

Abstract
The electron paramagnetic resonance (EPR) spectrum of an intermediate freeze trapped during the steady state of the reaction catalyzed by the adenosylcobalamin (AdoCbl)-dependent enzyme, methylmalonyl-CoA mutase, has been studied. The EPR spectrum is that of a hybrid triplet spin system created as a result of strong electron–electron spin coupling between an organic radical and the low-spin Co2+ in cob(II)alamin. The spectrum was analyzed by simulation to obtain the zero-field splitting (ZFS) parameters and Euler angles relating the radical-to-cobalt interspin vector to the g axis system of the low-spin Co2+. Labeling of the substrate with 13C and 2H was used to probe the identity of the organic radical partner in the triplet spin system. The patterns of inhomogeneous broadening in the EPR signals produced by [2′-13C]methylmalonyl-CoA and [2-13C]methylmalonyl-CoA as well as line narrowing resulting from deuterium substitution in the substrate were consistent with those expected for a succinyl-CoA radical wherein the unpaired electron was centered on the carbon α to the free carboxyate group of the rearranged radical. The interspin distance and the Euler angles were used to position this product radical into the active site of the enzyme.
AdoCbl1-dependent enzymes catalyze chemically challenging rearrangement reactions in which a hydrogen atom and a variable group on vicinal carbons switch positions (1–3). Methylmalonyl-CoA mutase (MCM, EC 5.4.99.2) is the only known AdoCbl-dependent enzyme present in mammals. This enzyme catalyzes the 1,2 rearrangement of methyl-malonyl-CoA, a catabolite of odd-chain fatty acids and branched-chain amino acids, to the Krebs cycle intermediate, succinyl-CoA (4).
In all AdoCbl-dependent isomerases, the cobalamin cofactor functions as a radical initiator, and the ensuing isomerization reactions proceed through a series of radical intermediates (Figure 1). The first and common step, among all of the enzymes, is homolysis of the cobalt–carbon σ-bond of the cofactor to generate a pair of radicals, cob(II)alamin [which contains a low-spin Co2+ (S = ½)] and the 5′-deoxyadenosyl radical (S = ½), a high-energy, primary alkyl radical. The 5′-deoxyadenosyl radical abstracts a hydrogen atom from the substrate, producing a substrate radical and 5′-deoxyadenosine. The substrate radical rearranges to a product radical which abstracts a hydrogen atom from the 5′-methyl of 5′-deoxyadenosine. The catalytic cycle closes upon recombination of the cofactor-based radicals to regenerate AdoCbl. In this mechanism, the cob(II)alamin, once generated, is a spectator until the final sector of the catalytic cycle. In contrast, the 5′-deoxyadenosyl radical is more proactive, being generated twice in each cycle and serving as a “radical shuttle” transferring the organic radical center to the substrate and recapturing the radical state from the product radical prior to re-formation of AdoCbl.
Figure 1.

(A) Proposed catalytic cycle of MCM showing the radical intermediates. (B) Positions of 13C labels in the substrate and in the product. (C) Positions of 2H labels used in this study.
The mechanism described above requires pairs of radicals or “biradical intermediates”, in which one of the radicals is organically based and the other is metal-based. Such intermediates have been detected by EPR spectroscopy of several AdoCbl-dependent isomerases trapped in steady-state catalytic turnover (5–8). Effects of electron–electron spin coupling typically dominate the EPR spectra of these trapped intermediates (9). In MCM, broad and rather featureless EPR spectra having axial line shapes of g⊥ = 2.11 and g|| = 2.0 have been observed in the presence either of substrates or of substrate analogues (6, 10, 11). Hyperfine splitting resulting from coupling of the unpaired electrons with the 59Co (I = 7/2) nuclear spin is evident in the high-field region of the spectrum. The substantial g anisotropy and the hyperfine multiplicity in the spectrum identified low-spin Co2+ as one component of the triplet spin system. However, the identity of the organic radical component was not established.
MCM belongs to the “His-on/base-off” class of AdoCbl enzymes in which the conformation of AdoCbl in the active site differs significantly from that in solution as demonstrated both by EPR spectroscopy (12, 13) and by the crystal structures of these enzymes (14, 15). In these enzymes, the intramolecular base, dimethylbenzimidazole, which serves as the lower axial ligand to Co in AdoCbl in solution at physiological pH, is “off” and is replaced by a side-chain imidazole (His610 in the Propionibacterium shermanii MCM) donated by the protein (14).
In this study, specifically deuterated or 13C-labeled substrates have been used to investigate isotopic perturbations of the EPR spectrum of the steady-state intermediate in MCM and thereby to identify the organic radical component of the triplet spin system. Simulations were employed to analyze the low-spin Co2+–organic radical triplet EPR patterns. The parameters arising from the analysis have been used to determine the position of the organic radical intermediate in the molecular axis of the cob(II)alamin.
EXPERIMENTAL PROCEDURES
Materials
AdoCbl, malonic acid, CoA, D2O, and CD3I were purchased from Sigma. [CD3]Methylmalonic acid was purchased from MSD Isotopes. [2-13C]Diethylmalonate and 13CH3I were purchased from Aldrich. All other chemicals were reagent-grade commercial products and were used without further purification.
Synthesis of [2′-2H3]Methylmalonyl-CoA and Perdeuterated Methylmalonyl-CoA
(R,S)-[2′-2H3]Methylmalonyl-CoA was synthesized and characterized as previously described (16). Perdeuteration of methylmalonyl-CoA was accomplished by exchanging the labile proton at C2 of [2′-2H3]-methylmalonyl-CoA in D2O. Exchange took 3–4 min for completion and was confirmed by 1H NMR of the sample in which disappearance of the methine proton signal at 3.7 ppm was followed. The EPR sample was then prepared in deuterated buffer, to ensure that the substrate remained perdeuterated.
Synthesis of [2′-13C]Methylmalonyl-CoA and [2-13C]M-ethylmalonyl-CoA
Methylmalonate labeled with 13C at the C2 or C2′ position was synthesized using [2-13C]malonate and unlabeled CH3I or unlabeled malonate and 13CH3I, respectively, using a previously described procedure (16). The presence of 13C-labeled substrates was confirmed by NMR analysis. The corresponding CoA esters were synthesized enzymatically using malonyl-CoA synthetase as follows. The reaction mixture (10 mL) containing 100 mM potassium phosphate buffer (pH 7.0), 2 mM methylmalonate, 8 mM ATP, 10 mM MgCl2, 2 mM CoA, and 1.5 mg of malonyl-CoA synthetase was incubated at room temperature for 4 h. The reaction was quenched with 0.1% trifluoroacetic acid, and the precipitated protein was removed by centrifugation. From the HPLC analysis of the supernatant, >90% of CoA was converted to methylmalonyl-CoA under these conditions. Methylmalonyl-CoA was purified by HPLC on a semipreparative C18 reverse phase column (Luna, 250 mm × 10 mm) and eluted with a gradient ranging from 0 to 30% acetonitrile in 0.1% trifluoroacetic acid over the course of 50 min at a flow rate of 5 mL/min. Fractions containing methylmalonyl-CoA were pooled, lyophilized, and stored at −80 °C.
Purification of Malonyl-CoA Synthetase
Recombinant malonyl-CoA synthetase was purified using pKW2 (generously provided by C. Khosla, Stanford University, Stanford, CA) expressed in Escherichia coli BL21 DE3 cells (17). Cells were cultured at 30 °C to an OD600 of ~0.8, then induced with 0.5 mM IPTG, and grown at 25 °C for 16 h before being harvested. The recombinant malonyl-CoA synthetase contains His6 tags at the N- and C-termini and was purified by Ni–NTA (Novagen) affinity chromatography on a 0.5 cm × 10 cm column. The column was washed with 10 mM imidazole in 50 mM potassium phosphate buffer (pH 7.0) containing 0.3 M NaCl. The enzyme was eluted with a linear gradient from 10 to 300 mM imidazole in 50 mM potassium phosphate adjusted to pH 7.0. Malonyl-CoA synthetase eluted at ~150 mM imidazole. The enzyme activity was measured as described previously (17). The specific activity of the purified enzyme was 0.35 IU at 30 °C.
Purification of Methylmalonyl-CoA Mutase and the Holoenzyme Preparation
Recombinant P. shermanii MCM was expressed using pMEX2/pGP1-2 (provided by P. Leadlay, Cambridge University, Cambridge, U.K.) in E. coli and was purified as described previously. The specific activity of the enzyme used in these studies was ~30 IU at 37 °C in the radiochemical assay (10). The holoenzyme was reconstituted by mixing ~10 mg of protein (~66 nmol) with a 4-fold molar excess of AdoCbl in 1 mL of 50 mM potassium phosphate (pH 7.5). The solution was kept on ice for ~30 min. Unbound AdoCbl was removed by repeated cycles of ultrafiltration in a Centricon-30 microconcentrator (Amicon) followed by dilution with 50 mM potassium phosphate (pH 7.5) until the filtrate was colorless (10).
EPR Spectroscopy
EPR spectra were recorded on a Bruker ESP 300E spectrometer equipped with an Oxford ITC4 temperature controller, a model 5340 automatic frequency counter from Hewlett-Packard, and an NMR gaussmeter. The specific conditions for spectral accumulation are provided in the figure legends. The samples were prepared as described in the figure legends.
Spectral Simulations
Fourier filtering methods were used to enhance the resolution of the EPR spectrum obtained with unlabeled methylmalonyl-CoA (18, 19). The resulting spectrum was then analyzed using the following spin Hamiltonian:
| (1) |
The first two terms in eq 1 represent the Zeeman interaction of the low-spin Co2+ and the succinyl-CoA radical with the external magnetic field, respectively. The third term is the isotropic exchange interaction. The fourth term is the electron spin–electron spin dipolar (ZFS) interaction. Hnuc represents the nuclear hyperfine interactions at the cobalt and organic radical centers:
| (2) |
In the case of the Co2+-succinyl–CoA triplet, only the 59Co nuclear spin of cob(II)alamin (I = 7/2) was included in the calculation. For the calculation of the cob(II)alamin spectrum (S = ½), only the first and last terms in eq 1 are necessary, with an additional nuclear spin, the 14N nuclear spin of the His610 lower axial ligand (I = 1), being included in Hnuc. In general, the principal axes of each tensoral quantity in eqs 1 and 2 need not be collinear. Euler rotations, using the convention given in ref 20, were therefore used to express each tensor in a common frame of reference, in this case the g-axis system of Co2+. The Euler angles are variables in the fitting procedure. Strategies for the diagonalization of the energy matrix and simulation of field-swept powder EPR spectra were described previously (21). In this analysis, the 59Co hyperfine interaction is treated to first-order, which effectively renders the energy matrix block-diagonal in mI. In strongly coupled triplets, hyperfine splitting measured from the spectra are half of their values in spectra of corresponding doublet states (22). This contraction of hyperfine splittings occurs naturally in the energy calculations, and the reported hyperfine splitting parameters relate to doublet values. Initial estimates of the ZFS parameters were taken from the measured splittings in the experimental spectra (22). These parameters were refined by trial and error until a reasonable fit was obtained. The parameters were then refined using a simulated annealing algorithm (23), wherein the objective function compared peak positions in experimental and simulated EPR spectra.
Analysis of the ZFS Tensor
The ZFS tensor obtained from simulation of the spectrum was analyzed in terms of the point-dipole approximation to obtain the distance and orientation of the succinyl-CoA radical with respect to cob-(II)alamin. The spin densities of the paramagnetic intermediates were assumed to be localized at the C3 nucleus of a succinyl-CoA radical and the Co2+ nucleus of cob(II)alamin.
In the point-dipole approximation, the interspin distance, r, is related to the axial ZFS parameter D by eq 3 (24):
| (3) |
The interspin vector can then be taken as the z-axis of the principal axis system of the ZFS tensor, and the Euler angle ζ corresponds to the polar angle of the interspin vector with respect to the normal to the plane of the corrin ring.
The structure of the succinyl-CoA radical was modeled into the active site of MCM, starting from the X-ray coordinates of succinyl-CoA bound to the enzyme, and replacing the substrate portion of succinyl-CoA with the geometry-optimized structure of a truncated version of the succinyl-CoA radical. In this truncated version, the sulfur and a methyl group approximated coenzyme A. The structure was optimized at the B3LYP/6–311+G(d,p) level using Gaussian98. The final geometry was then obtained by using a simulated annealing algorithm (23) to determine which torsional angles in the geometry-optimized portion of the radical best reproduced the interspin distance and Euler angle obtained from analysis of the EPR powder pattern.
RESULTS AND DISCUSSION
Analysis of the EPR Pattern
The experimental EPR spectrum (Figure 2, solid line) of the steady-state intermediate possesses the hallmarks of a hybrid triplet spin system in which one-half of the triplet is the inorganic ion, low-spin Co2+, and the other half is an organic radical. In particular, the apparent g values in the spectrum and the magnitude of the 59Co hyperfine splitting are similar to those found for other cob(II)alamin–organic radical triplets in AdoCbl-dependent enzymes (9). The appearance of the EPR spectra in these hybrid triplet systems is dependent on the separation between Co2+ and the organic radical and on the position of the organic radical in the g-axis system of Co2+ (9, 25, 26). The radical rearrangement that is outlined in Figure 1 includes the primary alkyl radicals, the 5′-deoxyadenosyl radical and the methylmalonyl-CoA radical, and a secondary alkyl radical, the succinyl-CoA radical. It is improbable that these distinct radicals would be equidistant from Co2+ and have the same positions in the g-axis system of Co2+. Hence, if the radicals were present at comparable concentrations, one would expect to find a superposition of spectra corresponding to the Co2+–methylmalonyl-CoA and Co2+–succinyl-CoA triplet species. The EPR spectrum is sufficiently complicated to require simulation to determine whether all of the transitions arise from a single pair of interacting spins having a unique geometry.
Figure 2.

Experimental (solid line) and calculated (dashed line) spectra of the steady-state intermediate in MCM. The samples were prepared by mixing an anaerobic solution of the holoenzyme (final concentration of 200 μM) in 50 mM potassium phosphate buffer (pH 7.5) with (R, S)-methylmalonyl-CoA (final concentration of 20–30 mM) prepared in the same buffer and rapidly freezing the mixture in liquid nitrogen. The spectra were recorded at 25 K, with a modulation amplitude of 10 G, a microwave power of 5 mW, and a microwave frequency of 9.437 GHz. The experimental spectrum was collected at a high S/N and was subjected to resolution enhancement. Parameters used in the calculated spectrum: radical g value, 2.00; Co2+ g tensor, 2.30, 2.21, 2.00; ZFS parameters, D = −130 G, E = −37 G; exchange coupling constant, J = 4 × 103 G; 59Co hyperfine tensor (G), 10, 6, and 105. Euler angles relating the interspin vector to the Co2+ g-axis: ζ = 50°, η = 33°, and ξ = 116°. These parameters provide a reasonable match of calculated and observed transition frequencies. A uniform line width of 7 G was used in the calculation. Uncertainty in D is estimated to be ±10%, and uncertainty in the Euler angle, ζ, is estimated to be ±2°. The calculated spectrum was somewhat less sensitive to J, resulting in a greater uncertainty (estimated to be 30%). The parameters are, however, correlated such that changes in one can be compensated in part by changes in others. The parameters relating to the Co2+ half of the triplet were obtained independently from the spectrum of cob(II)alamin bound to MCM (see the legend of Figure 3).
The overall appearance of the spectrum suggests that the radical and Co2+ are strongly exchange coupled such that the g values are an average of those for low-spin Co2+ and a carbon-centered radical (9). The spectrum was simulated (Figure 2, dashed line) by a combination of manual trial and error and automated fitting using simulated annealing, global minimization methods. EPR parameters for the low-spin Co2+ component of the triplet were obtained from simulation of the EPR spectrum of cob(II)alamin-bound MCM (Figure 3). Parameters used for the calculated spectra are provided in the legends. The match in positions of transitions in the experimental and calculated spectra indicates that the model of a single Co2+–radical pair interacting at fixed positions is adequate to explain the data. The number (>8) and location of 59Co hyperfine transitions that appear in the resolution-enhanced spectrum indicate that the interspin vector does not lie along a principle direction in the g-axis of Co2+ (26). The Euler angle (ζ = 50°) accounts for the position and multiplicity of the 59Co hyperfine transitions. An interspin distance of 6.0 Å results from application of eq 3 and the ZFS parameter, D.
Figure 3.

Experimental (solid line) and calculated (dashed line) spectra for cob(II)alamin bound to MCM. The experimental spectrum was taken from Figure 2 (top) in ref 12. The calculated spectrum included the following parameters: Co2+ g tensor, 2.30, 2.21, 2.00; 59Co hyperfine tensor (G), 10, 6, 105; axial 14N superhyperfine splitting constant, 20 G. An isotropic line width of 7 G was used. Errors in the hyperfine constants are estimated to be on the order of ±10% or less. Uncertainties in the elements of the g tensor are estimated to be ±0.01.
Identification of the Organic Radical
The pattern of line width effects from isotopic substitutions within the substrate provides a means of identifying the organic radical component of the triplet spin system provided that the radical resides on the carbon skeleton of the substrate. EPR spectra of the samples made up with deuterated methylmalonyl-CoA and unlabeled substrate are compared in Figure 4. Substitution of 2H for 1H is expected to decrease the line widths of the EPR signals if unresolved hyperfine splitting from the 1H contributes to inhomogeneous broadening of the signals. A decrease in the magnitude of hyperfine splitting is expected from the smaller magnetogyric ratio of 2H relative to 1H ( ). EPR signals from samples made up with [2,2′-2H4]methylmalonyl-CoA (Figure 4B) and with [2′-2H3]-methylmalonyl-CoA (Figure 4C) exhibit narrowing as a result of the isotopic substitution. As illustrated in Figure 1, protons on the 2′-methyl, which are α-protons in the methylmalonyl-CoA radical, become the β-methylene protons in the succinyl-CoA product radical. Protons in the α and β positions relative to the radical center can give rise to substantial hyperfine splitting (22). Likewise, the C2 proton in the methylmalonyl-CoA goes from a β position in the methyl-malonyl-CoA radical to an α position in the product succinyl-CoA radical. Hence, observations of narrowing of the EPR signals from both of the samples labeled with 2H, and the slightly sharper signals for the perdeuterated sample, confirm that the organic radical resides on the carbon skeleton of methylmalonyl-CoA. However, the results from the 2H labeling do not distinguish between substrate and product radical species.
Figure 4.

EPR spectra of the steady-state intermediate in MCM starting from the unlabeled substrate (A) and starting with 2H substitution in the substrate (B and C). The samples were prepared as described in the legend of Figure 2. (A) Sample made up with the unlabeled substrate. (B) Sample made up with [2,2′-2H4]-methylmalonyl-CoA. (C) Sample made up with [2′-2H3]methyl-malonyl-CoA. The spectra were subjected to resolution enhancement using the same parameters.
EPR spectra of samples made up with [2′-13C]methyl-malonyl-CoA and with [2-13C]methylmalonyl-CoA are compared with the spectrum of the unlabeled sample in Figure 5. In these spectra, one expects to observe splitting or inhomogeneous broadening from 13C (I = ½) that is largest for the carbon where the radical is centered (22). The hyperfine interaction with 13C at the radical center is also anisotropic (27). Carbons (13C) adjacent to the radical center are expected to give smaller hyperfine splitting that is isotropic in character (28, 29). The data depicted in Figure 5 show that the greatest broadening comes from the sample made up with [2-13C]methylmalonyl-CoA. As shown in Figure 1, this carbon is at the radical center, C3, in the product, the succinyl-CoA radical. Hence, results from the isotopic labeling experiments point to the succinyl-CoA radical as the organic partner in the Co2+–organic radical hybrid triplet system. It is expected that this radical would have greater stability than the substrate radical because of the potential for delocalization of spin onto the π system of the adjacent carboxylate group (30).
Figure 5.

EPR spectra of the steady-state intermediate made up from the unlabeled (A) and 13C-labeled (B and C) substrate prepared as described in the legend of Figure 2. The position of 13C labeling in the original substrate is shown. The spectra were subjected to resolution enhancement using the same parameters.
Position of the Succinyl-CoA Radical in the Active Site
Coordinates of the equilibrium mixture of the substrate and product with MCM provide a logical starting point for positioning the succinyl-CoA radical into the active site (31). The succinyl moiety of the succinyl-CoA was replaced with the geometry-optimized succinyl-CoA radical, and the position and conformation of the molecule were adjusted to accommodate the interspin distance and Euler angle, ζ, derived from the analysis described here. The results of this procedure are shown in the stereoview in Figure 6. The EPR-derived distance and angle are accommodated by the X-ray structural model without any gross changes in the positions of groups within the active site.
Figure 6.

Stereoview of the succinyl-CoA radical relative to cob-(II)alamin and 5′-deoxyadenosine in the active site of MCM. X-ray coordinates from Protein Data Bank entry 4REQ were used to position atoms other than those from the succinyl radical moiety. The distance from Co2+ to C3 of the succinyl-CoA radical is 6.0 Å. The distance between C5′ of 5′-deoxyadenosine and C3 of the succinyl-CoA radical is 3.5 Å.
Simulation of the triplet-state EPR spectrum provided Euler angles, ζ and η, that relate the interspin vector to the g-axis system of low-spin Co2+ in cob(II)alamin. The EPR spectrum of cob(II)alamin bound to MCM (Figure 3) exhibits an in-plane anisotropy (i.e., gx ≠ gy). The X-ray-derived structure provides the position of the substrate binding site with respect to the asymmetric corrin ring (31). This crystallographic and spectroscopic information allows positioning of gx and gy in the plane of the corrin macrocycle. This result is illustrated in Figure 7.
Figure 7.
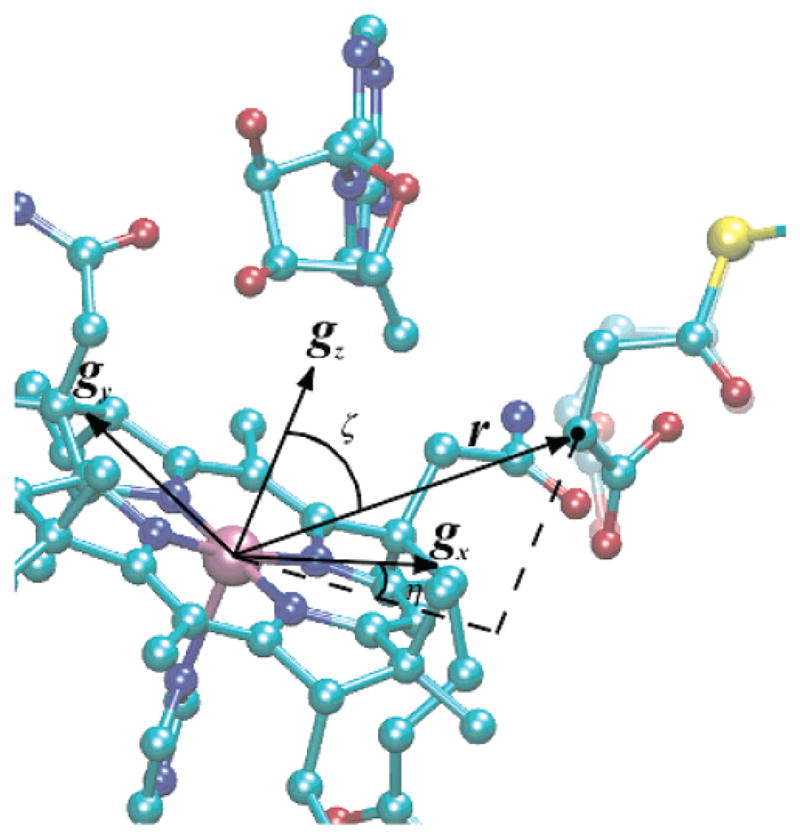
Model of the active site of MCM showing the position of the succinyl-CoA radical relative to cob(II)alamin and the inferred g-axis system of the low-spin Co2+ in the molecule. Euler angles (ζ 50° and η = 33°) are also shown. The faded image of the succinyl-CoA represents the position from X-ray coordinates (PDB entry 4REQ) (31).
Conclusions
The spectroscopic results with MCM provide a clear example of the structural information that is encoded by triplet EPR spectra (9, 25). Anisotropic magnetic interactions complicate the appearance of the EPR spectra and also their analysis. However, these same anisotropic terms contain the structural information that can often be directly related to a molecular axis system (26).
The α-radical of succinyl-CoA accumulates in the steady state of the reaction because it is more stable than other radicals in the catalytic cycle. This situation is akin to the radical rearrangement cycle catalyzed by lysine-2,3-amino-mutase wherein the steady-state radical is α to the carboxylate group of the product, β-lysine (32).
Footnotes
This work was supported by grants from the National Institutes of Health (DK 45776 to R.B. and GM 35752 G.H.R.). S.O.M. was supported by NIH Predoctoral Training Grant T32 GM 08293.
Abbreviations: AdoCbl, 5′-deoxyadenosylcobalamin; methyl-malonyl-CoA, methylmalonyl-coenzyme A; succinyl-CoA, succinyl-coenzyme A; MCM, methylmalonyl-CoA mutase; EPR, electron paramagnetic resonance; NMR, nuclear magnetic resonance; S/N, signal-to-noise ratio; ZFS, zero-field splitting.
References
- 1.Banerjee R. Radical peregrinations catalyzed by coenzyme B12-dependent enzymes. Biochemistry. 2001;40:6191–6198. doi: 10.1021/bi0104423. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R. Radical carbon skeleton rearrangements: Catalysis by coenzyme B12-dependent mutases. Chem Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee R, Ragsdale SW. The many faces of vitamin B12: Catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee R, Chowdhury S. Methylmalonyl-CoA Mutase. John Wiley and Sons; New York: 1999. [Google Scholar]
- 5.Babior BM, Moss TH, Orme-Johnson WH, Beinert H. The Mechanism of Action of Ethanolamine Ammonia Lyase, a B12-dependent Enzyme. The participation of paramagnetic species in the catalytic deamination of 2-aminopropanol. J Biol Chem. 1974;249:4537–4544. [PubMed] [Google Scholar]
- 6.Zhao Y, Such P, Retey J. Radical intermediates in the coenzyme-B12-dependent methylmalonyl-CoA mutase reaction shown by ESR spectroscopy. Angew Chem, Int Ed Engl. 1992;31:215–216. [Google Scholar]
- 7.Bothe H, Darley DJ, Albracht SP, Gerfen GJ, Golding BT, Buckel W. Identification of the 4-glutamyl radical as an intermediate in the carbon skeleton rearrangement catalyzed by coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. Biochemistry. 1998;37:4105–4113. doi: 10.1021/bi971393q. [DOI] [PubMed] [Google Scholar]
- 8.Gerfen GJ, Licht S, Willems JP, Hoffman BM, Stubbe J. Electron paramagnetic resonance investigations of a kinetically competent intermediate formed in ribonucleotide reduction: Evidence for a thiyl radical-cob(II)alamin interaction. J Am Chem Soc. 1996;118:8192–8197. [Google Scholar]
- 9.Gerfen GF. EPR spectroscopy of B12-dependent enzymes. John Wiley and Sons; New York: 1999. [Google Scholar]
- 10.Padmakumar R, Banerjee R. Evidence from EPR spectroscopy of the participation of radical intermediates in the reaction catalyzed by methylmalonyl-CoA mutase. J Biol Chem. 1995;270:9295–9300. doi: 10.1074/jbc.270.16.9295. [DOI] [PubMed] [Google Scholar]
- 11.Abend A, Illich V, Retey J. Further insights into the mechanism of action of methylmalonyl-CoA mutase by electron paramagnetic resonance studies. Eur J Biochem. 1997;249:180–186. doi: 10.1111/j.1432-1033.1997.00180.x. [DOI] [PubMed] [Google Scholar]
- 12.Padmakumar R, Taoka S, Padmakumar R, Banerjee R. Coenzyme B12 is coordinated by histidine and not dimethylbenzimidazole on methylmalnyl-CoA mutase. J Am Chem Soc. 1995;117:7033–7034. [Google Scholar]
- 13.Chang CH, Frey PA. Cloning, sequencing, heterologous expression, purification, and characterization of adenosylcobalamin-dependent D-lysine 5,6-aminomutase from Clostridium sticklandii. J Biol Chem. 2000;275:106–114. doi: 10.1074/jbc.275.1.106. [DOI] [PubMed] [Google Scholar]
- 14.Mancia F, Keep NH, Nakagawa A, Leadlay PF, Mc-Sweeney S, Rasmussen B, Bösecke P, Diat O, Evans PR. How coenzyme B(12 radicals are generated: The crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure. 1996;4:339–350. doi: 10.1016/s0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 15.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W, Kratky C. Glutamate mutase from Clostridium cochlearium: The structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Struct Folding Des. 1999;7:891–902. doi: 10.1016/s0969-2126(99)80116-6. [DOI] [PubMed] [Google Scholar]
- 16.Padmakumar R, Gantla S, Banerjee R. A Rapid Method for the synthesis of methylmalonyl-Coenzyme A and other CoA-esters. Anal Biochem. 1993;214:318–320. doi: 10.1006/abio.1993.1494. [DOI] [PubMed] [Google Scholar]
- 17.Pohl NL, Hans M, Lee HY, Kim YS, Cane DE, Khosla C. Remarkably broad substrate tolerance of malonyl-CoA synthetase, an enzyme capable of intracellular synthesis of polyketide precursors. J Am Chem Soc. 2001;123:5822–5823. doi: 10.1021/ja0028368. [DOI] [PubMed] [Google Scholar]
- 18.Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Fourier self-deconvolution: A method for resolving intrinsically overlapped bands. Appl Spectrosc. 1981;35:271–276. [Google Scholar]
- 19.Latwesen DG, Poe M, Leigh JS, Reed GH. Electron paramagnetic resonance studies of a ras p21-Mn(II)GDP complex in solution. Biochemistry. 1992;31:4946–4950. doi: 10.1021/bi00136a004. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein H, Poole CP, Jr, Safko JL. Classical Mechanics. 3. Addison-Wesley; San Francisco: 2002. [Google Scholar]
- 21.Bandarian V, Reed GH. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: Mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry. 1999;38:12394–12402. doi: 10.1021/bi990620g. [DOI] [PubMed] [Google Scholar]
- 22.Wertz JE, Bolton JR. Electron Spin Resonance. Chapman and Hall; New York: 1986. [Google Scholar]
- 23.Goffe WL, Ferrier GD, Rogers J. Global optimization of statistical functions with simulated annealing. J Econometrics. 1994;60:65–100. [Google Scholar]
- 24.Luckhurst GR. In: Spin Labeling: Theory and Applications. Berliner LJ, editor. Academic Press; New York: 1976. pp. 133–181. [Google Scholar]
- 25.Reed GH, Mansoorabadi SO. The positions of radical intermediates in the active sites of adenosylcobalamin-dependent enzymes. Curr Opin Struct Biol. 2003;13:716–721. doi: 10.1016/j.sbi.2003.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansoorabadi SO, Reed GH. In: Paramagnetic resonance of metallobiomolecules. Telser J, editor. American Chemical Society; Washington, DC: 2003. pp. 82–96. [Google Scholar]
- 27.Morton JR. Electron spin resonance spectra of oriented radicals. Chem Rev. 1964;64:453–471. [Google Scholar]
- 28.Laroff GP, Fessenden RW. 13C hyperfine interactions in radicals from some carboxylic acids. J Chem Phys. 1971;55:5000–5008. [Google Scholar]
- 29.Fischer H. In: Free Radicals. Kochi JK, editor. John Wiley and Sons; New York: 1973. pp. 435–491. [Google Scholar]
- 30.Carey FA, Sundberg RJ. Part A: Structure and Mechanisms. 3. Plenum Press; New York: 1990. Advanced Organic Chemistry. [Google Scholar]
- 31.Mancia F, Evans P. Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Structure. 1998;6:711–720. doi: 10.1016/s0969-2126(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 32.Ballinger MD, Frey PA, Reed GH. Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry. 1992;31:10782–10789. doi: 10.1021/bi00159a020. [DOI] [PubMed] [Google Scholar]