

Abstract
We describe a technique, sequence-tagged microsatellite profiling (STMP), to rapidly generate large numbers of simple sequence repeat (SSR) markers from genomic or cDNA. This technique eliminates the need for library screening to identify SSR-containing clones and provides an ∼25-fold increase in sequencing throughput compared to traditional methods. STMP generates short but characteristic nucleotide sequence tags for fragments that are present within a pool of SSR amplicons. These tags are then ligated together to form concatemers for cloning and sequencing. The analysis of thousands of tags gives rise to a representational profile of the abundance and frequency of SSRs within the DNA pool, from which low copy sequences can be identified. As each tag contains sufficient nucleotide sequence for primer design, their conversion into PCR primers allows the amplification of corresponding full-length fragments from the pool of SSR amplicons. These fragments permit the full characterisation of a SSR locus and provide flanking sequence for the development of a microsatellite marker. Alternatively, sequence tag primers can be used to directly amplify corresponding SSR loci from genomic DNA, thereby reducing the cost of developing a microsatellite marker to the synthesis of just one sequence-specific primer. We demonstrate the utility of STMP by the development of SSR markers in bread wheat.
INTRODUCTION
Microsatellites, or simple sequence repeats (SSRs), are molecular markers widely used for DNA fingerprinting, paternity testing, linkage map construction and population genetic studies. Based on tandem repeats of short (1–6 bp) DNA sequences, these markers are highly polymorphic due to variation in the number of repeat units (1). The repeat length at specific SSR loci is easily assayed by PCR using primers specific to conserved regions flanking the repeat array. The hypervariability and co-dominance of SSRs, their dispersion throughout genomes and suitability for automation are the principle reasons for their wide utility (2–4).
A major limitation of SSRs is the time and cost required to isolate and characterise each locus when pre-existing DNA sequence is not available. Typically, this process requires the construction and screening of a genomic library of size-selected DNA fragments with SSR-specific probes, followed by DNA sequencing of isolated positive clones, PCR primer synthesis and testing (5,6). Only after this can the copy number, informativeness and chromosomal position of SSR loci be determined. Significant cost and time savings could be made if library screening was eliminated and sequence information for more than one SSR locus was obtained from each plasmid clone. We report here a technique that employs the principles of serial analysis of gene expression (SAGE) (7) to generate a sequence tag library for the rapid characterisation of SSR loci within a genome of interest, from which microsatellite markers can be developed.
MATERIALS AND METHODS
Preparation of amplified PstI–MseI restriction fragments
One microgram of genomic DNA isolated from hexaploid wheat (Triticum aestivum L. cv. Halberd) was digested for 3 h at 37°C with 5 U each of MseI and PstI in 50 µl of 1× New England Biolabs (NEB) buffer II and 100 ng/µl BSA. Next, 5 and 50 pmol PstI and MseI adapters, respectively, were added and the mixture was heated at 45°C for 5 min to melt any cohesive adapter termini that had annealed. This step prevented self-ligation of the phosphorylated adapters before they could become attached to the restriction fragments. A solution containing 1× NEB buffer II, 100 ng/µl BSA, 6 mM ATP and 1 U T4 ligase was then added to give a total volume of 60 µl and incubation was continued for a further 1 h at 37°C, then at room temperature for 2 h. The DNA fragments were purified with a QiaQuick PCR purification kit (Qiagen), eluted in 50 µl of 1× NEB exonuclease III buffer containing 100 U exonuclease III and incubated overnight at 37°C. The mixture was heated at 70°C for 20 min to inactivate the exonuclease. Adapters were prepared by incubating equimolar amounts of two synthetic oligomers in annealing buffer (10 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA) at 65°C for 10 min, followed by slow cooling to room temperature over a 2 h period. The sequence of the PstI adapter sense strand was GCC TCG GAA GCC TCA GTC CCG ATG TCT TCG ATG CAC CTG TAT GGC CCT TAG CAT TGA ATT CGT GCA-OH and that of the antisense strand was phos-CGA ATT CAA TGC TAA GGG CCA TAC AGG TGC ATC GAA GAC ATC GGG ACT GAG GCT TCC* G*A*G* G-OH. The sequence of the MseI adapter sense strand was GAC GAG CAA GGC TCT CAC AGG AGC AGG CCA GGA G-OH and that of the antisense strand was phos-TAC TCC TGG CCT G*C*T* C*C-NH2. Nucleotide residues followed by an asterisk indicate phosphorothioate linkages. Oligomers were purified by PAGE to enrich for full-length product.
Amplification of PstI–MseI restriction fragments was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer (Advanced Biotechnologies), 1.5 mM MgCl2, 5 pmol each MseI (GAG CAA GGC TCT CAC A-OH) and PstI (CTC GGA AGC CTC AGT C-OH) adapter primers, 2 µl of digested/ligated template DNA and 1 U Taq DNA polymerase (Advanced Biotechnologies). PCR amplification was performed for 20 cycles (60 s at 92°C, 60 s at 56°C and 60 s at 72°C) in a Hybaid 96-well thermocycler. The reaction products were diluted 1:20 with TE buffer.
Sequence tag library construction
Amplification of SSR sequences from the pool of PstI–MseI restriction fragments was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 5 pmol biotinylated PstI adapter primer, 40 pmol PCT6 primer mix, 1 U Taq DNA polymerase and 2 µl of diluted PstI–MseI fragments as DNA template. The PCT6 primer mix consisted of equimolar amounts of two synthetic oligomers (KKV RVR VCT CTC TCT CTC T-OH and KKR VRV RTC TCT CTC TCT C-OH, where K is G or T, R is A or G, and V is A, C or G). These primers were designed to faithfully anchor to the 5′-ends of (CT)n and (TC)n repeat arrays (8). PCR was performed for 27 cycles with the profile 60 s at 92°C, 60 s annealing (see below) and 30 s at 72°C. The annealing temperature for the first cycle was 65°C, reducing by 1°C per cycle for the next seven cycles. The following five cycles were with annealing at 59°C. Following the twelfth cycle the denaturation and annealing steps were reduced to 30 s and the remaining 15 cycles were continued using annealing at 57°C. The reaction products were separated on a 2% low melting point agarose gel and fragments within the size range 170–470 bp were excised and purified using a QiaQuick gel purification kit (Qiagen).
Sequence tags were prepared by digesting the size-selected SSR fragments in 100 µl of 1× NEB buffer IV containing 80 mM S-adenosylmethionine and 25 U BsgI for 6 h at 37°C. The restriction endonuclease was inactivated by heating the mixture at 65°C for 20 min and 100 µg prewashed streptavidin-coated magnetic beads (Merck), suspended in 100 µl of 2× STEX (20 mM Tris–HCl pH 8.0, 200 mM NaCl, 2 mM EDTA, 0.2% Triton X-100), were added. The suspension was then incubated at room temperature for 30 min with gentle agitation. The beads were washed three times with 100 µl of 1× STEX to remove unbound DNA fragments, resuspended in 80 µl of 1× NEB ligase buffer and 400 pmol biotin and incubated at room temperature for 15 min with gentle agitation. The large molar excess of biotin ensured that the streptavidin-binding sites on the beads were completely saturated before attachment of the biotinylated BsgI adapter to the ends of the solid-phase restriction fragments in the subsequent ligation step. The BsgI adapter was prepared by annealing synthetic oligomers as described above (sense strand, biotin-GGC CGC TCT AGA ATT CNN-OH; antisense strand, GAA TTC TAG AGC GGC-OH). Aliquots of 100 pmol BsgI adapter and 12 U T4 ligase (NEB) were added to the bead suspension and the volume was adjusted to 100 µl with 1× NEB ligase buffer. The mixture was incubated at room temperature for 2 h with gentle agitation. The beads were washed three times with 100 µl of 1× STEX, resuspended in 50 µl of sterile water, immobilised by magnetic capture and the supernatant containing the eluted sequence adapter tags was transferred to a new tube.
The sequence adapter tags were amplified in a 100 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 20 pmol each biotinylated BsgI (biotin-GGC CGC TCT AGA ATT C-OH) and PstI adapter primers, 1 U Taq DNA polymerase and 50 µl of eluted sequence adapter tags. PCR amplification was performed using the following conditions: 92°C for 15 s and 56°C for 30 s for 20 cycles. The amplification products were purified with a QiaQuick PCR purification kit, eluted in 100 µl of 1× NEB EcoRI buffer containing 100 U EcoRI and incubated at 37°C for 3 h. Biotinylated adapter molecules were removed from the digestion mixture by affinity capture using streptavidin-coated magnetic beads as follows. One milligram of prewashed beads suspended in 100 µl of 2× STEX was added to the digestion mix, followed by incubation at room temperature for 30 min with gentle agitation. The beads were immobilised and the supernatant was transferred to a new tube. The beads were washed twice with 100 µl of 1× STEX, with the supernatant being preserved after each wash. The pooled supernatants were then ethanol precipitated with 15 µg Glyco Blue (Ambion) as carrier. The pelleted sequence tags were resuspended in 20 µl of 1× NEB ligase buffer containing 6 U T4 ligase and incubated overnight at room temperature. The concatemerised sequence tags were heated at 65°C for 15 min, chilled quickly on ice and size fractionated (>400 bp) through a SizeSelect-400 Spun Column (Pharmacia) according to the manufacturer’s instructions. The eluted sequence tag concatemers were quantified by spectrophotometry and ligated overnight at room temperature into the dephosphorylated EcoRI site of a modified pGEM-7Zf(+) vector at a 3:1 insert:vector ratio. The vector was modified by inserting a 20 bp linker with an EcoRI site at its centre to replace the original polylinker cloning region. This modification increased the amount of insert sequence obtained per sequencing reaction by reducing the length of polylinker region sequenced. The ligation mix was transformed into competent JM109 cells (Promega) according to the manufacturer’s instructions and plated on LB–ampillicin plates supplemented with IPTG-Xgal. Individual white colonies were picked for DNA sequence analysis using ABI Prism BigDye terminator chemistry and the M13 forward and reverse primers. Tag sequences were extracted from raw sequence data files using the STMP software package (available from the authors). This software builds a database of tag sequences for profile generation and performs frequency distribution analysis. Individual tag sequences are placed in the database in the PstI→SSR (sense) orientation to facilitate the design of PCR primers, irrespective of their orientation in the concatemer. Additional information is also stored to allow the origin of each tag to be followed and ambiguous tag sequences (those with uncalled bases, or Ns) to be identified and manually edited at a later time.
Amplification of SSR markers
Amplification of a target SSR fragment from the pool of amplified PstI–MseI fragments or directly from genomic DNA required the synthesis of just one additional primer, whose sequence was derived from a sequence tag. These primers were designed with NetPrimer (Premier Biosoft International) using the parameters: primer length 16–24 nt, 3′-end stability –5.5 to –9.0 kcal/mol, oligomer Tm 55–70°C, GC content 30–70% and primer rating >80. PCR was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 1 U Taq DNA polymerase, 5 pmol sequence tag primer (supplemented with 0.5 pmol 33P-end-labelled primer when using autoradiographic detection), 2 µl of DNA template (see below) and either 5 pmol MseI adapter primer or PCT6 primer mix. The choice of the latter primer depended on the type of template DNA used. Amplification of a target full-length SSR fragment from the pool of amplified PstI–MseI fragments employed the MseI adapter primer, while amplification of a target SSR locus directly from genomic DNA used the PCT6 primer mix. Each reaction used 2 µl of the 1:20 diluted pool of amplified PstI–MseI fragments or genomic DNA (50 ng/µl) as template DNA. To amplify target SSRs from genomic DNA, PCR was performed using the profile used to amplify SSR fragments from the pool of amplified PstI–MseI fragments, except that 20–35 cycles were performed at the annealing temperature of 57°C. A touchdown PCR profile was used to amplify target SSRs from the pool of amplified PstI–MseI fragments: 30 s at 92°C, 60 s at (Tm + 10)°C and 30 s at 72°C, where Tm was the predicted melting temperature of the sequence tag primer as determined by NetPrimer. The annealing temperature was reduced by 1°C per cycle for the first 10 cycles, with 25–35 cycles performed at the final annealing temperature. Microsatellite fragments were separated on 5% sequencing gels and visualised by silver staining or overnight exposure to X-ray film when radiolabelled STM primers were used.
RESULTS AND DISCUSSION
Principles of STMP
The technique reported here employs the principles of SAGE (7) to generate a comprehensive library of locus-specific tags for SSR sequences within genomic or cDNA. It is based on the amplification of restriction fragments containing SSR arrays by PCR. A type IIs restriction endonuclease is then used to generate a short but positionally defined nucleotide sequence tag for every fragment that is present within the amplicon pool. As each tag consists of 16 nt of fragment sequence, a characteristic tag will identify each unique amplicon, since a 16 nt sequence can distinguish more than 4.3 × 109 (416) fragments given random nucleotide distribution along the length of the tag. This number far exceeds estimates of the number of SSR loci present in plant species with extremely large genomes, such as hexaploid wheat (9,10) and pine (11,12). By ligating sequence tags together to form large concatemers and cloning into plasmids, a large number of sequence tags can be efficiently sequenced in a single reaction. Thus, the sequencing throughput of STMP vastly exceeds that of traditional library screening methods that characterise only one SSR locus per clone. The delineation of each tag by a unique punctuation sequence allows the identification and orientation of each tag within the concatemer. The tags contain sufficient nucleotide sequence to allow the design of a primer that can be used to directly amplify a specific SSR locus from genomic DNA or to isolate the corresponding full-length SSR fragment from the pool of amplified PstI–MseI fragments. STMP incorporates recent modifications to the SAGE technology that improve the efficiency of concatemerisation and increase the number of tags identified from each plasmid clone sequenced (13,14).
The major steps of the STMP technique are shown in Figure 1. Genomic DNA is digested with PstI and MseI and adapters are ligated to the ends of the DNA fragments. Adapter ligation is performed in the presence of active restriction endonucleases to prevent fragment–fragment concatemers, a step made possible by the design of the adapters, which destroy the restriction sites upon ligation. Ligation of the PstI adapter to a DNA fragment also creates a recognition domain for the type IIs restriction endonuclease BsgI at the position of the PstI restriction site. The adapter-ligated restriction fragments serve as template DNA for amplification, with the adapters functioning as primer-binding sites.
Figure 1.

Flow chart for STMP.
The design of the adapters permits amplification only of PstI–MseI fragments, a step essential for the isolation of full-length restriction fragments containing target SSR sequences. Amplification of MseI–MseI fragments is prevented by the 3′ modification on the antisense strand of the MseI adapter. Amination prevents its extension by DNA polymerase during the initial denaturation step of PCR and ensures that the end of the adapter remains single stranded (vectorette PCR) (15). As a consequence, a binding site for the MseI adapter primer is not created unless a complementary DNA strand is first formed by extension of a primer at the other end of the restriction fragment. As neither end of a MseI–MseI fragment contains an adapter primer-binding site, these fragments cannot undergo amplification. However, maintenance of the single-stranded region relies on covalent attachment of the antisense strand to the restriction fragment by a phosphodiester linkage. If this bond is not formed (phosphorylation of oligomers is never complete) then the 3′-end of the restriction fragment will be extended by polymerase activity to fill in the single-stranded region of the adapter, following dissociation of the non-ligated strand during the initial heating step (16). Formation of an adapter primer-binding site in these fragments will allow their exponential amplification. To guard against the amplification of such molecules, exonuclease III digestion was used to remove DNA fragments containing adapters that were only partially ligated. When added in excess, exonuclease III progressively degrades double-stranded DNA from nicked and recessed 3′-termini and, therefore, prevents polymerase extension of the restriction fragments. Exonuclease III-resistant phosphorothioate linkages at the 3′-termini of the MseI and PstI antisense adapter strands protect against degradation of correctly ligated DNA fragments (17). In experiments where exonuclease III digestion was not used, PCR amplification generally produced a smear of non-specific products during isolation of full-length restriction fragments containing target SSR sequences. These non-specific products were shown by PCR amplification using only the MseI adapter primer to result from amplification of MseI–MseI fragments (data not shown). Strong PCR suppression effects (18) generated by the long (65 bp) adapter sequences that are needed for enzymatic manipulation of the DNA fragments when attached to the streptavidin-coated magnetic beads inhibit amplification of PstI–PstI fragments (data not shown).
Following amplification of PstI–MseI restriction fragments the pool of amplicons is used to obtain SSR sequences for construction of the sequence tag library. This is achieved by PCR amplification of the pool of amplified PstI–MseI fragments with a biotinylated PstI adapter primer and a partially degenerate 5′-anchored SSR primer. The resulting amplicons are then size fractionated by gel electrophoresis and amplicons within an arbitrarily selected size distribution are isolated and purified. The size selection of SSR-containing amplicons predetermines the size distribution of subsequently developed microsatellite markers. For example, the selection of amplicons between 170 and 470 bp in length will generate SSR markers with a size distribution between ∼100 and 400 bp, as the selected amplicons include the length of the PstI adapter (65 bp). Digestion of the selected amplicons with the type IIs restriction endonuclease BsgI releases a sequence adapter tag for each amplicon, which consists of the PstI adapter itself and an additional 16 nt of unknown DNA sequence. Separating the adapter and unknown sequences is the BsgI recognition domain, as shown in Figure 1. The biotinylated sequence adapter tags are isolated by affinity capture on streptavidin-coated magnetic beads and a synthetic linker, the BsgI adapter, is ligated to the exposed BsgI sites. This adapter has a 16-fold degenerate 3′-overhang that renders it compatible with all possible DNA sequences released by BsgI and introduces an EcoRI restriction domain at the 3′-end of the sequence adapter tag. The resulting molecules are then amplified by PCR using biotinylated BsgI and PstI adapter primers, followed by digestion with EcoRI to release the sequence tags. The biotinylated adapter fragments are removed from the digestion mix by affinity capture on streptavidin-coated magnetic beads. These manipulations result in 22 bp fragments flanked at both ends by an EcoRI restriction site. A BsgI recognition domain located adjacent to the 5′-located EcoRI restriction site defines the orientation of the sequence tag in the SSR-containing amplicon from which it was derived (Fig. 1). The sequence tags are then concatemerised, size selected, cloned and sequenced.
As each tag is unique to the amplicon from which it was derived, the nucleotide sequence of a tag can be used to design a PCR primer (called a sequence-tagged microsatellite or STM primer) to amplify the corresponding SSR from either the original pool of amplified restriction fragments or directly from genomic DNA. Isolation of restriction fragments containing target SSR sequences is achieved by PCR amplification of the amplified PstI–MseI fragment pool with a STM and the MseI adapter primer. This approach is possible because the absence of MseI–MseI fragments in the DNA pool avoids non-specific amplification by the adapter primer alone (data not shown). A full-length restriction fragment allows complete characterisation of a microsatellite locus and provides a flanking sequence for the development of a SSR marker. Alternatively, STM primers can be used to amplify a target SSR directly from genomic DNA. Following the strategy of Fisher et al. (8), this is achieved by PCR amplification of genomic DNA with a STM primer and the 5′-anchored SSR primer used to construct the sequence tag library. This PCR produces an amplicon with the entire microsatellite sequence by faithful anchoring of the SSR primer to the 5′-end of the repeat array. Therefore, the cost of developing a SSR marker is reduced to the synthesis of just one primer specific to the conserved region flanking the repeat.
Application of STMP to develop SSR markers in bread wheat
To test and validate the STMP technique we constructed a sequence tag library of CT repeat arrays from PstI–MseI restriction fragments in the genome of bread wheat. The library contained more than 2000 clones with a concatemer insert size of 400 bp or greater. Plasmid DNA was prepared for 50 randomly picked single colonies and subjected to automated sequence analysis. The number of sequence tags in each concatemer ranged from 15 to 53, with an average of 24.7. Figure 2 shows a typical chromatogram for a plasmid clone and illustrates how the BsgI recognition sequence can be used to determine the orientation of the tag within the concatemer. A total of 1235 tags were obtained from the sequenced clones, of which 51.5% were unique. Whilst duplicate tags were observed (average copy number 2.96), the relative abundance of any tag did not exceed 1.5% of the total number of tags sequenced. As the PCR cycle number was kept to a minimum during library construction to reduce the possibility of non-representative amplification, the frequency distribution of the tags should be representative of the abundance of SSR sequences within the DNA pool from which they were derived (7,19–21). Our results therefore indicate that CT repeat sequences occur frequently in the genome of hexaploid wheat and at varying copy number, a finding in agreement with previous work using traditional methods for developing SSR markers (9,10,22,23). These results also indicate that the serial analysis of thousands of tags will be required to generate a comprehensive profile of the abundance and frequency of SSR sequences in bread wheat.
Figure 2.

ABI 377 sequence output for a typical plasmid clone. BsgI and EcoRI sites are represented by dashed and solid lines, respectively. The nucleotide sequences of individual tags are shown by dotted lines. The orientation of each sequence tag in the concatemer is defined by the sequence of the BsgI recognition domain, with GTGCAG and CTGCAC indicating sense and antisense orientation, respectively.
Despite containing only 16 nt of sequence, primers suitable for amplification of corresponding SSR fragments could be designed for 68.8% of the unique sequence tags. This high frequency was possible because the PstI recognition sequence used to positionally locate the tag within a restriction fragment could also be included to increase the length of the primer. This facilitated the design of primers with the specified parameters (see Materials and Methods) and allowed the 3′-end of a primer to be positioned for maximum specificity. Software-based analysis performed using Amplify (University of Wisconsin, Madison, WI) to test the specificity of each STM primer against the sequence tag library indicated that 54.2% should amplify a single SSR fragment from the pool of amplified PstI–MseI restriction fragments. The proportion of sequence tags for which primers could be designed was similar to those reported (62% on average) for primer pairs designed for wheat SSR sequences contained in plasmid clones (22,23).
To demonstrate the use of STM primers for the isolation of corresponding full-length restriction fragments from the pool of amplified PstI–MseI fragments and direct amplification of target SSR loci from genomic DNA we synthesised primers for 110 unique sequence tags. Table 1 shows the nucleotide sequence of typical STM primers and the tags from which they were designed. The isolation of full-length restriction fragments was achieved by PCR amplification using each of the STM primers in combination with the MseI adapter primer and the diluted pool of amplified PstI–MseI fragments as template DNA. Half of the reactions (51.8%) revealed a single strongly amplified fragment, 21.8% showed two or more discrete fragments and 26.4% produced a smear when visualised on agarose gels (Fig. 3). The presence of single fragments indicated that the corresponding restriction fragments were amplified, while the visualisation of reactions revealing multiple amplicons on sequencing gels generally allowed the identification of target fragments due to the presence of SSR band stuttering (data not shown). Non-specific fragment amplification could be minimised with the optimisation of PCR conditions for individual reactions and was believed to be a consequence of the large size and complexity of the bread wheat genome and the use of only one primer determining PCR specificity. It is expected that the specificity of isolating full-length fragments would improve significantly in the analysis of smaller genomes. Numerous reactions also revealed multiple SSR fragments, which were shown by DNA sequence analysis to result from the amplification of closely related microsatellite sequences (data not shown). Although unique tags were selected for primer design, the amplification of multiple SSR fragments was expected as: (i) the selection of only unique tags could not be guaranteed because too few tags were sequenced to provide a sufficiently comprehensive profile for SSR sequences in the DNA pool; (ii) SSR sequences closely related to those profiled in the tag library could occur in the population of microsatellite amplicons that were not used for library construction. As tags for these amplicons were not created, their abundance in the unselected pool of restriction fragments used as template DNA could not be predicted. However, the amplification of multiple SSR fragments is not a limitation of the technique, as the sequence of each fragment can be used to design a pair of primers for the specific amplification of each microsatellite locus.
Table 1. Sequence of typical STM primers and the tags from which they were derived.
| Tag no. |
Tag sequence (5′→3′) |
STM primer sequence |
Tm (°C)a |
Cycle no.b |
| 9 | GTGCAGAGACCATTGCGTTCAC | GCAGAGACCATTGCGTTCAC | 58.7 | 30 |
| 43 | GTGCAGCAGCGAACAGGTGGCG | CTGCAGCAGCGAACAGGTG | 60.6 | 20 |
| 46 | GTGCAGCAGGGCTAAACTCCAG | CCAGCAGGGCTAAACTCCAG | 60.0 | 30 |
| 62 | GTGCAGCGAGCAGGTGGCGTAG | CTGCAGCGAGCAGGTGGCGTAG | 69.8 | 20 |
| 67 | GTGCAGCGGCAAGAGGAGCAGG | CTGCAGCGGCAAGAGGAGCAG | 67.8 | 20 |
| 123 | GTGCAGACATCATGCGTGACTG | CTGCAGACATCATGCGTGACTG | 62.1 | 30 |
| 135 | GTGCAGATCGAGGTTTGCGTCC | CTGCAGATCGAGGTTTGCGTC | 62.9 | 30 |
| 189 | GTGCAGCCTTGTCTTTACTTGG | CTGCAGCCTTGTCTTTACTTG | 55.4 | 35 |
| 210 | GTGCAGCTCCACCACATCTCCG | CTGCAGCTCCACCACATCTCC | 62.5 | 35 |
| 297 | GTGCAGCAAAAGACTTCAGTGC | CTGCAGCAAAAGACTTCAGTGC | 60.2 | 30 |
| 298 | GTGCAGAGACCATTGCGTTCAC | CTGCAGAGACCATTGCGTTCAC | 62.8 | 30 |
| 313 | GTGCAGATCGTGATCCATGTTC | CTGCAGATCGTGATCCATGTTC | 60.1 | 35 |
| 639 | GTGCAGATTTTTCCAACTTCAG | CTGCAGATTTTTCCAACTTCAG | 57.0 | 35 |
| 764 | GTGCAGCAACACACGGGAAGAT | CTGCAGCAACACACGGGAAGA | 64.1 | 25 |
| 773 | GTGCAGCTGAGGACACTCTTCA | CTGCAGCTGAGGACACTCTTCA | 60.1 | 30 |
| 804 | GTGCAGCTGAGCTGGCGCTTTC | CTGCAGCTGAGCTGGCGCTTTC | 69.2 | 35 |
aMelting temperature of the STM primer, as determined by NetPrimer.
bNumber of PCR cycles performed with final annealing at 57°C to amplify target SSR loci from genomic DNA.
Figure 3.

Amplification of full-length restriction fragments corresponding to specific sequence tags from the pool of amplified PstI–MseI fragments. The STM primers used in each reaction are, from left to right: STM 43, 46, 62, 67, 123, 135, 189, 210, 297, 298, 313, 639, 764 and 804. pUC19/HpaII was used as a size marker.
The direct amplification of SSR loci corresponding to specific sequence tags from genomic DNA was achieved by employing the STM primers individually in PCR with the SSR primer used to construct the sequence tag library. Using a standard set of PCR conditions fragments displaying SSR band stuttering were obtained in 79% (87/110) of reactions visualised on sequencing gels. The complexity of the STM fingerprints were typical of those obtained for initial screening of primer pairs designed for SSR sequences in bread wheat (Fig. 4). Thirty-seven per cent amplified a single SSR locus, 42% revealed multiple SSR fragments and 23% gave no amplification. Of the 87 functional STM primers 62% were suitable for use as DNA markers in genetic studies. Importantly, the sizes of the SSR fragments amplified by locus-specific STM primers were within the expected range of 100–400 bp. This demonstrated that the size selection of SSR amplicons prior to library construction permits predetermination of the size distribution of subsequently developed markers. Also noteworthy was the low level of ISSR amplification, indicated by the virtual absence of fragments that were common between reactions (Fig. 4). It is assumed that the parameters used to design STM primers ensured their participation first in PCR, giving preferential amplification of target microsatellite fragments over amplification of ISSR sequences by the SSR primer alone. The visualisation of ISSRs could be completely avoided by labelling the STM primer.
Figure 4.

Amplification of SSR loci corresponding to specific sequence tags from genomic DNA of bread wheat (cv. Halberd). The STM primers used in each reaction are, from left to right: STM 43, 46, 62, 67, 123, 135, 189, 210, 297, 298, 313, 639, 764 and 804. A 25 bp DNA ladder (Gibco BRL) was used as a size marker. DNA fragments were visualised with silver staining.
The proportion of STM primers amplifying a scorable SSR marker (49% of the total number of STM primers synthesised) compared favorably with those reported for traditional hybridisation-based techniques. In three recently published works (9,22,23) the proportion of usable primer pairs designed for wheat SSR sequences contained in clones isolated from several different genomic libraries was reported to range from 31 to 67%, with an average of 37%. However, we believe, for the reasons discussed above, that the proportion of usable STM markers could be significantly increased. More extensive profiling of the SSR amplicon pool would facilitate the selection of unique tags for primer design and thereby provide a higher proportion of markers amplifying a single locus. Optimisation of PCR conditions for individual STM primers or the development of primer pairs for the conserved regions flanking target SSR sequences that cannot be specifically amplified from genomic DNA with a single STM primer would also increase the proportion of locus-specific markers. Nevertheless, the present results demonstrate that SSR markers can be developed from sequence tags with the design and synthesis of only one sequence-specific primer. STMs are robust and informative Mendelian markers (Fig. 5) and are distributed throughout the bread wheat genome (M.J.Hayden, S.Khatkar and P.J.Sharp, manuscript in preparation).
Figure 5.
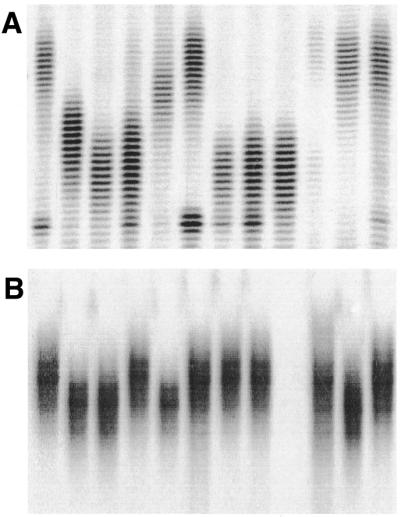
Polymorphisms revealed among 12 current Australian bread wheat cultivars by (A) STM 764 and (B) STM 773. DNA fragments were visualised by autoradiography.
Technical considerations of the STMP method
While the use of other restriction enzyme combinations for STMP are possible, we have used PstI as one of the enzymes for generating a pool of amplified restriction fragments. There are several reasons for this. (i) The recognition sequence for PstI can be easily manipulated by site-directed mutagenesis to create a restriction site for BsgI, thereby maximising the length of the sequence tags generated. The use of other restriction enzymes will generally require that the type IIs restriction site be located further upstream of the adapter–restriction fragment junction. (ii) The use of a common restriction enzyme to define the position of the sequence tags in the genome will allow tags from different libraries to be pooled. For example, if two libraries are generated independently from PstI–MseI and PstI–TaqI fragments then tag data from both could be pooled because SSR sequences common to both libraries will be identified by the same tag sequences. Without a common restriction enzyme to positionally define the location of each sequence tag in the genome, the pooling of such information would be impossible. (iii) PstI is methylation sensitive and creates an amplifiable fraction that is highly enriched for low and single copy DNA (24,25). Moreover, PstI significantly increases the proportion of primer pairs that amplify target microsatellite sequences when it is used to construct the genomic library from which the SSR-containing clones are derived. Libraries prepared using restriction enzymes that lack methylation sensitivity are often reported to produce a high proportion of non-functional primers that amplify multiple, rather than specific, SSR loci (22,26).
Amplification of an informative SSR marker using only one primer specific to the conserved region flanking a microsatellite relies on faithful anchoring of the SSR primer to the 5′-end of the repeat array in template DNA. The SSR primer design reported by Fisher et al. (8) has proven suitable for this purpose and can be extended to different types of di-, tri- and tetranucleotide repeats. Importantly, the proportion of informative markers was improved when individual primers targeting the same repeat motif (e.g. those targeting CT and TC repeat arrays) were combined in a single primer mix (M.J.Hayden, unpublished data). This presumably resulted from the increase in degeneracy of the 5′-anchor sequence of the SSR primer, which meant that they should be complementary to one in three of all possible random sequences adjacent to CT repeats. However, STMP is not restricted to the use of 5′-anchored SSR primers. When used to isolate full-length target SSR fragments from the pool of amplified restriction fragments both 3′-anchored and compound SSR primers can also be used because incorporation of the entire repeat array in the initial amplicon is unnecessary. Alternatively, primers targeting other repetitive elements in eukaryotic genomes, such as retrotransponsons (27), could be employed to provide a rich source of binary markers.
CONCLUSION
STMP offers a cost-efficient and rapid approach to developing SSR markers. By eliminating the need for library screening to identify SSR-containing clones and providing an ∼25-fold increase in sequencing throughput, STMP offers significant advantages over traditional techniques for developing SSR markers. The quantitative relationship between the abundance of individual tags and SSR copy number in the genome provides a tool to help select low copy or unique SSR sequences for primer design, thereby increasing the proportion of locus-specific markers. Furthermore, the capacity to develop SSR markers with the synthesis of just one primer specific to the conserved region flanking the repeat offers the opportunity to significantly reduce the cost of developing microsatellite markers. STMP is a flexible technique that can be adapted to a large number of restriction enzyme combinations and used with both genomic and cDNA.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Nigel Haynes for writing and supporting the STMP software package. This research was supported by Quality Wheat CRC and the Grain and Research Development Corporation, Australia.
References
- 1.Tautz D., Trick,M. and Dover,G. (1986) Cryptic simplicity in DNA is a major source of genetic variation. Nature, 322, 652–656. [DOI] [PubMed] [Google Scholar]
- 2.Powell W., Morgante,M., Andre,C., Hanafey,M., Vogel,J., Tingey,S. and Rafalsky,A. (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed., 2, 225–238. [Google Scholar]
- 3.Jarne P. and Lagoda,P.J.G. (1996) Microsatellites, from molecules to populations and back. Trends Ecol. Evol., 11, 424–429. [DOI] [PubMed] [Google Scholar]
- 4.Gupta P.K., Balyan,H.S., Sharma,P.C. and Ramesh,B. (1996) Microsatellites in plants: a new class of molecular markers. Curr. Sci., 70, 45–54. [Google Scholar]
- 5.Edwards K.J., Barker,J.H.A., Daly,A., Jones,C. and Karp,A. (1996) Microsatellite libraries enriched for several microsatellite sequences in plants. Biotechniques, 20, 759–760. [DOI] [PubMed] [Google Scholar]
- 6.Ostrander E.A., Jong,P.M., Rine,J. and Duyk,G. (1992) Construction of small-insert genomic DNA libraries highly enriched for microsatellite sequences. Proc. Natl Acad. Sci. USA, 89, 3419–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Velculescu V.E., Zhang,L., Vogelstein,B. and Kinzler,K.W. (1995) Serial analysis of gene expression. Science, 270, 484–487. [DOI] [PubMed] [Google Scholar]
- 8.Fisher P.J., Gardner,R.C. and Richardson,T.E. (1996) Single locus microsatellites isolated using 5′ anchored PCR. Nucleic Acids Res., 24, 4369–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roder M.S., Plaschke,J., Konig,S.U., Borner,A., Sorrells,M.E., Tanksley,S.D. and Ganal,M.W. (1995) Abundance, variability and chromosomal location of microsatellites in wheat. Theor. Appl. Genet., 246, 327–333. [DOI] [PubMed] [Google Scholar]
- 10.Ma Z.Q., Roder,M.S. and Sorrells,M.E. (1996) Frequencies and sequence characteristics of di-, tri- and tetra-nucleotide microsatellites in wheat. Genome, 39, 123–130. [DOI] [PubMed] [Google Scholar]
- 11.Pfeiffer A., Olivieri,A.M. and Morgante,M. (1997) Identification and characterisation of microsatellites in Norway spruce (Picea abies K.). Genome, 40, 411–419. [DOI] [PubMed] [Google Scholar]
- 12.Hicks M., Adams,S., O’Keefe,S., Macdonald,E. and Hodgetts,R. (1998) The development of RAPD and microsatellite markers in lodgepole pine (Pinus contorta var. latifolia). Genome, 41, 797–805. [Google Scholar]
- 13.Powell J. (1998) Enhanced concatemer cloning—a modification to the SAGE (Serial Analysis of Gene Expression) technique. Nucleic Acids Res., 26, 3445–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenzelmann M. and Muhlemann,K. (1999) Substantially enhanced cloning efficiency of SAGE (Serial Analysis of Gene Expression) by adding a heating step to the original protocol. Nucleic Acids Res., 27, 917–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagerstrom M., Parik,J., Malmgren,H., Stewart,J., Pettersson,U. and Landegren,U. (1991) Capture PCR: efficient amplification of DNA fragments adjacent to a known sequence in human and YAC DNA. PCR Methods Appl., 1, 111–119. [DOI] [PubMed] [Google Scholar]
- 16.Vos P., Hogers,R., Bleeker,M., Reijans,M., van de Lee,T., Hornes,M., Frijters,A., Pot,J., Peleman,J., Kuiper,M. and Zabeau,M. (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res., 23, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Putney S.D., Benkovic,S.J. and Schimmel,P.R. (1981) A DNA fragment with an α-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc. Natl Acad. Sci. USA, 78, 7350–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukyanov S.A., Gurskaya,N.K., Luklanov,V.S., Tarabukin,V.S. and Sverdlov,E.D. (1994) Highly efficient subtractive hybridisation of cDNA. Bioinorg. Chem., 20, 701–704. [Google Scholar]
- 19.Velculescu V.E., Zhang,L., Zhou,W., Vogelstein,J., Basrai,M.A., Bassett,D.E., Hieter,P., Vogelstein,B. and Kinzler,K.W. (1997) Characterisation of the yeast transcriptome. Cell, 88, 243–251. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L., Zhou,W., Velculescu,V.E., Kern,S.E., Hruban,R.H., Hamilton,S.R., Vogelstein,B. and Kinzler,K.W. (1997) Gene expression profiles in normal and cancer cells. Science, 276, 1268–1272. [DOI] [PubMed] [Google Scholar]
- 21.Neilson L., Andalibi,A., Kang,D., Coutifaris,C., Strauss,J.F., Stanton,J.L. and Green,D.P.L. (2000) Molecular phenotype of the human oocyte by PCR–SAGE. Genomics, 63, 13–24. [DOI] [PubMed] [Google Scholar]
- 22.Roder M.S., Korzun,V., Wendehake,K., Plaschke,J., Tixier,M.-H., Leroy,P. and Ganal,M.W. (1998) A microsatellite map of wheat. Genetics, 149, 2007–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryan G.J., Collins,A.J., Stephenson,P., Orry,A., Smith,J.B. and Gale,M.D. (1997) Isolation and characterisation of microsatellites from hexaploid bread wheat. Theor. Appl. Genet., 94, 557–563. [Google Scholar]
- 24.Burr B., Burr,F.A., Thompson,K.H., Albertson,M.C. and Stuber,C.W. (1988) Gene mapping with recombinant inbreds in maize. Genetics, 118, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gill K.S., Lubbers,E.L., Gill,B.S., Raupp,W.J. and Cox,T.S. (1991) A genetic linkage map of Triticum tauschii (DD) and its relationship to the D genome of bread wheat (AABBDD). Genome, 34, 362–374. [Google Scholar]
- 26.Plaschke J., Borner,A., Wendehake,K., Ganal,M.W. and Roder,M.S. (1996) The use of wheat aneuploids for the chromosomal assignment of microsatellite loci. Euphytica, 89, 33–40. [Google Scholar]
- 27.Voytas D.F., Cummings,M.P., Konieczny,A., Ausubel,F.M. and Rodermel,S.R. (1992) Copia-like retrotransposons are ubiquitous among plants. Proc. Natl Acad. Sci. USA, 89, 7124–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]