

Abstract
The renin-angiotensin system (RAS), a key regulator of the blood pressure and fluid/electrolyte homeostasis, also plays a critical role in kidney development. All the components of the RAS are expressed in the developing metanephros. Moreover, mutations in the genes encoding components of the RAS in mice or humans are associated with a broad spectrum of congenital anomalies of the kidney and urinary tract (CAKUT). These forms of CAKUT include renal papillary hypoplasia, hydronephrosis, duplicated collecting system, renal tubular dysgenesis, renal vascular abnormalities, and aberrant glomerulogenesis. Emerging evidence indicates that (pro)renin receptor (PRR), a novel component of the RAS, is essential for proper kidney development and that aberrant PRR signaling is causally linked to cardiovascular and renal disease. This paper describes the role of the RAS in kidney development and highlights emerging insights into the cellular and molecular mechanisms by which the PRR may regulate this critical morphogenetic process.
1. Introduction
1.1. Brief Overview of Kidney Development
During embryogenesis, the nephric duct (ND) is formed from the intermediate mesoderm on embryonic (E) day E22 in humans and E8 in mice [1]. The ND extends caudally and induces adjacent intermediate mesoderm to form two transient kidney types, pronephros and mesonephros. The pronephros degenerates in mammals, whereas the mesonephros involutes in females, but gives rise to male reproductive organs [1]. On 5th week of gestation in humans (E10.5 in mice), the caudal portion of the ND forms an epithelial outgrowth called the ureteric bud (UB). The metanephric kidney arises from two embryonic tissues: the UB and the metanephric mesenchyme (MM) [2, 3] (Figures 1(a)–1(d)). UB grows out from the ND, elongates, invades the MM, and then branches repeatedly within the mesenchyme to form the renal collecting system (the ureter, pelvis, calyces, and collecting ducts) [3–5]. Linear arrays of inner (medullary) collecting ducts converge centrally to form the papilla. Distal ureter subsequently translocates from the ND to fuse with the bladder which originates from the urogenital sinus (Figures 1(e)–1(g)) [6, 7]. Terminal UB tips induce surrounding MM-derived nephron progenitors to condense and then differentiate into nephrons (from the glomerulus to the distal tubule), thus forming the metanephric kidney (Figures 1(a)–1(d)) [3, 4].
Figure 1.
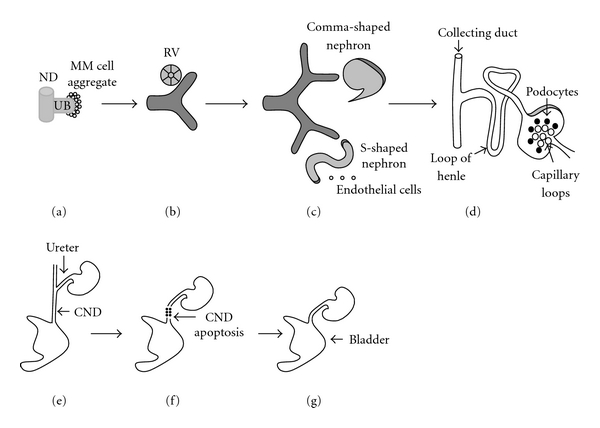
Schematic representation of normal development of the kidney and urinary tract. (a): Invasion of the metanephric mesenchyme (MM) by the ureteric bud (UB) on weeks 5-6 of gestation induces MM cells to aggregate around the UB tip. (a)–(c): UB outgrowth from the nephric duct (ND), its subsequent iterative branching (branching morphogenesis), and continuous condensation of the MM cells around emerging UB tips are induced primarily by reciprocal interactions among glial-derived neurotrophic factor (GDNF), its receptor c-Ret, and coreceptor GFRα1. (b): MM cell aggregates undergo mesenchymal-to-epithelial transformation (MET) to form the renal vesicle (RV) on weeks 6–36 of gestation. (c): RV elongates along the proximal-distal axis to form comma-shaped and then S-shaped nephron. Distal ends of S-shaped nephrons fuse with UB-derived collecting ducts, whereas proximal clefts form glomeruli. Endothelial cells migrate into the proximal cleft. UB branching occurs on weeks 6–22 of gestation. Formation of nascent nephrons and their patterning occur on weeks 6–36 of gestation. (d): Patterning of the S-shaped nephron and UB result in formation of mature nephron which contains glomerulat capillary tuft, podocytes, proximal tubule, loop of Henle, distal tubule, and collecting duct. (e). Ureter becomes patent and common ND (CND) fuses with cloaca on weeks 4-5 of gestation. (f): Apoptosis of the CND accounts for the positioning of the ureter (derived from proximal UB) in proximity of the urogenital sinus on weeks 5-6 of gestation. (g): Ureter fuses with the bladder by 6 weeks of gestation (with kind permission from Springer Science + Business Media: [82]). Please see text for details.
1.2. Wnt Signaling in Metanephric Kidney Development
Metanephric kidney development depends on reciprocal interactions of transcription and growth factors expressed in the metanephric mesenchyme, stroma and the UB [3, 8, 9]. The wingless (Wnts) are secreted glycoproteins fundamental for proper kidney development. Wnt ligands bind to extracellular domain of frizzled (Fz) seven trans-membrane domain receptors and, in some cases, the low-density lipoprotein (LRP) 5 and 6 coreceptors to activate distinct intracellular signaling cascades [10–12]. Wnt signaling regulates metanephric kidney development via canonical or noncanonical signaling pathways [13]. Binding of Wnt to its receptor leads to accumulation of β-catenin in the cytoplasm followed by translocation to the nucleus and interaction with the T-cell factor/lymphoid-enhancing factor (Tcf/Lef) family of transcription factors to regulate gene transcription [14]. In addition, β-catenin is an important component of cell-cell adherens junctions and interacts with the actin cytoskeleton [15]. The noncanonical Wnt signaling pathway consists of the planar cell polarity/convergent extension (PCP/CE) pathway and the Ca2+-releasing pathway [13]. PCP controls polarization of cells within the plane of the tissue, whereas CE directs intercalation of cells in an epithelial sheet to form a longer and narrower strip of the tissue [16]. Several Wnts are expressed in the discrete domains of the developing mouse kidney and play a critical role in proper metanephric organogenesis. Wnt6, Wnt7b, Wnt9b, and Wnt11 are expressed in the UB [17–19]. Wnt4 is expressed in the MM and Wnt2b in the cortical stroma [17, 20]. Of the Wnts expressed in the metanephros, Wnt2b, Wnt4, Wnt7b, and Wnt9b activate canonical pathway. Wnt signaling is essential for UB branching, nephrogenesis, and medullary patterning. Available data suggest that UB signals to the MM by secreting Wnt9b, a soluble growth factor, which acts via the canonical β-catenin to induce expression of fibroblast growth factor 8 (FGF8), LIM homeobox 1 (Lhx1) and Wnt4 in the MM [18, 21]. In turn, Wnt4 induces MM cells to undergo mesenchymal-to-epithelial transformation (MET) and differentiate into the nephron epithelia [21]. Genetic inactivation of Wnt9b or Wnt4 in mice leads to arrest of nephrogenesis at renal vesicle stage, and deficiency of Wnt9b causes severe defects in UB branching [18, 21]. UB-specific inactivation of β-catenin, the central effector of the canonical Wnt signaling pathway, causes aberrant UB branching and premature differentiation of collecting duct cells and results in renal hypodysplasia [22, 23]. In addition to directing UB branching and nephrogenesis via the canonical pathway, Wnt9b acts via a noncanonical Wnt pathway to induce PCP in UB-derived collecting ducts. Available evidence suggests that longitudinally oriented cell division (OCD) leads to collecting duct elongation without a change in diameter. In conditions in which collecting ducts dilate to form cysts (e.g., polycystic kidney disease), OCD is randomized, leading to a progressive increase in collecting duct diameter [24]. Mice that lack Wnt9b exhibit dilated collecting ducts, aberrant OCD of the collecting duct cells and develop renal cysts postnatally [25]. Wnt7b-mutant mice do not form renal medulla and papilla [26]. These defects are likely due to aberrant OCD and decreased survival of the medullary collecting duct cells [26].
2. The Renin-Angiotensin System
The renin-angiotensin system (RAS) plays a fundamental role in the regulation of arterial blood pressure, fluid/electrolyte homeostasis, and kidney development [27]. In the RAS, renin cleaves angiotensinogen (AGT) to generate Ang I [Ang-(1–10)] (Figure 2). Ang I is converted by angiotensin-converting enzyme (ACE) to Ang II [Ang-(1–8)], the most powerful effector peptide hormone of the RAS [28–30]. Ang II acts via two major G protein-coupled receptors (GPCR): AT1R and AT2R [29]. Most of hypertensinogenic and sodium-retaining actions of Ang II are attributed to the AT1R [31]. In contrast to the AT1R, the AT2R elicits vasodilation, promotes renal sodium excretion, and inhibits proliferation in mesangial cells [32–34]. ACE2 is a homologue of ACE which is abundantly expressed in the kidney and acts to counterbalance ACE activity by promoting Ang II degradation to the vasodilator peptide Ang-(1–7) [35, 36]. Ang-(1–7) acts via the GPCR Mas encoded by Mas protooncogene to oppose Ang II-AT1R-mediated effects [37, 38]. Renin is synthesized in juxtaglomerular cells of the afferent arterioles of the kidney as preprorenin [39]. The human renin gene encoding preprorenin is located on chromosome 1 [40]. Cleavage of a 23 amino acid signal peptide at carboxyl terminus of preprorenin generates prorenin. Prorenin is then converted to active renin by cleavage of 43-amino acid N-terminal prosegment by proteases [41, 42]. The kidney secretes both renin and prorenin into the peripheral circulation. Plasma levels of prorenin are approximately 10-fold higher than those of renin [43].
Figure 2.
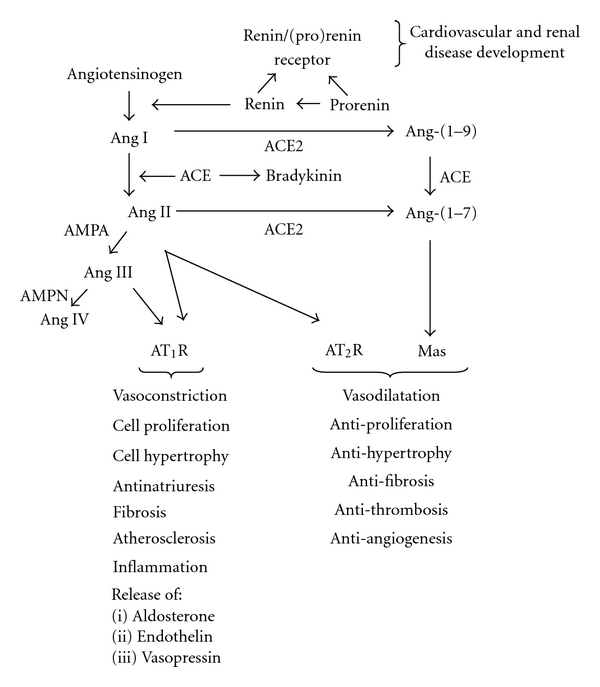
The Renin-Angiotensin System. Ang: angiotensin II. ACE: angiotensin-convertin enzyme, ACE2: angiotensin-converting enzyme 2, AMPA: aminopeptidase A. Please see text for details.
In addition to cleaving AGT, renin binds the (pro)renin receptor (PRR) [44, 45]. The PRR protein is a seven trans-membrane domain receptor encoded for by the ATP6AP2 (ATPase-associated protein2)/PRR gene (subsequently referred to as PRR) located on the X chromosome in humans [44]. The PRR protein exists in three forms: (1) A full-length 35–39 kDa form composed of 3 domains: extracellular, a single transmembrane, and a cytoplasmic, (2) A 28 kDa soluble form found in plasma and urine, and (3) A truncated form containing transmembrane and cytoplasmic domains [42, 45]. In addition to proteolytic activation via cleavage of the prosegment by proteases, prorenin may be activated by binding to the PRR and undergoing a conformational change that does not require removal of the prosegment [44].
2.1. Expression of the RAS Components in the Developing Metanephros
The developing metanephros expresses all the components of the RAS [46–48]. In the fetal mouse kidney, renin mRNA is first detected on E14.5 by in situ hybridization in a few scattered foci of cells [46]. By E15.5, renin is widely expressed in branches of the renal artery, interlobar, and arcuate arteries. In the human kidney, renin mRNA is detected at the vascular pole of the glomerulus and in arteries located next to glomeruli [48]. With fetal maturation, renin expression shifts to its mature localization in the juxtaglomerular cells [46, 48]. Studies in renin knockin reporter mice have demonstrated that renin-producing cells may originate from the mesenchyme at E11-E12 before vessel development has occurred [49]. Ontogeny studies have demonstrated that renal renin synthesis is highly activated during early postnatal development in rodents [47]. Because immunoreactive Ang II levels are higher in the fetal and newborn than adult mouse kidney [50, 51], renin is considered to be the rate-limiting factor for Ang II generation during metanephric development. In the adult rat kidney, PRR mRNA and protein are expressed in the collecting duct and the distal nephron [52]. In the CD, the PRR is most abundant at the apical surface of type A intercalated cells where it colocalizes with the vacuolar H+-ATPase [52]. In addition, PRR immunoreactivity is also detected in the podocytes, renal mesangial, vascular smooth muscle, and endothelial cells [53–55]. Even though PRR is expressed in Xenopus pronephros [56], the expression of the PRR gene and PRR protein during metanephric development remains to be determined.
2.2. Effect of Pharmacological or Genetic Interruption of the RAS on Kidney Development
Treatment of several animal species or humans with ACE inhibitors or AT1R antagonists during gestation or postnatal metanephrogenesis leads to renal tissue dysplasia [57]. A decrease in the number and size of glomeruli, delay in glomerulogenesis, a reduction in the number and length of the renal arteries accompanied by arterial thickening, tubular dilation and a hypoplastic papilla are observed [58]. Nephrotoxic effects of ACE inhibitors and AT 1R antagonists in humans include oligohydramnios and anuria [59, 60]. AGT-, renin-, ACE-, or AT 1R-deficient mice exhibit virtually identical phenotypes characterized by vascular hyperplasia, hydronephrosis, hypoplastic medulla, and papilla [61–66]. Functionally, Renin-, ACE-, and AT 1R-null animals are polyuric and have a reduced ability to concentrate urine [63–65]. Deletion of the AT 2R in mice causes ectopic UB budding from the nephric duct, duplicated collecting systems, and hydronephrosis [67]. Mutations in the genes encoding for AGT, renin, ACE, or AT 1R in humans are associated with renal tubular dysgenesis (RTD) [68, 69]. Kidneys of patients with RTD demonstrate reduced number of proximal tubules, collapsed collecting ducts, enlarged glomeruli, and thickened arteries [69]. Attempts to generate knockout mice with global deletion of the Prr failed [70]. Even though this observation precludes so far determination of the specific role for the PRR in kidney organogenesis, it also indicates that the PRR is essential during development. Targeted inactivation of the Prr gene in mouse cardiomyocytes causes cardiac tissue fibrosis, cardiomyocyte apoptosis, and death within the first several weeks of postnatal life from heart failure [71].
2.3. Signaling Pathways Downstream of the PRR
Even though the cytoplasmic domain of the PRR has no intrinsic kinase activity [48], in vitro studies demonstrate that binding of the PRR by renin or prorenin leads to activation of mitogen-activated protein (MAP) kinases such as Erk1/2 or p38 in renal mesangial and vascular smooth muscle (VSMC) cells and induces phosphorylation of phosphatidylinositol 3-kinase/Akt (PI3K/Akt) in human embryonic kidney (HEK293T) cells [72, 73]. The effect of the PRR on Erk1/2 phosphorylation in human monocytes is independent of Ang II or transactivation of the EGF receptor [74]. In contrast, induction of Erk1/2 and Akt phosphorylation by recombinant rat prorenin in cultured rat VSMCs is independent of Ang II, but dependent on phosphorylation of EGF receptor [75]. Since these changes are blocked by the PRR siRNA, activation of the EGF receptor, Erk1/2, and Akt in VSMC depends on PRR. The importance of the PRR-dependent Erk1/2 activation in brain development is evident from the observations that hypomorphic mutation in the PRR is causally linked to the absence of Erk1/2 phosphorylation and the development of X-linked mental retardation in humans [76].
2.4. Role of the PRR in Kidney and Cardiovascular Disease
A direct pathological role of the PRR in renal injury and cardiovascular disease is suggested by the findings of glomerulosclerosis, proteinuria, and elevated blood pressure in rats with ubiquitous transgenic overexpression of the human PRR (Table 1) [91]. Targeted overexpression of the Prr in the rat vasculature under the control of the mouse smooth muscle myosin heavy chain gene causes mild hypertension after six months of age [77]. Although transgenic overexpression of the prorenin, a major ligand for the PRR, in rats does not cause renal fibrosis, it leads to myocardial hypertrophy, proteinuria, and hypertension [92, 93]. Of interest, since hypertension is controlled by ACE inhibition, it may be due to the increased formation of Ang II [92, 93]. Double-transgenic mice that overexpress human prorenin in the liver and human angiotensinogen in the heart display a selective increase in Ang I content in the heart (but not the plasma) as compared to the single-transgenic mice [94]. These results suggest that circulating prorenin is taken up by tissues where it can contribute to the local synthesis of Ang peptides and tissue damage. Expression of the PRR mRNA and protein is increased in the myocardium and renal tubular, VSMC, and endothelial cells in rats with congestive heart failure due to coronary ligation [54]. Moreover, treatment of spontaneously hypertensive rats (SHRs) with a synthetic peptide that blocks prorenin binding to the PRR reduces renal and cardiac fibrosis [78, 95]. These data demonstrate that the PRR may contribute to the pathogenesis of heart failure and kidney tissue damage. An important role for the PRR in the pathogenesis of hypertension in humans is supported by the findings that a polymorphism in the PRR gene is associated with a high blood pressure in men (IVS5+169C>T) and left ventricular hypertrophy in women (+1513A>G) [80, 81].
Table 1.
Role of (pro)renin receptor (PRR) signaling in kidney development and disease.
| Model | Renal phenotype | Cardiovascular phenotype | Other phenotype | References |
|---|---|---|---|---|
| Global overexpression of human PRR in the rat | Glomerulosclerosis Proteinuria | Elevated blood pressure | Unknown | [54] |
| Targeted overexpression of human PRR in rat vasculature | Unknown | Elevated blood pressure and heart rate | Unknown | [77] |
| SHR rats treated with PRR blocker | Attenuation of renal fibrosis | Attenuation of cardiac fibrosis | Unknown | [78] |
| Mice with targeted deletion of Prr in cardiomyocytes | Unknown | Cardiac tissue Fibrosis Cardiomyocyt opoptosis | Unknown | [71] |
| Xenopus embryos treated with anti-PRR morpholino | Unknown | Unknown | Short body axis Impaired CE Small head Short tail Defects in eye pigmentation |
[79] [56] |
| Genetic polymorphism of PRR in humans | Unknown | Hypertension in men LVH in women | Unknown | [80] |
| Unknown | [81] | |||
| Hypomorphic PRR mutations in humans | Unknown | Unknown | X-linked mental retardation Epilepsy | [76] |
SHR: spontaneously hypertensive rats, CE: convergent extension, LVH: left ventricular hypertrophy, PRR: human PRR gene, PRR: human PRR protein, Prr: mouse PRR gene.
Although rare cases of human hypertension or CAKUT are due to mutations in single genes, the contribution of the genetic determinants in the vast majority of subjects with high blood pressure or CAKUT remains unknown [82, 96]. If common diseases such as hypertension or nonsyndromic cases of CAKUT are due to multiple gene variants with small effects, large study samples are needed to identify them. Within the last few years, several genome-wide association studies (GWAS), in which thousands of common genetic variants are analyzed for disease association, identified significant association of a limited number of genes with primary hypertension [97]. Further studies are needed to understand how the implicated genes contribute to such a complex multifactorial disease as primary hypertension. Despite identification of significant association of hypertension with variants of renin, ACE and AGTR1 genes in a study by Zhu et al. [98], the results of RAS gene-association studies are inconsistent [99]. With respect to CAKUT, broad phenotypic spectrum of renal system anomalies and variability in genotype-phenotype correlation demonstrate that pathogenesis of CAKUT is a complex process that depends on interplay of many factors [82, 100]. It is likely that well-powered studies utilizing total human exome capture and next-generation sequencing will identify single-gene defects leading to CAKUT.
2.5. Potential Roles for the PRR in Kidney Development
The mechanisms by which the PRR may regulate kidney development may involve changes in the expression of genes or transcription factors that are critical for metanephric organogenesis, physical interaction between the PRR and other receptors or proteins with established roles in renal ontogeny or function, activation of the intracellular signaling pathways, or other mechanisms (Figure 3).
Figure 3.
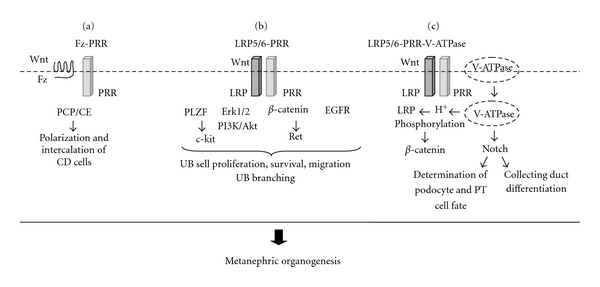
Proposed mechanisms mediating the effect of the (pro)renin receptor (PRR) in metanephric kidney development. (a): PRR interacts with Wnt receptor frizzled (Fz) to regulate polarization and intercalation of collecting duct (CD) cells via planar cell polarity (PCP)/convergent extension (CE) Wnt signaling pathway [16, 25, 79]. (b): PRR may regulate UB branching by (1) inhibiting c-kit transcription via promyelocytic zinc finger transcription factor (PLZF) [73, 83, 84] (2) induction of Erk1/2, PI3K/Akt or epidermal growth factor receptor (EGFR) phosphorylation [52, 72, 73, 75, 85–87], and (3) interaction with LRP5/6 Wnt coreceptor leading to activation of β-catenin and Ret gene expression [22, 23, 56]. (c): PRR interacts with LRP5/6 and V-ATPase to form a complex at the plasma membrane. Following endocytosis, V-ATPase generates a gradient of H+ ions that is essential for LRP5/6 phosphorylation and activation of β-catenin [56]. V-ATPase stimulates Notch signaling [88]. Notch acts to define the podocyte and proximal tubular cell fates [89] and regulates differentiation of the CD cells [90].
2.5.1. Interaction with the Promyelocytic Zinc Finger Transcription Factor (PLZF)
PLZF is a nuclear phosphoprotein which belongs to the POZ/zinc-finger family of transcription factors and is encoded by the Zfp145 gene. Zfp145-null mice exhibit aberrant expression of Hox genes, defects in limb, and axial skeletal patterning, whereas the kidney phenotype of these mice was not described [101]. Several observations support potential role for PLZF in kidney development. For example, Hox (Hox11) genes are necessary to specify the metanephric kidney identity from the intermediate mesoderm [102]. In patients with acute promyelocytic leukemia, PLZF fuses with the retinoic acid receptor α (RARα) and recruits histone deacetylase 1 (HDAC1) to render retinoic acid-target genes unresponsive to retinoic acid, an active form of vitamin A [103]. Both HDAC1 and retinoic acid are essential for embryo development. Genetic inactivation of HDAC1 in mice results in embryonic lethality before E10.5 due to severe proliferation defects [104], whereas RAR-mutant mice display renal dysplasia [105]. Concerning the RAS, inhibition of HDAC activity induces renin expression in the embryonic kidney [106, 107]. PRR interacts with the PLZF protein in HEK293 cells in vitro [73]. Moreover, treatment of HEK293 cells with renin causes nuclear translocation of PLZF followed by recruitment of PLZF to the promoters of the PRR and PI3K-p85αgenes. This leads to repression of PRR transcription and induction of PI3K gene expression [73]. Notably, inhibition of PI3K/Akt blocks UB branching induced by glial-derived neurotrophic factor (GDNF)/rearranged during transfection (Ret) ligand-receptor pair or by Ang II [85, 86].
In addition, PLZF inhibits transcription of c-kit in CD34+ hematopoietic progenitor cells (HPC) and in spermatogonia [83, 108]. Functionally, increased c-kit expression is necessary to sustain differentiation of these cells. Zfp145-null mice exhibit depletion of the proliferative spermatogonial compartment in the testis due to deregulated expression of c-kit, which controls a tight balance between spermatogonial self-renewal and differentiation [108]. c-kit is a receptor tyrosine kinase (RTK) for the stem cell factor (SCF), a key regulator of HPC proliferation, survival, and differentiation [83]. Notably, c-kit is expressed in the interstitial cell and angioblasts of the developing metanephros [84]. Moreover, antagonism of c-kit RTK activity inhibits UB branching and reduces the number of nephrons and renal angioblasts [84]. Since PLZF represses c-kit expression, loss of PLZF can cause dysregulation of the differentiation of c-kit-positive progenitors of renal interstitial cells and lead to renal hypodysplasia.
2.5.2. Interactions with the Canonical Wnt/β-Catenin Signaling
Another mechanism by which the PRR may control metanephric development is by the regulation of the canonical Wnt/β-catenin signaling. Inhibition of the PRR by small inhibitory RNA (siRNA) in HEK293T cells in vitro or by the PRR antisense morpholino in Xenopus embryos in vivo decreases luciferase reporter activity stimulated by canonical Wnt signaling [56]. The PRR binds to Fz8 and LRP6 in HEK293T cells transfected with the PRR expression vector. Activation of LRP6 and β-catenin signaling depends on extracellular, but not on cytoplasmic or transmembrane, domain of the PRR [56]. Given that canonical Wnt signaling is critical in metanephric organogenesis [22, 23], it is conceivable that direct interaction between the PRR and LRP6 plays an important role in the activation of canonical Wnt signaling via β-catenin to regulate kidney development. This possibility is supported by the observations that mutations in LRP4, which is known to antagonize LRP6-mediated activation of canonical Wnt signaling, are associated with CAKUT in humans with Cenani-Lenz syndrome (OMIM# 604270) [109].
2.5.3. Interactions with Noncanonical Wnt/PCP Signaling
In addition to its function in canonical Wnt signaling, PRR modulates Wnt/PCP pathway of the noncanonical Wnt signaling. Treatment of Xenopus embryos with anti-PRR morpholinos causes a short body axis and a broader expression domain of the notochord marker Xnot, a hallmark of impaired convergent extension movements [79]. Aberrant convergent extension (lateral intercalation) of the collecting duct and renal tubular cells may be causally linked to polycystic kidney disease [25]. In addition, Drosophila PRR interacts biochemically with Fz in HEK293T cells [79]. The Drosophila Fz receptor is required for PCP. Collectively, PRR mediates both the Wnt/PCP and the Wnt/β-catenin signaling pathways. These effects of the PRR are independent of renin [56, 79].
2.5.4. Interactions with V-ATPase
PRR may regulate kidney development or function via the V-ATPase (Figure 3). V-ATPase is a multiprotein complex localized in the kidney in intracellular organelles and at the plasma membrane of the intercalated collecting duct cells. Its major function is to pump protons to promote endocytosis [110]. Mutations in the genes encoding the kidney-specific isoforms B1 or A4 of V-ATPase in humans are responsible for inheritable forms of distal renal tubular acidosis, a disease characterized by elevated H+ concentrations in the plasma due to the impaired renal excretion of acid [111, 112]. Critical role for V-ATPase in development is evident from the observation that mutations in the genes encoding subunits C or D of V-ATPase in mice result in embryonic lethality [113, 114]. Mutations in subunits B1 or A3 of V-ATPase in mice cause metabolic acidosis and osteopetrosis, respectively [115, 116]. Variability in defects observed in these mice indicates that different subunits of V-ATPase have different functions and that subunits A3, A4, and B1 may be important in bone remodeling or differentiation of collecting duct cells involved in acid-base homeostasis.
In vitro studies demonstrate that the endogenous PRR binds to the endogenous V-ATPase subunits ATP6V0D1 and ATP6V0C in HEK293T cells [56]. Injection of a dominant-negative V-ATPase subunit E in Xenopus embryos synergizes with anti-PRR morpholino in inhibition of Wnt signaling. Phosphorylation of LRP6, which correlates with LRP6 activation, is inhibited in mouse P19 embryonal carcinoma cells treated with PRR- or V-ATPase siRNA [56]. These findings indicate that the PRR and V-ATPase interact functionally to inhibit Wnt signaling during Xenopus embryonic development in vivo.
Genetic inactivation of the Prr in cardiomyocytes in mice decreases expression of the V(O) subunits of V-ATPase, resulting in deacidification of the intracellular vesicles [71]. Thus, the PRR is also essential for vacuolar H+-ATPase assembly in murine cardiomyocytes in vivo. Mutations in VhaAC39, a V-ATPase subunit, in Drosophila are associated with the loss of Notch signaling [88]. Ligand binding to Notch receptor induces proteolytic cleavage and release of the intracellular domain of Notch which enters the cell nucleus to regulate target gene expression. Notch signaling exerts dual function during mouse metanephric development. Notch activity defines the podocyte and proximal tubular cell fates during segmentation of the S-shaped body [89]. In the collecting duct, Notch acts to increase the ratio of principal-to-intercalated cells and regulate urinary concentrating ability [90]. Thus, Notch signaling is required for the differentiation and functional maturation of the principal cells in murine renal collecting duct. Mutations in Notch2 in humans result in Alagille syndrome (OMIM# 610205) [117]. The renal phenotype in Alagille syndrome is characterized by hypodysplasia. Since PRR is most abundant at the apical surface of type A intercalated cells of the collecting duct, where it colocalizes with the vacuolar H+-ATPase, in the rat [52], the PRR may act in an autocrine fashion to regulate H+ transport. During metanephric development, PRR may promote differentiation of H+-secreting intercalated cells in the collecting duct. PRR located on the apical membrane of intercalated cells may be activated in a paracrine fashion by prorenin released by adjacent principal cells [118]. Additional evidence to implicate regulated intracellular acidification in mediating Wnt signaling is provided by the observations that Nhe2, a sodium/proton exchanger in Drosophila and a homologue to human NHE3, interacts genetically with Fz receptor to regulate Wnt/PCP signaling [119]. Together, these findings suggest that PRR cross-talks with V-ATPase in a renin-independent fashion to regulate both canonical Wnt/β-catenin and noncanonical Wnt/PCP signaling.
2.5.5. Regulation of Intracellular Signaling Pathways Critical for Metanephric Kidney Development
Another mechanism by which the PRR may modulate metanephric morphogenesis is via activation of downstream signaling pathways such as PI3K or Erk1/2 [72, 73]. This possibility is supported by an attenuation of Erk1/2 phosphorylation with PRR siRNA in collecting duct/distal tubule lineage Madin-Darby Canine Kidney (MDCK) cells in vitro [52]. An important role for Erk1/2 and PI3K in kidney development is demonstrated by the findings that inhibition of Erk1/2 decreases UB branching [120] and that antagonism of PI3K/Akt blocks directional migration of Ret-transfected MDCK cells in response to GDNF in vitro [85]. Critical role of GDNF and Ret in UB morphogenesis is evident from the findings that targeted inactivation of GDNF or Ret in mice results most frequently in bilateral renal agenesis due to a failure of UB outgrowth [121, 122]. Notably, tyrosine phosphorylation of the EGF receptor and activation of PI3K/Akt and Erk1/2 are also essential for Ang II-induced UB branching [86, 87].
3. Conclusions and Perspectives
The PRR is emerging as potential critical player in normal metanephric kidney development. It has become evident that the PRR has an evolutionary conserved role in bridging V-ATPases with canonical and noncanonical Wnt signaling. Acting via these or other intracellular pathways (e.g., Erk1/2, PI3K), the PRR may regulate segmentation of the proximal nephron and direct functional maturation of UB-derived collecting ducts. Even though significant progress has been made in defining the potential role of the PRR and specific contribution of PRR-dependent signal transduction in metanephric development, further evidence supporting the direct role for the PRR in metanephric organogenesis is needed. What are the perspectives for system-wide approaches to understand the role of the PRR in kidney organogenesis? In this regard, application of new genetic tools, such as conditional/tissue/cell-specific gene knockouts, genetic lineage tracing and fluorescent in vivo reporters of cell signaling, genome-wide analysis of gene regulatory networks (including epigenetic regulation) that control different aspects of kidney development (e.g., microarray, ChIP-Seq) should provide important insights in understanding molecular mechanisms by which the PRR may direct normal and abnormal metanephric kidney development. Defining molecular aberrations leading to CAKUT in animals and humans with mutations in the PRR will uncover biomarkers that can be used for early diagnosis or prevention of renal system anomalies in children. Finally, establishment of shared large biorepositories of patients encompassing a wide spectrum of CAKUT phenotypes for molecular, genetic, and translational studies will define clinically relevant mutations in the PRR, its ligands, and interacting genes. The evolution of our understanding of the cellular and molecular mechanisms by which intact PRR controls metanephric kidney development may provide targets for improved diagnosis and prevention of CAKUT in children.
References
- 1.Saxen L. Organogenesis of the Kidney. Cambridge, UK: Cambridge University Press; 1987. [Google Scholar]
- 2.Schedl A. Renal abnormalities and their developmental origin. Nature Reviews Genetics. 2007;8(10):791–802. doi: 10.1038/nrg2205. [DOI] [PubMed] [Google Scholar]
- 3.Costantini F, Kopan R. Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Developmental Cell. 2010;18(5):698–712. doi: 10.1016/j.devcel.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekblom P. Developmentally regulated conversion of mesenchyme to epithelium. FASEB Journal. 1989;3(10):2141–2150. doi: 10.1096/fasebj.3.10.2666230. [DOI] [PubMed] [Google Scholar]
- 5.Grobstein C. Inductive epithlio mesenchymal interaction in cultured organ rudiments of the mouse metanephros. Science. 1953;118(2):52–55. doi: 10.1126/science.118.3054.52. [DOI] [PubMed] [Google Scholar]
- 6.Batourina E, Choi C, Paragas N, et al. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nature Genetics. 2002;32(1):109–115. doi: 10.1038/ng952. [DOI] [PubMed] [Google Scholar]
- 7.Batourina E, Tsai S, Lambert S, et al. Apoptosis induced by vitamin A signaling is crucial for connecting the ureters to the bladder. Nature Genetics. 2005;37(10):1082–1089. doi: 10.1038/ng1645. [DOI] [PubMed] [Google Scholar]
- 8.Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Seminars in Nephrology. 2009;29(4):321–337. doi: 10.1016/j.semnephrol.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dressler GR. The specification and maintenance of renal cell types by epigenetic factors. Organogenesis. 2009;5(2):73–82. doi: 10.4161/org.5.2.8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CM, Struhl G. Wingless transduction by the Frizzled and Frizzled2 proteins of Drosophila. Development. 1999;126(23):5441–5452. doi: 10.1242/dev.126.23.5441. [DOI] [PubMed] [Google Scholar]
- 11.Bhanot P, Brink M, Samos CH, et al. A new member of the frizzled family from Drosophila functions as a wingless receptor. Nature. 1996;382(6588):225–231. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 12.Bhanot P, Fish M, Jemison JA, Nusse R, Nathans J, Cadigan KM. Frizzled and DFrizzled-9 function as redundant receptors for wingless during Drosophila embryonic development. Development. 1999;126(18):4175–4186. doi: 10.1242/dev.126.18.4175. [DOI] [PubMed] [Google Scholar]
- 13.Merkel CE, Karner CM, Carroll TJ. Molecular regulation of kidney development: is the answer blowing in the Wnt? Pediatric Nephrology. 2007;22(11):1825–1838. doi: 10.1007/s00467-007-0504-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arce L, Yokoyama NN, Waterman ML. Diversity of LEF/TCF action in development and disease. Oncogene. 2006;25(57):7492–7504. doi: 10.1038/sj.onc.1210056. [DOI] [PubMed] [Google Scholar]
- 15.Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Developmental Cell. 2006;11(5):601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karner C, Wharton KA, Carroll TJ. Planar cell polarity and vertebrate organogenesis. Seminars in Cell and Developmental Biology. 2006;17(2):194–203. doi: 10.1016/j.semcdb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Kispert A, Vainio S, Shen L, Rowitch DH, McMahon AP. Proteoglycans are required for maintenance of Wnt-11 expression in the ureter tips. Development. 1996;122(11):3627–3637. doi: 10.1242/dev.122.11.3627. [DOI] [PubMed] [Google Scholar]
- 18.Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Developmental Cell. 2005;9(2):283–292. doi: 10.1016/j.devcel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 19.Itäranta P, Lin Y, Peräsaari J, Roël G, Destrée O, Vainio S. Wnt-6 is expressed in the ureter bud and induces kidney tubule development in vitro. Genesis. 2002;32(4):259–268. doi: 10.1002/gene.10079. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y, Liu A, Zhang R, et al. Induction of ureter branching as a response to Wnt-2b signaling during early kidney organogenesis. Developmental Dynamics. 2001;222(1):26–39. doi: 10.1002/dvdy.1164. [DOI] [PubMed] [Google Scholar]
- 21.Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1996;372(6507):679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 22.Marose TD, Merkel CE, McMahon AP, Carroll TJ. β-catenin is necessary to keep cells of ureteric bud/Wolffian duct epithelium in a precursor state. Developmental Biology. 2008;314(1):112–126. doi: 10.1016/j.ydbio.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bridgewater D, Cox B, Cain J, et al. Canonical WNT/β-catenin signaling is required for ureteric branching. Developmental Biology. 2008;317(1):83–94. doi: 10.1016/j.ydbio.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Fischer E, Legue E, Doyen A, et al. Defective planar cell polarity in polycystic kidney disease. Nature Genetics. 2006;38(1):21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 25.Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nature Genetics. 2009;41(7):793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, Carroll TJ, Rajagopal J, Kobayashi A, Ren Q, McMahon AP. A Wnt7b-dependent pathway regulates the orientation of epithelial cell division and establishes the cortico-medullary axis of the mammalian kidney. Development. 2009;136(1):161–171. doi: 10.1242/dev.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobori H, Ozawa Y, Suzaki Y, et al. Young scholars award lecture: intratubular angiotensinogen in hypertension and kidney diseases. American Journal of Hypertension. 2006;19(5):541–550. doi: 10.1016/j.amjhyper.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brasier AR, Li J. Mechanisms for inducible control of angiotensinogen gene transcription. Hypertension. 1996;27(3):465–475. doi: 10.1161/01.hyp.27.3.465. [DOI] [PubMed] [Google Scholar]
- 29.Navar LG. The kidney in blood pressure regulation and development of hypertension. Medical Clinics of North America. 1997;81(5):1165–1178. doi: 10.1016/s0025-7125(05)70573-3. [DOI] [PubMed] [Google Scholar]
- 30.Paul M, Mehr AP, Kreutz R. Physiology of local renin-angiotensin systems. Physiological Reviews. 2006;86(3):747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 31.Ito M, Oliverio MI, Mannon PJ, et al. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(8):3521–3525. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siragy HM, Carey RM. The subtype-2 (AT) angiotensin receptor mediates renal production of nitric oxide in conscious rats. Journal of Clinical Investigation. 1997;100(2):264–269. doi: 10.1172/JCI119531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goto M, Mukoyama M, Suga SI, et al. Growth-dependent induction of angiotensin II type 2 receptor in rat mesangial cells. Hypertension. 1997;30(3):358–362. doi: 10.1161/01.hyp.30.3.358. [DOI] [PubMed] [Google Scholar]
- 34.Gross V, Schunck WH, Honeck H, et al. Inhibition of pressure natriuresis in mice lacking the AT receptor. Kidney International. 2000;57(1):191–202. doi: 10.1046/j.1523-1755.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- 35.Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circulation Research. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 36.Brosnihan KB, Li P, Ferrario CM. Angiotensin-(1-7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension. 1996;27(3):523–528. doi: 10.1161/01.hyp.27.3.523. [DOI] [PubMed] [Google Scholar]
- 37.Santos RAS, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos RA, Ferreira AJ. Angiotensin-(1-7) and the renin-angiotensin system. Current Opinion in Nephrology and Hypertension. 2007;16(2):122–128. doi: 10.1097/MNH.0b013e328031f362. [DOI] [PubMed] [Google Scholar]
- 39.Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiological Reviews. 1990;70(4):1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- 40.Miyazaki H, Fukamizu A, Hirose S. Structure of the human renin gene. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(19):5999–6003. doi: 10.1073/pnas.81.19.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schweda F, Friis U, Wagner C, Skott O, Kurtz A. Renin release. Physiology. 2007;22(5):310–319. doi: 10.1152/physiol.00024.2007. [DOI] [PubMed] [Google Scholar]
- 42.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009;53(6):1077–1082. doi: 10.1161/HYPERTENSIONAHA.108.127258. [DOI] [PubMed] [Google Scholar]
- 43.Jan Danser AH, Derkx FHM, Schalekamp MADH, Hense HW, Riegger GAJ, Schunkert H. Determinants of interindividual variation of renin and prorenin concentrations: evidence for a sexual dimorphism of (pro)renin levels in humans. Journal of Hypertension. 1998;16(6):853–862. doi: 10.1097/00004872-199816060-00017. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. Journal of Clinical Investigation. 2002;109(11):1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen G, Muller DN. The biology of the (pro)renin receptor. Journal of the American Society of Nephrology. 2010;21(1):18–23. doi: 10.1681/ASN.2009030300. [DOI] [PubMed] [Google Scholar]
- 46.Jones CA, Sigmund CD, McGowan RA, Kane-Haas CM, Gross KW. Expression of murine renin genes during fetal development. Molecular Endocrinology. 1990;4(3):375–383. doi: 10.1210/mend-4-3-375. [DOI] [PubMed] [Google Scholar]
- 47.Gomez RA, Lynch KR, Sturgill BC, et al. Distribution of renin mRNA and its protein in the developing kidney. American Journal of Physiology. 1989;257(5):F850–F858. doi: 10.1152/ajprenal.1989.257.5.F850. [DOI] [PubMed] [Google Scholar]
- 48.Schütz S, Le Moullec JM, Corvol P, Gasc JM. Early expression of all the components of the renin-angiotensin-system in human development. American Journal of Pathology. 1996;149(6):2067–2079. [PMC free article] [PubMed] [Google Scholar]
- 49.Sequeira Lopez MLS, Pentz ES, Robert B, Abrahamson DR, Gomez RA. Embryonic origin and lineage of juxtaglomerular cells. American Journal of Physiology. 2001;281(2):F345–F356. doi: 10.1152/ajprenal.2001.281.2.F345. [DOI] [PubMed] [Google Scholar]
- 50.Yosipiv IV, El-Dahr SS. Activation of angiotensin-generating systems in the developing rat kidney. Hypertension. 1996;27(2):281–286. doi: 10.1161/01.hyp.27.2.281. [DOI] [PubMed] [Google Scholar]
- 51.Miyazaki Y, Tsuchida S, Nishimura H, et al. Angiotensin induces the urinary peristaltic machinery during the perinatal period. Journal of Clinical Investigation. 1998;102(8):1489–1497. doi: 10.1172/JCI4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Advani A, Kelly DJ, Cox AJ, et al. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-atpase in the kidney. Hypertension. 2009;54(2):261–269. doi: 10.1161/HYPERTENSIONAHA.109.128645. [DOI] [PubMed] [Google Scholar]
- 53.Sakoda M, Ichihara A, Kurauchi-Mito A, et al. Aliskiren inhibits intracellular angiotensin ii levels without affecting (Pro)renin Receptor Signals in Human Podocytes. American Journal of Hypertension. 2010;23(5):575–580. doi: 10.1038/ajh.2009.273. [DOI] [PubMed] [Google Scholar]
- 54.Hirose T, Mori N, Totsune K, et al. Gene expression of (pro)renin receptor is upregulated in hearts and kidneys of rats with congestive heart failure. Peptides. 2009;30(12):2316–2322. doi: 10.1016/j.peptides.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 55.Batenburg WW, Krop M, Garrelds IM, et al. Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. Journal of Hypertension. 2007;25(12):2441–2453. doi: 10.1097/HJH.0b013e3282f05bae. [DOI] [PubMed] [Google Scholar]
- 56.Cruciat CM, Ohkawara B, Acebron SP, et al. Requirement of prorenin receptor and vacuolar H-ATPase-mediated acidification for Wnt signaling. Science. 2010;327(5964):459–463. doi: 10.1126/science.1179802. [DOI] [PubMed] [Google Scholar]
- 57.Yoo KH, Wolstenholme JT, Chevalier RL. Angiotensin-converting enzyme inhibition decreases growth factor expression in the neonatal rat kidney. Pediatric Research. 1997;42(5):588–592. doi: 10.1203/00006450-199711000-00006. [DOI] [PubMed] [Google Scholar]
- 58.Tufro-McRreddie A, Romano LM, Harris JM, Ferder L, Gomez RA. Angiotensin II regulates nephrogenesis and renal vascular development. American Journal of Physiology. 1995;269(1):F110–F115. doi: 10.1152/ajprenal.1995.269.1.F110. [DOI] [PubMed] [Google Scholar]
- 59.Schaefer C. Angiotensin II-receptor-antagonists: further evidence of fetotoxicity but not teratogenicity. Birth Defects Research A. 2003;67(8):591–594. doi: 10.1002/bdra.10081. [DOI] [PubMed] [Google Scholar]
- 60.Tabacova S, Little R, Tsong Y, Vega A, Kimmel CA. Adverse pregnancy outcomes associated with maternal enalapril antihypertensive treatment. Pharmacoepidemiology and Drug Safety. 2003;12(8):633–646. doi: 10.1002/pds.796. [DOI] [PubMed] [Google Scholar]
- 61.Nagata M, Tanimoto K, Fukamizu A, et al. Nephrogenesis and renovascular development in angiotensinogen-deficient mice. Laboratory Investigation. 1996;75(5):745–753. [PubMed] [Google Scholar]
- 62.Niimura F, Labosky PA, Kakuchi J, et al. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. Journal of Clinical Investigation. 1995;96(6):2947–2954. doi: 10.1172/JCI118366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi N, Lopez MLSS, Cowhig JE, et al. Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. Journal of the American Society of Nephrology. 2005;16(1):125–132. doi: 10.1681/ASN.2004060490. [DOI] [PubMed] [Google Scholar]
- 64.Esther CR, Jr., Howard TE, Marino EM, Goddard JM, Capecchi MR, Bernstein KE. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Laboratory Investigation. 1996;74(5):953–965. [PubMed] [Google Scholar]
- 65.Oliverio MI, Kim HS, Ito M, et al. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15496–15501. doi: 10.1073/pnas.95.26.15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsuchida S, Matsusaka T, Chen X, et al. Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. Journal of Clinical Investigation. 1998;101(4):755–760. doi: 10.1172/JCI1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Srougi M, Nesrallah LJ, Kauffmann JR, Nesralleh A, Leite KRM, Soloway MS. Angiotensin type II receptor expression and ureteral budding. Journal of Urology. 2001;166(5):1848–1852. [PubMed] [Google Scholar]
- 68.Gribouval O, Gonzales M, Neuhaus T, et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nature Genetics. 2005;37(9):964–968. doi: 10.1038/ng1623. [DOI] [PubMed] [Google Scholar]
- 69.Lacoste M, Cai Y, Guicharnaud L, et al. Renal tubular dysgenesis, a not uncommon autosomal recessive disorder leading to oligohydramnios: role of the renin-angiotensin system. Journal of the American Society of Nephrology. 2006;17(8):2253–2263. doi: 10.1681/ASN.2005121303. [DOI] [PubMed] [Google Scholar]
- 70.Sihn G, Rousselle A, Vilianovitch L, Burckle C, Bader M. Physiology of the (pro)renin receptor: wnt of change? Kidney International. 2010;78(3):246–256. doi: 10.1038/ki.2010.151. [DOI] [PubMed] [Google Scholar]
- 71.Kinouchi K, Ichihara A, Sano M, et al. The (pro)renin receptor/atp6ap2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circulation Research. 2010;107(1):30–34. doi: 10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
- 72.Huang Y, Wongamorntham S, Kasting J, et al. Renin increases mesangial cell transforming growth factor-β1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney International. 2006;69(1):105–113. doi: 10.1038/sj.ki.5000011. [DOI] [PubMed] [Google Scholar]
- 73.Schefe JH, Menk M, Reinemund J, et al. A novel signal transduction cascade involving direct physical interaction of the renin/prorenin receptor with the transcription factor promyelocytic zinc finger protein. Circulation Research. 2006;99(12):1355–1366. doi: 10.1161/01.RES.0000251700.00994.0d. [DOI] [PubMed] [Google Scholar]
- 74.Feldt S, Batenburg WW, Mazak I, et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension. 2008;51(3):682–688. doi: 10.1161/HYPERTENSIONAHA.107.101444. [DOI] [PubMed] [Google Scholar]
- 75.Liu G, Hitomi H, Hosomi N, et al. Prorenin induces vascular smooth muscle cell proliferation and hypertrophy via epidermal growth factor receptor-mediated extracellular signal-regulated kinase and Akt activation pathway. Journal of Hypertension. 2011;29(4):696–705. doi: 10.1097/HJH.0b013e328343c62b. [DOI] [PubMed] [Google Scholar]
- 76.Ramser J, Abidi FE, Burckle CA, et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Human Molecular Genetics. 2005;14(8):1019–1027. doi: 10.1093/hmg/ddi094. [DOI] [PubMed] [Google Scholar]
- 77.Burcklé CA, Danser AHJ, Müller DN, et al. Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension. 2006;47(3):552–556. doi: 10.1161/01.HYP.0000199912.47657.04. [DOI] [PubMed] [Google Scholar]
- 78.Ichihara A, Kaneshiro Y, Takemitsu T, et al. Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension. 2006;47(5):894–900. doi: 10.1161/01.HYP.0000215838.48170.0b. [DOI] [PubMed] [Google Scholar]
- 79.Buechling T, Bartscherer K, Ohkawara B, et al. Wnt/Frizzled signaling requires dPRR, the Drosophila homolog of the prorenin receptor. Current Biology. 2010;20(14):1263–1268. doi: 10.1016/j.cub.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 80.Hirose T, Hashimoto M, Totsune K, et al. Association of (pro)renin receptor gene polymorphism with blood pressure in Japanese men: the Ohasama study. American Journal of Hypertension. 2009;22(3):294–299. doi: 10.1038/ajh.2008.357. [DOI] [PubMed] [Google Scholar]
- 81.Hirose T, Hashimoto M, Totsune K, et al. Association of (pro)renin receptor gene polymorphisms with lacunar infarction and left ventricular hypertrophy in Japanese women: the Ohasama study. Hypertension Research. 2011;34(4):530–535. doi: 10.1038/hr.2010.274. [DOI] [PubMed] [Google Scholar]
- 82.Song R, Yosypiv IV. Genetics of congenital anomalies of the kidney and urinary tract. Pediatric Nephrology. 2011;26(3):353–364. doi: 10.1007/s00467-010-1629-4. [DOI] [PubMed] [Google Scholar]
- 83.Spinello I, Quaranta MT, Pasquini L, et al. PLZF-mediated control on c-kit expression in CD34(+) cells and early erythropoiesis. Oncogene. 2009;28(23):2276–2288. doi: 10.1038/onc.2009.87. [DOI] [PubMed] [Google Scholar]
- 84.Schmidt-Ott KM, Masckauchan TNH, Chen X, et al. β-catenin/TCF/Lef controls a differentiation-associated transcriptional program in renal epithelial progenitors. Development. 2007;134(17):3177–3190. doi: 10.1242/dev.006544. [DOI] [PubMed] [Google Scholar]
- 85.Tang MJ, Cai Y, Tsai SIJ, Wang YK, Dressler GR. Ureteric bud outgrowth in response to RET activation is mediated by phosphatidylinositol 3-kinase. Developmental Biology. 2002;243(1):128–136. doi: 10.1006/dbio.2001.0557. [DOI] [PubMed] [Google Scholar]
- 86.Song R, Spera M, Garrett C, Yosypiv IV. Angiotensin II-induced activation of c-Ret signaling is critical in ureteric bud branching morphogenesis. Mechanisms of Development. 2010;127(1-2):21–27. doi: 10.1016/j.mod.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yosypiv IV, Schroeder M, El-Dahr SS. AT1R-EGFR crosstalk regulates ureteric bud branching morphogenesis. Journal of the American Society of Nephrology. 2006;17(4):1005–1014. doi: 10.1681/ASN.2005080803. [DOI] [PubMed] [Google Scholar]
- 88.Yan Y, Denef N, Schüpbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Developmental Cell. 2009;17(3):387–402. doi: 10.1016/j.devcel.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng HT, Kim M, Valerius MT, et al. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134(4):801–811. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jeong HW, Un SJ, Koo BK, et al. Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. Journal of Clinical Investigation. 2009;119(11):3290–3300. doi: 10.1172/JCI38416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaneshiro Y, Ichihara A, Sakoda M, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. Journal of the American Society of Nephrology. 2007;18(6):1789–1795. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 92.Peters B, Grisk O, Becher B, et al. Dose-dependent titration of prorenin and blood pressure in Cyp1a1ren-2 transgenic rats: absence of prorenin-induced glomerulosclerosis. Journal of Hypertension. 2008;26(1):102–109. doi: 10.1097/HJH.0b013e3282f0ab66. [DOI] [PubMed] [Google Scholar]
- 93.Mercure C, Prescott G, Lacombe MJ, Silversides DW, Reudelhuber TL. Chronic increases in circulating prorenin are not associated with renal or cardiac pathologies. Hypertension. 2009;53(6):1062–1069. doi: 10.1161/HYPERTENSIONAHA.108.115444. [DOI] [PubMed] [Google Scholar]
- 94.Prescott G, Silversides DW, Reudelhuber TL. Tissue activity of circulating prorenin. American Journal of Hypertension. 2002;15(3):280–285. doi: 10.1016/s0895-7061(01)02284-1. [DOI] [PubMed] [Google Scholar]
- 95.Ichihara A, Kaneshiro Y, Takemitsu T, et al. Contribution of nonproteolytically activated prorenin in glomeruli to hypertensive renal damage. Journal of the American Society of Nephrology. 2006;17(9):2495–2503. doi: 10.1681/ASN.2005121278. [DOI] [PubMed] [Google Scholar]
- 96.Lifton RP. Genetic determinants of human hypertension. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(19):8545–8551. doi: 10.1073/pnas.92.19.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Newton-Cheh C, Johnson T, Gateva V, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nature Genetics. 2009;41(6):666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu X, Chang YPC, Yan D, et al. Associations between hypertension and genes in the renin-angiotensin system. Hypertension. 2003;41(5):1027–1034. doi: 10.1161/01.HYP.0000068681.69874.CB. [DOI] [PubMed] [Google Scholar]
- 99.Schmidt S, Van Hooft IMS, Grobbee DE, Ganten D, Ritz E. Polymorphism of the angiotensin I converting enzyme gene is apparently not related to high blood pressure: Dutch hypertension and offspring study. Journal of Hypertension. 1993;11(4):345–348. doi: 10.1097/00004872-199304000-00003. [DOI] [PubMed] [Google Scholar]
- 100.Hildebrandt F. Genetic kidney diseases. The Lancet. 2010;375(9722):1287–1295. doi: 10.1016/S0140-6736(10)60236-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barna M, Hawe N, Lee N, Pandolfi PP. Plzf regulates limb and axial skeletal patterning. Nature Genetics. 2000;25(2):166–172. doi: 10.1038/76014. [DOI] [PubMed] [Google Scholar]
- 102.Dressler GR. Advances in early kidney specification, development and patterning. Development. 2009;136(23):3863–3874. doi: 10.1242/dev.034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391(6669):815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 104.Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO Journal. 2002;21(11):2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rosselot C, Spraggon L, Chia I, et al. Non-cell-autonomous retinoid signaling is crucial for renal development. Development. 2010;137(2):283–292. doi: 10.1242/dev.040287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pentz ES, Sequeira Lopez MLS, Cordaillat M, Gomez RA. Identity of the renin cell is mediated by cAMP and chromatin remodeling: an in vitro model for studying cell recruitment and plasticity. American Journal of Physiology. 2008;294(2):H699–H707. doi: 10.1152/ajpheart.01152.2007. [DOI] [PubMed] [Google Scholar]
- 107.Song R, Van Buren T, Yosypiv IV. Histone deacetylases are critical regulators of the renin-angiotensin system during ureteric bud branching morphogenesis. Pediatric Research. 2010;67(6):573–578. doi: 10.1203/PDR.0b013e3181da477c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Filipponi D, Hobbs RM, Ottolenghi S, et al. Repression of kit expression by Plzf in germ cells. Molecular and Cellular Biology. 2007;27(19):6770–6781. doi: 10.1128/MCB.00479-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li Y, Pawlik B, Elcioglu N, et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. American Journal of Human Genetics. 2010;86(5):696–706. doi: 10.1016/j.ajhg.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Toei M, Saum R, Forgac M. Regulation and isoform function of the V-ATPases. Bochemistry. 2010;49(23):4715–4723. doi: 10.1021/bi100397s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nature Genetics. 1999;21(1):84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 112.Smith AN, Skaug J, Choate KA, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nature Genetics. 2000;26(1):71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- 113.Inoue H, Noumi T, Nagata M, Murakami H, Kanazawa H. Targeted disruption of the gene encoding the proteolipid subunit of mouse vacuolar H-ATPase leads to early embryonic lethality. Biochimica et Biophysica Acta. 1999;1413(3):130–138. doi: 10.1016/s0005-2728(99)00096-1. [DOI] [PubMed] [Google Scholar]
- 114.Miura GI, Froelick GJ, Marsh DJ, Stark KL, Palmiter RD. The d subunit of the vacuolar ATPase (Atp6d) is essential for embryonic development. Transgenic Research. 2003;12(1):131–133. doi: 10.1023/a:1022118627058. [DOI] [PubMed] [Google Scholar]
- 115.Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nature Genetics. 1999;23(4):447–451. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 116.Finberg KE, Wagner GA, Bailey MA, et al. The B1-subunit of the H ATPase is required for maximal urinary acidification. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(38):13616–13621. doi: 10.1073/pnas.0506769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. American Journal of Human Genetics. 2006;79(1):169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG. Collecting duct renin: a major player in angiotensin II-dependent hypertension. Journal of the American Society of Hypertension. 2009;3(2):96–104. doi: 10.1016/j.jash.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Simons M, Gault WJ, Gotthardt D, et al. Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nature Cell Biology. 2009;11(3):286–294. doi: 10.1038/ncb1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fisher CE, Michael L, Barnett MW, Davies JA. Erk MAP kinase regulates branching morphogenesis in the developing mouse kidney. Development. 2001;128(21):4329–4338. doi: 10.1242/dev.128.21.4329. [DOI] [PubMed] [Google Scholar]
- 121.Sánchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382(6586):70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 122.Schuchardt A, D’Agati V, Pachnis V, Costantin F. Renal agenesis and hypodysplasia in ret-k mutant mice result from defects in ureteric bud development. Development. 1996;122(6):1919–1929. doi: 10.1242/dev.122.6.1919. [DOI] [PubMed] [Google Scholar]