

Abstract
The endoplasmic reticulum (ER) is the site of synthesis and maturation of proteins designed for secretion or for localization on the cell membrane. Various types of stress from both inside and outside cells disturb ER function, thus causing unfolded or misfolded proteins to accumulate in the ER. To improve and maintain the ER functions against such stresses, the ER stress response pathway is activated. However, when the stress is prolonged or severe, apoptosis pathways are activated to remove damaged cells. It was recently reported that the ER stress pathway is also involved in the inflammatory response, whereby inflammation induces ER stress, and ER stress induces an inflammatory response. Therefore, the ER stress response pathway is involved in various diseases, including cardiovascular diseases such as atherosclerosis and ischemic diseases, in various ways. The ER stress pathway may represent a novel target for the treatment of these diseases.
1. Introduction
The endoplasmic reticulum (ER) is the site of synthesis and maturation of proteins destined for secretion, the cell membrane, Golgi apparatus, and lysosomes [1–4]. The ER has several additional important functions, including assisting the maturation processes of newly synthesized proteins, such as cleavage, glycosylation, formation of disulfide bonds, folding, and assembly. These processes are assisted by ER chaperone proteins, such as BiP (an ER-resident Hsp70 family molecule), Grp94 (an ER-resident Hsp90 family molecule), calreticulin, calnexin, and protein disulfide isomerase (PDI). The protein maturation processes are dependent on intraluminal circumstances such as the Ca2+ concentration, oxygen supply, and redox homeostasis. High concentrations of Ca2+ in the ER are necessary for the functions of several ER chaperones, such as calreticulin, calnexin, and PDI. The formation of disulfide bonds is influenced by the redox circumstances in the ER. Unfolded or misfolded proteins cannot be delivered to the Golgi apparatus and are retained in the ER. The accumulation of these abnormal proteins in the ER disturbs the ER function, and cell survival can thus be threatened. When cells are subjected to ER stress due to accumulation of unfolded proteins in the ER, the ER stress response (also known as unfolded protein response) pathways are induced to protect cells [1] (Figure 1). However, when the ER functions are severely impaired, apoptosis occurs to remove damaged cells and protect the surrounding cells. When ER stress-induced apoptosis causes the loss of a large number of cells, the functions of tissues or organs are impaired, resulting in various pathological conditions, such as diabetes mellitus, hepatic steatosis, inflammatory diseases, ischemic diseases, and neurodegenerative diseases [2–21]. Interestingly, it was recently shown that the ER stress pathway is directly involved in the induction of not only apoptosis but also inflammation and autophagy [4–9].
Figure 1.
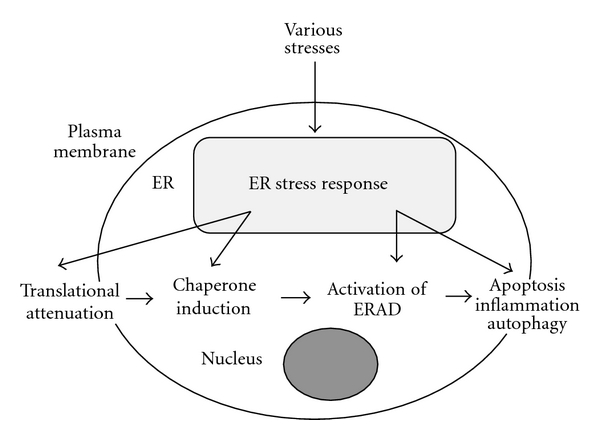
An overview of the ER stress response pathway. See the text for details.
Inflammation is a reaction of the host following exposure to various stimuli, and it is an attempt to eliminate the causal factors. There are two types of inflammation, acute inflammation and chronic inflammation [4, 22–24]. Chronic inflammation is caused by the failure to eliminate the causal factors and/or by the failure to restore the damaged tissues to normal conditions. Macrophages and lymphocytes play major roles in chronic inflammation. Recently, chronic inflammation has been considered as the underlying pathology of various diseases including cardiovascular diseases, cancer, and metabolic diseases. It has been demonstrated that the ER stress response pathway is involved in the pathogenesis of various chronic inflammatory diseases [2, 4, 8, 9, 20].
ER stress-induced transcription factor CHOP/GADD153 is involved in ER stress-mediated cell damage, and it is also involved in the metabolic and inflammatory processes [6, 10, 11, 20, 25] (Figure 2). Therefore, CHOP plays crucial roles in the development and progression of cardiovascular and metabolic diseases. In this paper, the inflammation-related roles of the ER stress pathway including CHOP in cardiovascular diseases are discussed.
Figure 2.
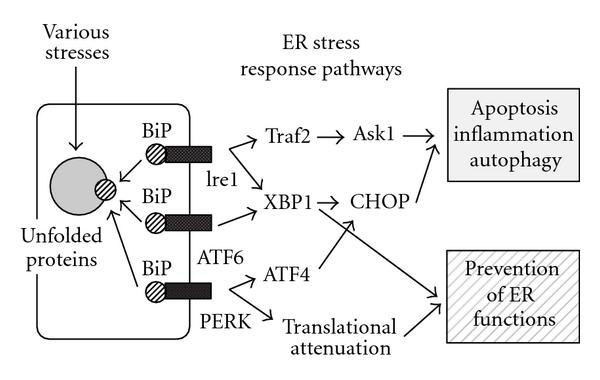
The major signal transduction pathway in the ER stress response. See the text for details.
2. Molecular Mechanisms of the ER Stress Response
There are three main ER stress sensors (Ire1, ATF6, and PERK) on the ER membrane [1–4, 9, 26] (Figure 2). Various stresses, including hypoxia and oxidative stresses, disturb ER function, and unfolded or misfolded proteins are accumulated in the ER. When unfolded or misfolded proteins are accumulated in the ER, Ire1 and PERK are activated by dimer formation and then are autophosphorylated. The inactive form of ATF6 (p90ATF6) is transported to the Golgi apparatus and is activated by a two-step cleavage by Site-1 protease (S1P) and Site-2 protease (S2P), to produce the active form of ATF6 (p50ATF6). The active form is then transported to the nucleus and functions as a transcriptional activator for ER stress-related genes [3, 27]. Four distinct ER stress response phases have been identified (Figure 1). (1) The first phase involves translational attenuation to reduce the load of newly synthesized proteins through phosphorylation of eIF-2α (α-subunit of eukaryotic initiation factor 2) by activated PERK. (2) The second involves the induction of ER chaperones such as BiP to enhance folding activity in the ER. (3) The third is degradation of unfolded or misfolded proteins in the ER through the ubiquitin-proteasome system in the cytosol. This system is called the ER-associated degradation (ERAD). (4) When ER stress-inducing stresses are too severe or prolonged to allow for recovery of ER function, the apoptosis pathway is activated to remove damaged cells [2, 10, 28–30]. At least three pathways are involved in the ER stress-mediated apoptosis [2, 10, 28–31]. The first is transcriptional activation of the gene for CHOP. The second is activation of the Ire1-TRAF2-ASK1-MAP kinase pathway. The third is activation of ER-associated caspase-12.
CHOP is expressed at low levels under physiological conditions, but it is strongly induced at the transcriptional level in response to ER stress [1, 2, 10, 25, 32]. The transcription of the chop gene is activated by all three ER stress sensors (Ire1, ATF6 and PERK) signaling pathways (Figure 2). The PERK-ATF4 signaling pathway plays a dominant role in the induction of CHOP over that of the ATF6 and Ire1 signaling pathways, although the presence of all three signaling pathways is required to achieve the maximal induction of CHOP [10, 25]. ER stress-induced apoptosis is suppressed in both CHOP null cells and chop knockout mice. Marciniak et al. reported that CHOP inhibits ER stress-induced attenuation of protein synthesis by dephosphorylation of eIF-2α through the induction of GADD34 [33]. They showed that the de novo protein synthesis, induced by CHOP under ER stress conditions, led to the accumulation of a high molecular weight protein complex in the ER and impaired ER function. It is also reported that the apoptosis signal induced by CHOP is transmitted to the mitochondria through activation of the proapoptotic molecule Bax and induction of the proapoptotic BH3-only type Bcl-2 family member Bim [34, 35].
The c-Jun N-terminal kinase (JNKs) and p38 MAP kinases are classified as members of the stress-responsive MAP kinase family [1, 36]. Apoptosis signal-regulating kinase 1 (ASK1) belongs to the MAPKKK family and activates both JNK and p38 pathways by directly phosphorylating and activating MAPKK family molecules, SEK1/MKK7 and MKK3/MKK6 (Figure 2). ASK1 is activated by the treatment of cells with TNFα or Fas ligand. In the case of activation by TNFα treatment, this activation is regulated by TNF receptor-associated factor 2 (TRAF2). TRAF2 directly interacts with ASK1, activating ASK1 in a redox-dependent manner. Under unstressed conditions, thioredoxin (Trx) directly binds to ASK1 and inhibits its kinase activity. Treatment with ROS, including H2O2, oxidizes Trx, thus leading to its dissociation from ASK1. Under ER stress conditions, activated Ire1, one of the ER stress sensors, recruits TRAF2; then ASK1 directly binds to TRAF2 and is activated. It is thought that the mitochondria pathway is involved in ASK1-mediated apoptosis.
Caspase-12 belongs to the ICE (caspase-1) subfamily of caspase cysteine protease family [1, 28, 29]. Pro-caspase-12 is localized on the cytosolic side of the ER membrane and is activated by ER stress. However, the mechanism underlying the caspase-12 activation under ER stress conditions has not been confirmed. Caspase-12 knockout cells are partially resistant to ER stress-induced apoptosis [28]. However, the human caspase-12 gene is nonfunctional (pseudogene). Therefore, caspase-12 does not appear to be involved in the ER stress-induced apoptosis pathway in human cells.
As mentioned above, there are several signaling pathways involved in ER stress-induced apoptosis. It has been speculated that these pathways function in parallel under ER stress conditions, and the pathway that plays the major role is cell type and stress dependent [1].
Autophagy is a degradation pathway responsible for the bulk turnover of intracellular proteins and organelles via lysosomal degradation [21, 37]. Autophagy is induced by proteasome inhibition under ER stress condition. It has been reported that autophagy is directly induced through the Ire1-JNK/p38 pathway and the ATF4 pathway under ER stress [7].
3. Inflammation-Related Roles of the ER Stress Pathway
Inducible type NO synthase (iNOS) is induced in activated inflammation-related cells such as macrophages, and NO is involved in various inflammation-related diseases [2, 38]. Treatment with NO activates the ER stress pathways including CHOP, probably through disturbance of Ca2+ homeostasis in the ER, or enhancing ROS production by inhibiting mitochondrial respiration system through nitrosylation of cytochrome c oxidase, or inhibiting PDI through NO-induced S-nitrosylation [10, 38, 39]. The production of other ROS is also elevated under inflammatory conditions and plays a role as an ER stress inducer in inflammatory lesions [40]. The electrons generated by disulfide-bond formation in the ER do not recycle to reduce compounds in the cytoplasm but are transferred from reduced ERO1 to molecular oxygen [41]. ERO1 is an ER-resident oxidase and is involved in the formation of disulfide bonds. Therefore, biosynthesis of secreted proteins causes a net loss of reducing equivalents in the ER. In addition, many proteins synthesized in the ER are retrotransported to the cytoplasm to be degraded in a process of ERAD, because of the protein quality system in the ER. In this process, disulfide bonds are reduced to go through membrane channel. ERAD system is therefore a net consumer of reducing equivalents. Totally, the ER is a net consumer of reducing power. As already mentioned, ERAD is enhanced in the ER stress response to preserve ER function. It is also reported that treatment with ROS disturbs ER functions including intraluminal Ca2+ homeostasis and protein maturation processes [42, 43]. Therefore, oxidative stress induces ER stress, and ER stress produces oxidative stress.
Recently, it is reported that Toll-like receptor (TLR) 2 and 4-mediated signals specifically activate Ire1-XBP-1 pathway and are involved in host defense against bacterial pathogen [44]. XBP-1 is an ER stress-induced transcription factor and is involved in ER function-protective factors. We also reported that treatment with LPS activates Ire1-XBP1 pathway, but not PERK pathway [6, 45]. Therefore, the ER stress pathway is involved in various inflammatory diseases. Not only does inflammation induce the ER stress pathway, but the ER stress pathway is also involved in the induction of inflammatory responses. When mice are given LPS intratracheally, the ER stress-CHOP pathway is induced in activated macrophages in the lungs [6]. Under this condition, caspase-11, which is needed for the activation of procaspase-1 and pro-IL-1β, was induced by LPS in a CHOP-dependent manner, both in the lungs and in primary cultured macrophages [6]. In fact, LPS-induced IL-1β secretion and inflammatory responses in lung tissues and primary cultured macrophages were evidently suppressed in chop knockout mice. Moreover, Park et al. reported that the CHOP is involved in regulating the expression of proinflammatory cytokine IL-8 through activation of NF-κB [46]. As already mentioned, activated ER stress sensor Ire1 activates JNK/p38 MAP kinase through the Ire1-TRAF2-ASK1 pathway [30]. The activated JNK/p38 MAP kinase phosphorylate c-Jun, a component of AP-1 transcription factor, then AP-1 activity is enhanced. Activated AP-1 is involved in the transcriptional induction of inflammation-related genes, such as TNF-α, IL-6, IL-8, and MCP-1 [47]. The IRE1α·TRAF2·IKK complex induces degradation of IκBα, activation of NF-κB, and the transcription of inflammatory genes such as TNF-α, TGF-β, IL-2, IL-6, and IL-8 [5, 48, 49]. Therefore, the ER stress pathway plays crucial roles in inflammatory response.
As mentioned above, severe or prolonged ER stress-inducing stresses activate the ER stress-CHOP pathway, and CHOP induces apoptosis. Under inflammation-related ER stress conditions, CHOP induces caspase-11 and activates the IL-1β activation pathway, but not the apoptosis pathway [6]. Therefore, CHOP plays different roles in response to different types of stimulation. We recently reported the differences between the precise induction of the ER stress-CHOP pathway by inflammatory stimuli and the typical ER stress-induction stimulus [45]. The transcription of the chop gene is regulated through all three ER stress sensor (ATF6, Ire1, and PERK) signaling pathways under ER stress-mediated apoptosis [10, 41]. When cells are exposed to inflammatory stimuli, ATF6 and Ire1 are activated, but PERK is not activated. Therefore, the induction of CHOP by LPS is delayed in comparison to that induced by the ER stress-inducer, thapsigargin, even though ER function-protective molecules such as BiP are induced during the early stage of response [45]. XBP-1 is an ER stress-induced transcription factor. The active form of XBP1 is induced by the combination of the ATF6 and Ire1 pathways. The active forms of ATF6 and XBP1 play crucial roles in the induction of the protective factors for ER functions, including BiP and ERAD-related molecules. Therefore, the ER stress-enhancing and apoptosis-inducing effect of CHOP is suppressed in LPS-treated macrophages, because the ER protective system is already enhanced before CHOP expression is induced. More detailed molecular analyses of the ER stress sensors are needed to elucidate the precise mechanisms of the starting point of the ER stress response pathway and how the three pathways are activated and regulated [50].
4. Roles of the ER Stress Pathway in Atherosclerogenesis
Atherosclerosis is mainly caused by metabolic disorders and is deeply involved in life-threatening ischemic diseases, such as myocardial infarction and cerebral infarction [20, 51–53]. Therefore, clarification of atherosclerogenesis is one of the most emergent issues in the field of medical science. For infarction, we need to distinguish the mechanisms of the progression of atherosclerosis and the disruption of blood flow. Acute coronary syndrome (ACS), including myocardial infarction and unstable angina, is most frequently caused by occlusive coronary thrombosis at the site of a preexisting atherosclerotic plaque. The formation of coronary thrombosis is generally the result of the rupture of an atherosclerotic plaque, followed by the aggregation of platelets and the formation of fibrin. It is thought that metalloproteinases secreted by macrophages and apoptosis of macrophage-derived foam cells affect the stability of plaques. Therefore, monocytes/macrophages play a key role in the instability of atherosclerotic plaques. Activations of the ER stress pathways including CHOP are detected in atherosclerotic lesion at all stage of atherosclerogenesis, especially in macrophage-derived cells [54] (Figure 3(a)). Myoishi et al. reported the induction of apoptosis and the activation of the ER stress pathway, including the induction of CHOP, to be detected in macrophages and smooth muscle cells within ruptured plaques, but not in stable fibrous plaques in humans [52]. They also reported the levels of ER stress in atherectomy specimens from patients with unstable angina pectoris to be higher than those from patients with stable angina pectoris. These results suggest that ER stress-induced apoptosis of macrophages affects the vulnerability of plaques to rupture. Thorp et al. reported the progression of atherosclerosis to be suppressed in two distinct models of advanced atherosclerosis in the mice lacking CHOP [55]. These results show that the ER stress-CHOP pathway plays crucial roles in atherosclerogenesis, which is a specific type of chronic inflammation of arterial wall induced by high levels of intracellular cholesterol, fatty acids, oxidative stress, and NO. Accumulation of free cholesterol in ER membrane is thought to be one of the main causes of the activation of the ER stress response pathway in atherosclerotic lesion [20, 56]. During the process of atherosclerogenesis, the iNOS expressed by macrophages invading the arterial walls plays a crucial role [2, 38]. Therefore, iNOS-derived NO is one of the major candidate ER stress-inducers in atherosclerotic lesions. TLR-mediated signals are also involved in atherosclerogenesis [57]. As already mentioned, TLR-mediated stimulation and ROS also can be the cause of the activation of the ER stress response pathway. The ER stress-CHOP pathway is activated in macrophage-derived cells at all stage of atherosclerosis, but apoptosis becomes prominent at the late stage (partly because of the rapid disposal of apoptotic cells at the early stage). Therefore, it can be speculated that the role of CHOP changes at the late stage. Li et al. showed the induction of the ER stress pathway by the accumulation of free cholesterol to be involved in the induction of TNF-α and IL-6 [5]. They also showed that the CHOP branch of the ER stress response is required for IL-6 induction and demonstrated that other ER stress-related pathways are responsible for the activation of p38, JNK1/2, NF-κB, and TNF-α. Therefore, the ER stress pathway contributes to the progress of atherosclerogenesis, through activation of inflammatory cells in arterial wall. Although it appears that CHOP plays a crucial role in atherosclerogenesis, its role in the final rupture of the plaque and subsequent thrombosis is still undefined. We recently showed that the ER stress-induced apoptotic pathway in macrophages contributes to the formation of the rupture of atherosclerotic plaques [56]. Large numbers of CHOP-expressing macrophages showed apoptosis in advanced ruptured atherosclerosis lesions in wild-type mice, while few apoptotic cells were observed in chop knockout mice. The rupture of atherosclerotic plaques was significantly reduced in high cholesterol-fed chop/apoe double knockout mice compared to chop wt, apoe knockout mice. Using primary cultured macrophages, we further showed that the unesterified free cholesterol derived from incorporated denatured LDL was accumulated in the ER and induced ER stress-mediated apoptosis in a CHOP-Bax pathway-dependent manner. We concluded that the ER stress-CHOP-Bax-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. In addition, the formation of a fibrous cap of plaques in the brachiocephalic artery was enhanced in chop/apoe double knockout mice compared to chop wt, apoe knockout mice. As already mentioned, CHOP is involved in the induction of the inflammatory response, and the CHOP-mediated extracellular matrix degeneration pathway may be induced in the final stage of atherosclerotic lesion rupture.
Figure 3.
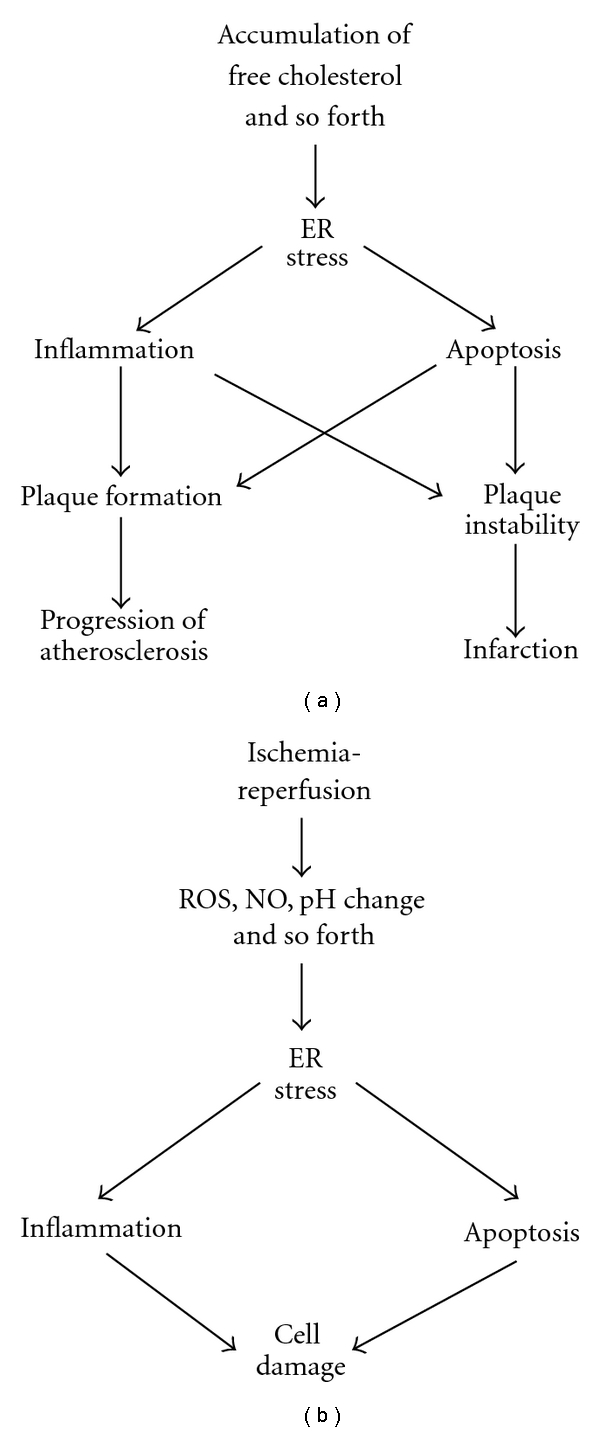
Involvement of ER stress response in atherosclerosis (a) and ischemia-reperfusion injury (b). See the text for details.
It has been suggested that the suppression of acyl–coenzyme A and cholesterol acyltransferase- (ACAT-) mediated cholesterol esterification and/or activation of neutral cholesteryl esterase, are induced during the progression of atherosclerosis [58–60], and unesterified free cholesterol is accumulated in foam cells. Under physiological conditions, the cholesterol content of the ER membrane is low, and it is thought that increases in the cholesterol content of the ER membrane may alter the stiffness of the membrane, thus leading to a disturbance in the function of ER membrane proteins, including Ca2+ pumps such as sarcoplasmic reticulum ATPase (SERCA) [59]. ACAT is an ER membrane protein, therefore, accumulation of free cholesterol in the ER membrane may further disturbs the ACAT activity [59]. As a result, the ER stress pathway may be activated by ER dysfunction. We directly showed that free cholesterol is transported to the ER and activates the ER stress pathway in primary cultured macrophages [56]. Seimon et al. reported that oxidized phospholipids, saturated fatty acids, and lipoprotein (a) induce apoptosis in ER-stressed macrophages via the CD36 and TLR2-mediated pathway [61]. As already mentioned, ER stress-mediated inflammatory pathway is induced by the TLR2-mediated pathway. Therefore, it can be speculated that TLR-ER stress-mediated inflammation is also involved in the process of atherosclerogenesis.
The ER stress pathway therefore may be a key therapeutic target related not only to atherosclerotic lesion progression, but also to the instability of existing atherosclerotic plaques.
5. Roles of the ER Stress Pathway in Ischemia-Reperfusion Injury
Ischemic stress, including hypoxia and hypoglycemia, is an inducing factor for the ER stress pathway, probably because of the disturbance of protein modification processes such as disulfide bond formation and N-glycosylation of newly synthesized proteins in the ER [3, 7, 40] (Figure 3(b)). Therefore, the ER stress pathway is also involved in the pathogenesis of ischemic diseases, such as cerebral infarction and myocardial infarction, most likely through induction of apoptosis or inflammation. Azfer et al. reported that ER stress-associated genes, including CHOP, are induced in the heart of an ischemic heart disease mouse model [62]. Szegezdi et al. reported that the ER stress pathway is induced by deprivation of serum glucose and oxygen in primary cultured rat neonatal cardiomyocytes [63]. Under those conditions, CHOP was induced by prolonged treatment alone, which induced apoptosis. Therefore, they concluded that CHOP was involved in ischemia-induced cardiomyocyte loss. The ER stress-CHOP or JNK/p38 MAP kinase pathway can be induced and play negative roles in ischemic tissues during the preapoptotic period, and it is also likely involved in the induction of inflammation. Mutant KDEL receptor (a retrieval receptor for ER chaperones in the early secretory pathway) transgenic mice show dilated cardiomyopathy, enhanced expression of CHOP, and apoptosis in cardiomyocytes [64]. Therefore, the normal function of the ER is essential for the physiological function of the heart.
In the case of cerebral infarction and acute myocardial infarction, early and successful reperfusion is the most effective therapy to reduce infarct size and improve the clinical outcome. However, the recanalization of the occluded artery can paradoxically reduce the beneficial effects by inducing further cellular damage, termed ischemia-reperfusion (I/R) injuries, and the clarification of the precise mechanism underlying this damage is one of the major urgent issues in the field of cardiovascular medicine [19, 21, 65, 66]. Several potential mediators have been proposed as the cause of I/R injury, such as ROS generation, intracellular Ca2+ disturbance, rapid pH changes, and inflammation. It is likely that all of these factors are involved in mediating neuronal cell and cardiomyocyte death, and inflammation, and they are also thought to be the potential causes of ER stress. Then, the ER stress response leads to further apoptosis and inflammation. In the case of cerebral infarction, the ER stress responses were shown to be activated by I/R in mouse brains, and I/R injury was suppressed in the chop knockout mouse brain [13]. Several studies have demonstrated that the ER stress response is activated in rodent models of myocardial I/R [66–69]. Moreover, overexpression of ATF6, one of the ER stress sensors on the ER membrane, induced the expression of ER function-protective ER stress-related genes such as BiP and Grp94 without a significant induction of CHOP, showing protective effects against reperfusion injury [67]. In addition, the general inhibition of the ER stress response by a δ-protein kinase C (δPKC) inhibitor also resulted in reduced injury size or cardiomyocyte apoptosis [69]. Therefore, ER stress appears to regulate myocardial I/R injury variably, either attenuating or exacerbating the condition, possibly depending on the balance between prosurvival and proapoptotic ER stress responses. It is reported that ischemic preconditioning or postconditioning reduces cardiac damage, probably through induction of ER function-protective molecules including PDI [70]. Recently, Miyazaki et al. showed that myocardial I/R activates the ER stress-induced, CHOP-mediated pathway partially through ROS overproduction occurring early after reperfusion, and that the pathway subsequently exacerbates myocardial I/R injury not only by inducing cardiomyocyte apoptosis but also by enhancing tissue inflammation [66]. Furthermore, an in vitro analysis showed the potential and complexity of the CHOP-related proinflammatory signaling under inflammatory conditions. Using a myocardial I/R model (not a chronic ischemia model), it was demonstrated that reperfusion-dependent ROS production is upstream of ER stress and that CHOP exhibits proinflammatory activities, such as induction of IL-1β and IL-6, in the reperfused tissue [66]. Additionally, the time-dependent activation of ER stress-related molecules, such as BiP, CHOP, and XBP-1 has been directly demonstrated in vivo. It is therefore suggested that the CHOP-mediated pathway enhances myocardial inflammation, possibly by the transcriptional induction of specific cytokine genes, such as IL-6. It was demonstrated that CHOP deficiency almost completely suppressed cardiomyocyte apoptosis in the reperfused myocardium and neuronal cell death in the reperfused mice brain, thus suggesting that the CHOP-mediated pathway also plays a potentially important role in inducing cardiomyocyte and neuronal cell apoptosis after I/R [13, 66]. Another study showed that either a deficiency in stress-activated MAP kinase JNK1 or JNK2 reduces cardiomyocyte apoptosis by approximately 40% [71]. It was also shown that intravenous administration of edaravone (a free radical scavenger) immediately before reperfusion significantly suppresses the superoxide overproduction and subsequent activation of the ER stress pathway, leading to reduced injury size in a mouse I/R model heart [66].
These experimental results suggest that the ER stress pathway may represent a new target for the prevention of I/R injuries, which include the apoptosis pathway and inflammation pathway, and that scavengers of ROS can be the promising agents that can reduce ER stress and suppress I/R injuries.
6. Conclusions
The ER stress-related Inflammation and apoptosis are involved in the pathogenesis of various diseases, including cardiovascular diseases. Therefore, the ER stress pathway can be a new target for the treatment of those diseases. However, the molecular mechanisms underlying the activation, regulation, and execution of the ER stress response have not been fully clarified. Recently, Iwawaki et al. reported that ER stress sensor IRE1α in the placenta is essential for placental development through induction of VEGF-A and other factors [72]. IRE1α-deficient mouse show embryonic lethality, because of reduced proliferation of trophoblasts. They also showed that IRE1α-deficient embryos supplied with functionally normal placentas can be born alive. The molecular mechanisms how IRE1α is involved in the secretion of VEGF-A and the placental angiogenesis remain to be elucidated. It is therefore necessary to investigate the molecular mechanisms by which the ER stress pathway exerts different effects in different situations, to elucidate the pathological roles of this pathway and the potential of targeting this pathway for the prevention or treatment of human diseases.
Acknowledgments
The authors thank their colleagues for their valuable suggestions and discussion, and Brian Quinn for comments on the paper. This work was supported in part by Grants-in-Aid (no. 20590310 to Tomomi Gotoh and no. 19790563 to Motoyoshi Endo) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and grants (to Tomomi Gotoh) from the Takeda Science Foundation and Mitsubishi Pharma Research Foundation.
References
- 1.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annual Review of Biochemistry. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 2.Gotoh T, Mori M. Nitric oxide and endoplasmic reticulum stress. Arteriosclerosis, Thrombosis & Vascular Biology. 2006;26(7):1439–1446. doi: 10.1161/01.ATV.0000223900.67024.15. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida H. ER stress and diseases. FEBS Journal. 2007;274(3):630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 4.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Schwabe RF, DeVries-Seimon T, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: model of NF-κB- and map kinase-dependent inflammation in advanced atherosclerosis. Journal of Biological Chemistry. 2005;280(23):21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 6.Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. Journal of Immunology. 2006;176(10):6245–6253. doi: 10.4049/jimmunol.176.10.6245. [DOI] [PubMed] [Google Scholar]
- 7.Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle. 2009;8(23):3838–3847. doi: 10.4161/cc.8.23.10086. [DOI] [PubMed] [Google Scholar]
- 8.McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. American Journal of Physiology. 2010;298(6):G820–G832. doi: 10.1152/ajpgi.00063.2010. [DOI] [PubMed] [Google Scholar]
- 9.Martinon F, Glimcher LH. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Current Opinion in Immunology . 2011;23(1):35–40. doi: 10.1016/j.coi.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death & Differentiation. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 11.Namba T, Tanaka KI, Ito Y, et al. Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. American Journal of Pathology. 2009;174(5):1786–1798. doi: 10.2353/ajpath.2009.080864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suyama K, Ohmuraya M, Hirota M, et al. C/EBP homologous protein is crucial for the acceleration of experimental pancreatitis. Biochemical & Biophysical Research Communications. 2008;367(1):176–182. doi: 10.1016/j.bbrc.2007.12.132. [DOI] [PubMed] [Google Scholar]
- 13.Tajiri S, Oyadomari S, Yano S, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death & Differentiation. 2004;11(4):403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 14.Ogawa S, Kitao Y, Hori O. Ischemia-induced neuronal cell death and stress response. Antioxidants & Redox Signaling. 2007;9(5):573–587. doi: 10.1089/ars.2006.1516. [DOI] [PubMed] [Google Scholar]
- 15.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metabolism. 2008;7(6):520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishitoh H, Kadowaki H, Nagai A, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes & Development. 2008;22(11):1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. Journal of Neuroinflammation. 2009;6, article 41 doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circulation Research. 2010;107(9):1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 20.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation Research. 2010;107(7):839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groenendyk J, Sreenivasaiah PK, Kim DH, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circulation Research. 2010;107(10):1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 23.Tabata M, Kadomatsu T, Fukuhara S, et al. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metabolism. 2009;10(3):178–188. doi: 10.1016/j.cmet.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Scrivo R, Vasile M, Bartosiewicz I, Valesini G. Inflammation as “common soil” of the multifactorial diseases. Autoimmunity Reviews. 2011;10(7):369–374. doi: 10.1016/j.autrev.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51(supplement 3):S455–S461. doi: 10.2337/diabetes.51.2007.s455. [DOI] [PubMed] [Google Scholar]
- 26.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 27.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Structure & Function. 2008;33(1):75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families: activation of caspase-12 by calpain in apoptosis. Journal of Cell Biology. 2000;150(4):887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. Journal of Biological Chemistry. 2002;277(37):34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 30.Homma K, Katagiri K, Nishitoh H, Ichijo H. Targeting ASK1 in ER stress-related neurodegenerative diseases. Expert Opinion on Therapeutic Targets. 2009;13(6):653–664. doi: 10.1517/14728220902980249. [DOI] [PubMed] [Google Scholar]
- 31.Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Communication & Signaling. 2009;7(9):1–10. doi: 10.1186/1478-811X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. Journal of Clinical Investigation. 2008;118(10):3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & Development. 2004;18(24):3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gotoh T, Terada K, Oyadomari S, Mori M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death & Differentiation. 2004;11(4):390–402. doi: 10.1038/sj.cdd.4401369. [DOI] [PubMed] [Google Scholar]
- 35.Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 36.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochimica et Biophysica Acta. 2008;1780(11):1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 37.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ignarro LJ. Nitric Oxide; Biology and Pathology. London, UK: Academic Press; 2000. [Google Scholar]
- 39.Uehara T, Nakamura T, Yao D, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441(7092):513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 40.Schröder M. Endoplasmic reticulum stress responses. Cellular and Molecular Life Sciences. 2008;65(6):862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 42.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Molecular Immunology. 2002;38(10):713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 43.Görlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants & Redox Signaling. 2006;8(9-10):1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 44.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunology. 2010;11(5):411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakayama Y, Endo M, Tsukano H, et al. Molecular mechanisms of the LPS-induced non-apoptotic ER stress-CHOP pathway. Journal of Biochemistry. 2010;147(4):471–483. doi: 10.1093/jb/mvp189. [DOI] [PubMed] [Google Scholar]
- 46.Park S-H, Choi HJ, Yang H, et al. Endoplasmic reticulum stress-activated C/EBP homologous protein enhances nuclear factor-κB signals via repression of peroxisome proliferator-activated receptor γ. Journal of Biological Chemistry. 2010;285(46):35330–35339. doi: 10.1074/jbc.M110.136259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. International Union of Biochemistry & Molecular Biology Life. 2005;57(4-5):283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 48.Gargalovic PS, Gharavi NM, Clark MJ, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arteriosclerosis, Thrombosis, & Vascular Biology. 2006;26(11):2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 49.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Molecular & Cellular Biology. 2006;26(8):3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimata Y, Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Current Opinion in Cell Biology. 2011;23(2):135–142. doi: 10.1016/j.ceb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 51.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine. 2005;352(16):1685–1626. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 52.Myoishi M, Hao H, Minamino T, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116(11):1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 53.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circulation Journal. 2010;74(2):213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- 54.Zhou J, Lhoták S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111(14):1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 55.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe − /− and Ldlr − /− mice lacking CHOP. Cell Metabolism. 2009;9(5):474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsukano H, Gotoh T, Endo M, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arteriosclerosis, Thrombosis & Vascular Bioliology. 2010;30(10):1925–1932. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 57.Cole JE, Georgiou E, Monaco C. The expression and functions of toll-like receptors in atherosclerosis. Mediators of Inflammation. 2010;2010:18 pages. doi: 10.1155/2010/393946. Article ID 393946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Small DM, Bond MG, Waugh D, Prack M, Sawyer JK. Physicochemical and histological changes in the arterial wall of nonhuman primates during progression and regression of atherosclerosis. Journal of Clinical Investigation. 1984;73(6):1590–1605. doi: 10.1172/JCI111366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. Journal of Clinical Investigation. 2002;110(7):905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. Journal of Lipid Research. 2009;50(supplement 11):S382–S387. doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seimon TA, Nadolski MJ, Liao X, et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metabolism. 2010;12(5):467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. American Journal of Physiology. 2006;291(3):H1411–H1420. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szegezdi E, Duffy A, O’Mahoney ME, et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochemical and Biophysical Research Communications. 2006;349(4):1406–1411. doi: 10.1016/j.bbrc.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 64.Hamada H, Suzuki M, Yuasa S, et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Molecular & Cellular Biology. 2004;24(18):8007–8017. doi: 10.1128/MCB.24.18.8007-8017.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elahi MM, Kong YX, Matata BM. Oxidative stress as a mediator of cardiovascular disease. Oxidative Medicine & Cellular Longevity. 2009;2(5):259–269. doi: 10.4161/oxim.2.5.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyazaki Y, Kaikita K, Endo M, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(5):1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- 67.Martindale JJ, Fernandez R, Thuerauf D, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circulation Research. 2006;98(9):1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 68.Natarajan R, Salloum FN, Fisher BJ, Smithson L, Almenara J, Fowler AA. Prolyl hydroxylase inhibition attenuates post-ischemic cardiac injury via induction of endoplasmic reticulum stress genes. Vascular Pharmacology. 2009;51(2-3):110–118. doi: 10.1016/j.vph.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 69.Qi X, Vallentin A, Churchill E, Mochly-Rosen D. δPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. Journal of Molecular & Cellular Cardiology. 2007;43(4):420–428. doi: 10.1016/j.yjmcc.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toldo S, Severino A, Abbate A, Baldi A. The role of PDI as a survival factor in cardiomyocyte ischemia. Methods in Enzymology. 2011;489:47–65. doi: 10.1016/B978-0-12-385116-1.00003-0. [DOI] [PubMed] [Google Scholar]
- 71.Kaiser RA, Liang Q, Bueno O, et al. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. Journal of Biological Chemistry. 2005;280(38):32602–32608. doi: 10.1074/jbc.M500684200. [DOI] [PubMed] [Google Scholar]
- 72.Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(39):16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]