. Val1146Met and Ile1148Thr in exon 16 have been identified in Greek population [62–65]. Along with these relatively common mutations, several other very rare mutations in Mediterraneans have been described in the exons object of our pilot investigation [62–65].
The ATP7B gene sequence analysis of exons 2, 5, 8, 14, 16 in 180 AD chromosomes—and 662 chromosomes only for exon 10—did not reveal any other sequence change than c.1216 T>G and c.2495 A>G ATP7B SNPs. As a result we focused our study on these 2 SNPs.
Our main observation is that c.2495 A>G ATP7B SNP as either the G allele frequency or the rate of distribution of the GG genotype is higher in AD patients than in healthy controls. The c.1216 T>G SNP was also differently distributed between AD and healthy controls but the significance did not reach the statistical threshold, probably because of the small size of the patient's sample analyzed. However, it has to be noted that the potential role as AD risk factor of the considered ATP7B SNPs could have been masked by the difference of 7 years between our AD patients and controls. In the attempt to reduce this potential confounder we took into account an age effect in our statistical analyses. However, the possibility that some controls might convert to AD while they age another 7 years makes controls and cases more close to each other, and thus our estimate of the statistical association between AD and these SNPs should be considered conservative.
Genotypes and allele distributions for both SNPs found in our panel were coherent with those reported in HapMap for general populations of European origin [47, 48]. While c.1216 T>G and c.2495 A>G SNPs are in LD in TSI and in CEU samples, in our panel they resulted in a lower LD degree.
Exon 2 encodes for a region containing metal-binding domains in ATPase 7B protein. This region encompasses amino acids 1-481. The genetic change in c.1216 T>G SNP corresponds to a substitution ser 406 ala, within this region. Exon 10 encodes for a region within the ATP binding domain region in the protein which encompasses amino acids 820–967. The genetic variation in c.2495 A>G corresponds to a lys 832 arg substitution [43] within this region. Thus it could be argued that these amino acids changes can have an a disturbing effect on ATPase7B function in terms of metal binding properties or ATP hydrolysis which can eventually result in copper homeostasis abnormalities.
This study has a number of limitations which include the small size of the sample, 7 years between cases and controls, the restriction of the sequence analysis to a limited number of ATP7B exons, the need for AD selection with possible sampling bias, and surely it needs confirmation in a larger subject population (in progress). Despite these limitations, this pilot investigation opens new routes—genetic rather than biochemical—for the study of free copper deregulation in AD and strengthens the concept that properly tuning the redistribution of metals via metal complexing or ligand agents, as successfully tested for WD, may positively affect the natural history of AD, at least for the ATP7B c.2495 GG AD carriers [1, 28, 69].
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
This study was partially supported by the following grants: Italian Health Department: “Profilo Biologico e Genetico della Disfunzione dei Metalli nella Malattia di Alzheimer e nel “Mild Cognitive Impairment” [RF 2006 conv.58]; Programma Strategico 2007 conv. PS39; “Ricerca Corrente” grants IRCCS Brescia, Italy, ERA-Net NEURON (JTC 2008 nEUROsyn), AFaR Foundation-Osp. Fatebenefratelli “AAMS l gioco sicuro.”
References
- 1.Bush AI, Tanzi RE. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics. 2008;5(3):421–432. doi: 10.1016/j.nurt.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith MA, Harris PLR, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(18):9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnal N, Cristalli DO, de Alaniz MJT, Marra CA. Clinical utility of copper, ceruloplasmin, and metallothionein plasma determinations in human neurodegenerative patients and their first-degree relatives. Brain Research. 2010;1319:118–130. doi: 10.1016/j.brainres.2009.11.085. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal R, Kushwaha SS, Tripathi CB, Singh N, Chhillar N. Serum copper in Alzheimer’s disease and vascular dementia. Indian Journal of Clinical Biochemistry. 2008;23(4):369–374. doi: 10.1007/s12291-008-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bocca B, Forte G, Petrucci F, et al. Monitoring of chemical elements and oxidative damage in patients affected by Alzheimer’s disease. Annali dell’Istituto Superiore di Sanita. 2005;41(2):197–203. [PubMed] [Google Scholar]
- 6.González C, Martín T, Cacho J, et al. Serum zinc, copper, insulin and lipids in Alzheimer’s disease epsilon 4 apolipoprotein E allele carriers. European Journal of Clinical Investigation. 1999;29(7):637–642. doi: 10.1046/j.1365-2362.1999.00471.x. [DOI] [PubMed] [Google Scholar]
- 7.Sevym SUO, Tamer I, Doğu O, Ozge A. Can serum levels of copper and zinc distinguish Alzheimer’s patients from normal subjects? Journal of Neurological Sciences (Turkish) 2007;24(3):197–205. [Google Scholar]
- 8.Smorgon C, Mari E, Atti AR, et al. Trace elements and cognitive impairment: an elderly cohort study. Archives of Gerontology and Geriatrics. Supplement. 2004;(9):393–402. doi: 10.1016/j.archger.2004.04.050. [DOI] [PubMed] [Google Scholar]
- 9.Squitti R, Barbati G, Rossi L, et al. Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF β-amyloid, and h-tau. Neurology. 2006;67(1):76–82. doi: 10.1212/01.wnl.0000223343.82809.cf. [DOI] [PubMed] [Google Scholar]
- 10.Squitti R, Bressi F, Pasqualetti P, et al. Longitudinal prognostic value of serum “free” copper in patients with Alzheimer disease. Neurology. 2009;72(1):50–55. doi: 10.1212/01.wnl.0000338568.28960.3f. [DOI] [PubMed] [Google Scholar]
- 11.Squitti R, Pasqualetti P, Cassetta E, et al. Elevation of serum copper levels discriminates Alzheimer’s disease from vascular dementia. Neurology. 2003;60(12):2013–2014. doi: 10.1212/01.wnl.0000068013.27968.29. [DOI] [PubMed] [Google Scholar]
- 12.Squitti R, Ventriglia M, Barbati G, et al. ‘Free’ copper in serum of Alzheimer’s disease patients correlates with markers of liver function. Journal of Neural Transmission. 2007;114(12):1589–1594. doi: 10.1007/s00702-007-0777-6. [DOI] [PubMed] [Google Scholar]
- 13.Zappasodi F, Salustri C, Babiloni C, et al. An observational study on the influence of the APOE-ε4 allele on the correlation between ‘free’ copper toxicosis and EEG activity in Alzheimer disease. Brain Research. 2008;1215(C):183–189. doi: 10.1016/j.brainres.2008.03.066. [DOI] [PubMed] [Google Scholar]
- 14.Basun H, Forssell LG, Wetterberg L, Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal ageing and Alzheimer’s disease. Journal of Neural Transmission. 1991;3(4):231–258. [PubMed] [Google Scholar]
- 15.Baum L, Chan IHS, Cheung SKK, et al. Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. BioMetals. 2010;23(1):173–179. doi: 10.1007/s10534-009-9277-5. [DOI] [PubMed] [Google Scholar]
- 16.Gerhardsson L, Lundh T, Minthon L, Londos E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2008;25(6):508–515. doi: 10.1159/000129365. [DOI] [PubMed] [Google Scholar]
- 17.Jeandel C, Nicolas MB, Dubois F, Nabet-Belleville F, Penin F, Cuny G. Lipid peroxidation and free radical scavengers in Alzheimer’s disease. Gerontology. 1989;35(5-6):275–282. doi: 10.1159/000213037. [DOI] [PubMed] [Google Scholar]
- 18.Kapaki E, Segditsa J, Zournas C, Xenos D, Papageorgiou C. Determination of cerebrospinal fluid and serum lead levels in patients with amyotrophic lateral sclerosis and other neurological diseases. Experientia. 1989;45(11-12):1108–1110. doi: 10.1007/BF01950171. [DOI] [PubMed] [Google Scholar]
- 19.Mattiello G, Gerotto M, Favarato M, et al. Plasma microelement analysis from Alzheimer’s and multi-infartual dementia patients. Alzheimer’s Diseases: Advances in Clinical and Basic Research. 1993 [Google Scholar]
- 20.Molina JA, Jiménez-Jiménez FJ, Aguilar MV, et al. Cerebrospinal fluid levels of transition metals in patients with Alzheimer’s disease. Journal of Neural Transmission. 1998;105(4-5):479–488. doi: 10.1007/s007020050071. [DOI] [PubMed] [Google Scholar]
- 21.Ozcankaya R, Delibas N. Malondialdehyde, superoxide dismutase, melatonin, iron, copper, and zinc blood concentrations in patients with Alzheimer disease: cross-sectional study. Croatian Medical Journal. 2002;43(1):28–32. [PubMed] [Google Scholar]
- 22.Sedighi B, Shafa MA, Shariati M. A study of serum copper and ceruloplasmin in Alzheimer’s disease in Kerman, Iran. Neurology Asia. 2006;11:107–109. [Google Scholar]
- 23.Snaedal J, Kristinsson J, Gunnarsdóttir S, et al. Copper, ceruloplasmin and superoxide dismutase in patients with Alzheimer’s disease. A case-control study. Dementia and Geriatric Cognitive Disorders. 1998;9(5):239–242. doi: 10.1159/000017067. [DOI] [PubMed] [Google Scholar]
- 24.Vural H, Demirin H, Kara Y, Eren I, Delibas N. Alterations of plasma magnesium, copper, zinc, iron and selenium concentrations and some related erythrocyte antioxidant enzyme activities in patients with Alzheimer’s disease. Journal of Trace Elements in Medicine and Biology. 2010;24(3):169–173. doi: 10.1016/j.jtemb.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Brewer GJ, Kanzer SH, Zimmerman EA, et al. Copper and ceruloplasmin abnormalities in Alzheimer’s disease. American Journal of Alzheimer’s Disease and Other Dementias. 2010;25(6):490–497. doi: 10.1177/1533317510375083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bucossi S, Ventriglia M, Panetta V, et al. Copper in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. Journal of Alzheimer’s Disease. 2011;24(1):175–185. doi: 10.3233/JAD-2010-101473. [DOI] [PubMed] [Google Scholar]
- 27.Walshe JM. Wilson’s disease: the importance of measuring serum caeruloplasmin non-immunologically. Annals of Clinical Biochemistry. 2003;40, part 2:115–121. doi: 10.1258/000456303763046021. [DOI] [PubMed] [Google Scholar]
- 28.Squitti R, Salustri C. Agents complexing copper as a therapeutic strategy for the treatment of Alzheimer’s disease. Current Alzheimer Research. 2009;6(6):476–487. doi: 10.2174/156720509790147133. [DOI] [PubMed] [Google Scholar]
- 29.Althaus JS, Quinn JF, Kaye JA, et al. Free copper measured directly in serum using a novel device is elevated in Alzheimer’s disease. Investigative Ophthalmology & Visual Science. 2008;49, E-abstract:p. 5218. [Google Scholar]
- 30.Hoogenraad TU. Measuring hypercupremia in blood of patients with Alzheimer’s disease is logical, but the utility of measuring free-copper has to be proven. In: Frijns CJM, Kappelle LJ, Klijn CJM, Wokke JHJ, editors. Neurologie. Utrechts: Tijdschrift voor; 2007. pp. 111–112. [Google Scholar]
- 31.Linder MC, Houle PA, Isaacs E, et al. Copper regulation of ceruloplasmin in copper-deficient rats. Enzyme. 1979;24(1):23–35. doi: 10.1159/000458625. [DOI] [PubMed] [Google Scholar]
- 32.Chutkow JG. Evidence for uptake of nonceruloplasminic copper in the brain: effect of ionic copper and amino acids. Proceedings of the Society for Experimental Biology and Medicine. 1978;158(1):113–116. doi: 10.3181/00379727-158-40152. [DOI] [PubMed] [Google Scholar]
- 33.Hartter DE, Barnea A. Brain tissue accumulates copper by two ligand-dependent saturable processes. A high affinity, low capacity and a low affinity, high capacity process. Journal of Biological Chemistry. 1988;263(2):799–805. [PubMed] [Google Scholar]
- 34.Choi BS, Zheng W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Research. 2009;1248:14–21. doi: 10.1016/j.brainres.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoogenraad TU, Van Den Hamer CJA, Koevoet R, De Ruyter Korver EGW. Oral zinc in Wilson’s disease. Lancet. 1978;2(8102):p. 1262. doi: 10.1016/s0140-6736(78)92141-4. [DOI] [PubMed] [Google Scholar]
- 36.Scheinberg IH, Sternlieb I. Wilson’s disease. In: Smith LH, editor. Problems in Internal Medicine. Philadelphia, Pa, USA: Saunders; 1984. [Google Scholar]
- 37.Siotto M, Bucossi S, Squitti R. Copper status abnormalities and how to measure them in neurodegenerative disorders. Recent Patents in CNS Drug Discovery. 2010;5(3):182–194. doi: 10.2174/157488910793362395. [DOI] [PubMed] [Google Scholar]
- 38.Squitti R, Lupoi D, Pasqualetti P, et al. Elevation of serum copper levels in Alzheimer’s disease. Neurology. 2002;59(8):1153–1161. doi: 10.1212/wnl.59.8.1153. [DOI] [PubMed] [Google Scholar]
- 39.Babiloni C, Squitti R, Del Percio C, et al. Free copper and resting temporal EEG rhythms correlate across healthy, mild cognitive impairment, and Alzheimer’s disease subjects. Clinical Neurophysiology. 2007;118(6):1244–1260. doi: 10.1016/j.clinph.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 40.Squitti R, Pasqualetti P, Dal Forno G, et al. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology. 2005;64(6):1040–1046. doi: 10.1212/01.WNL.0000154531.79362.23. [DOI] [PubMed] [Google Scholar]
- 41.Folstein MF, Folstein SE, McHugh PR. “Mini mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 42.Petrukhin K, Fischer SG, Pirastu M, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nature Genetics. 1993;5(4):338–343. doi: 10.1038/ng1293-338. [DOI] [PubMed] [Google Scholar]
- 43.Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nature Genetics. 1993;5(4):344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 44.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurology. 2007;6(8):734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 45.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 46.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. Journal of Lipid Research. 1990;31(3):545–548. [PubMed] [Google Scholar]
- 47.Gupta A, Aikath D, Neogi R, et al. Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Human Genetics. 2005;118(1):49–57. doi: 10.1007/s00439-005-0007-y. [DOI] [PubMed] [Google Scholar]
- 48.Gupta A, Maulik M, Nasipuri P, et al. Molecular diagnosis of Wilson disease using prevalent mutations and informative single-nucleotide polymorphism markers. Clinical Chemistry. 2007;53(9):1601–1608. doi: 10.1373/clinchem.2007.086066. [DOI] [PubMed] [Google Scholar]
- 49.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 50.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nature Genetics. 1993;5(4):327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 51.DiDonato M, Narindrasorasak S, Forbes JR, Cox DW, Sarkar B. Expression, purification, and metal binding properties of the N-terminal domain from the Wilson disease putative copper-transporting ATPase (ATP7B) Journal of Biological Chemistry. 1997;272(52):33279–33282. doi: 10.1074/jbc.272.52.33279. [DOI] [PubMed] [Google Scholar]
- 52.DiDonato M, Sarkar B. Copper transport and its alterations in Menkes and Wilson diseases. Biochimica et Biophysica Acta. 1997;1360(1):3–16. doi: 10.1016/s0925-4439(96)00064-6. [DOI] [PubMed] [Google Scholar]
- 53.Didonato M, Zhang J, Que L, Jr., Sarkar B. Zinc binding to the NH-terminal domain of the Wilson disease copper-transporting ATPase. Implications for in vivo metal ion-mediated regulation of ATPase activity. Journal of Biological Chemistry. 2002;277(16):13409–13414. doi: 10.1074/jbc.M111649200. [DOI] [PubMed] [Google Scholar]
- 54.Terada K, Schilsky ML, Miura N, Sugiyama T. ATP7B (WND) protein. International Journal of Biochemistry and Cell Biology. 1998;30(10):1063–1067. doi: 10.1016/s1357-2725(98)00073-9. [DOI] [PubMed] [Google Scholar]
- 55.Roelofsen H, Wolters H, Van Luyn MJA, Miura N, Kuipers F, Vonk RJ. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology. 2000;119(3):782–793. doi: 10.1053/gast.2000.17834. [DOI] [PubMed] [Google Scholar]
- 56.Schaefer M, Gitlin JD. Genetic disorders of membrane transport IV. Wilson’s disease and Menkes disease. American Journal of Physiology. 1999;276(2, part 1):G311–G314. doi: 10.1152/ajpgi.1999.276.2.G311. [DOI] [PubMed] [Google Scholar]
- 57.Bielli P, Calabrese L. Structure to function relationships in ceruloplasmin: a ’moonlighting’ protein. Cellular and Molecular Life Sciences. 2002;59(9):1413–1427. doi: 10.1007/s00018-002-8519-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Capo CR, Arciello M, Squitti R, et al. Features of ceruloplasmin in the cerebrospinal fluid of Alzheimer’s disease patients. BioMetals. 2008;21(3):367–372. doi: 10.1007/s10534-007-9125-4. [DOI] [PubMed] [Google Scholar]
- 59.Squitti R, Quattrocchi CC, Salustri C, Rossini PM. Ceruloplasmin fragmentation is implicated in ’free’ copper deregulation of Alzheimer’s disease. Prion. 2008;2(1):23–27. doi: 10.4161/pri.2.1.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chuang LM, Wu HP, Jang MH, et al. High frequency of two mutations in codon 778 in exon 8 of the ATP7B gene in Taiwanese families with Wilson disease. Journal of Medical Genetics. 1996;33(6):521–523. doi: 10.1136/jmg.33.6.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curtis D, Durkie M, Balac P, et al. A study of Wilson disease mutations in Britain. Human Mutation. 1999;14(4):304–311. doi: 10.1002/(SICI)1098-1004(199910)14:4<304::AID-HUMU5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 62.Figus A, Angius A, Loudianos G, et al. Molecular pathology and haplotype analysis of Wilson disease in Mediterranean populations. American Journal of Human Genetics. 1995;57(6):1318–1324. [PMC free article] [PubMed] [Google Scholar]
- 63.Loudianos G, Dessi V, Lovicu M, et al. Mutation analysis in patients of Mediterranean descent with Wilson disease: identification of 19 novel mutations. Journal of Medical Genetics. 1999;36(11):833–836. [PMC free article] [PubMed] [Google Scholar]
- 64.Loudianos G, Dessì V, Lovicu M, et al. Haplotype and mutation analysis in Greek patients with Wilson disease. European Journal of Human Genetics. 1998;6(5):487–491. doi: 10.1038/sj.ejhg.5200219. [DOI] [PubMed] [Google Scholar]
- 65.Loudianos G, Dessì V, Lovicu M, et al. Further delineation of the molecular pathology of Wilson disease in the Mediterranean population. Human Mutation. 1998;12(2):89–94. doi: 10.1002/(SICI)1098-1004(1998)12:2<89::AID-HUMU3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 66.Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. American Journal of Human Genetics. 1997;61(2):317–328. doi: 10.1086/514864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nature Genetics. 1995;9(2):210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- 68.Thomas GR, Roberts EA, Walshe JN, Cox DW. Haplotypes and mutations in Wilson disease. American Journal of Human Genetics. 1995;56(6):1315–1319. [PMC free article] [PubMed] [Google Scholar]
- 69.Squitti R, Zito G. Anti-copper therapies in Alzheimer’s disease: new concepts. Recent Patents on CNS Drug Discovery. 2009;4(3):209–219. doi: 10.2174/157488909789104802. [DOI] [PubMed] [Google Scholar]