

Abstract
Background
Exposure to particulate matter (PM) air pollution may be an important environmental factor leading to exacerbations of inflammatory illnesses in the GI tract. PM can gain access to the gastrointestinal (GI) tract via swallowing of air or secretions from the upper airways or mucociliary clearance of inhaled particles.
Methods
We measured PM-induced cell death and mitochondrial ROS generation in Caco-2 cells stably expressing oxidant sensitive GFP localized to mitochondria in the absence or presence of an antioxidant. C57BL/6 mice were exposed to a very high dose of urban PM from Washington, DC (200 μg/mouse) or saline via gastric gavage and small bowel and colonic tissue were harvested for histologic evaluation, and RNA isolation up to 48 hours. Permeability to 4kD dextran was measured at 48 hours.
Results
PM induced mitochondrial ROS generation and cell death in Caco-2 cells. PM also caused oxidant-dependent NF-κB activation, disruption of tight junctions and increased permeability of Caco-2 monolayers. Mice exposed to PM had increased intestinal permeability compared with PBS treated mice. In the small bowel, colocalization of the tight junction protein, ZO-1 was lower in the PM treated animals. In the small bowel and colon, PM exposed mice had higher levels of IL-6 mRNA and reduced levels of ZO-1 mRNA. Increased apoptosis was observed in the colon of PM exposed mice.
Conclusions
Exposure to high doses of urban PM causes oxidant dependent GI epithelial cell death, disruption of tight junction proteins, inflammation and increased permeability in the gut in vitro and in vivo. These PM-induced changes may contribute to exacerbations of inflammatory disorders of the gut.
Background
In human populations, investigators have associated PM exposure with an increasing number of adverse health outcomes including all-cause mortality, cardiovascular mortality, accelerated atherosclerosis in postmenopausal women, loss of lung function in healthy adults, impaired lung development in children, exacerbations of obstructive lung disease, pneumonia and increased risk of venous thromboembolism [1-11]. Recently, Kaplan et al reported that individuals younger than 23 years were more likely to be diagnosed with Crohn's disease if they lived in regions with higher NO2 concentrations [12], a marker of traffic related pollution that correlates with the levels of PM and an independent group of investigators reported that exposure to PM was associated with hospitalizations in patients with inflammatory bowel disease [13]. PM exposure has also been reported to increase the risk of appendicitis,[14] the development of gastroenteritis in children [15] and colorectal cancer [16-19].
Many of the adverse health consequences of PM are thought to be caused by the ability of PM to induce cellular oxidative stress, which activates signaling pathways that contribute to cytokine release and barrier dysfunction [20]. As the bulk of PM enters the body by inhalation, investigators have used radiolabeled particles to examine the fate of inhaled particles in rodents [21]. They found that the overwhelming majority of the particles (> 95%) are taken up by alveolar macrophages, brought to the oropharynx by mucociliary clearance and excreted in the feces [21].
PM may also enter the GI tract directly through swallowed air or through the ingestion of foods such as vegetables and fruits coated with PM. These findings suggest that the steady state concentrations of PM in the GI tract may be similar to those encountered in the lung. In this paper, we sought to test the hypothesis that PM exposure can induce oxidant dependent epithelial dysfunction of the GI tract in mice. We tested our hypothesis in cell based systems and in mice using a very high dose of a well characterized urban PM.
Methods
Particulate Matter (PM)
We used an urban PM collected from ambient air in Washington, DC (National Institute of Standards and Technology Standard Reference Material, SRM 1649a). The characteristics of PM have been previously described [22,23].
Animals and administration of PM via gavage
The protocol for the use of mice was approved by the Animal Care and Use Committee at Northwestern University and Rush University in Chicago, IL. We used eight to twelve weeks old, (20-25 g), male, C57BL/6 mice (Jackson Laboratories). Mice were anesthetized with isoflurane 2-3%. After adequate anesthesia is achieved, the esophagus was intubated using a mouse gastric gavage tube and 200 μg of PM suspended in sterile PBS or PBS only (control) was administered via gavage tube followed by 100 μl of air to clear the liquid in the gavage tube. Particulate matter does not dispersed well in aqueous solutions because it forms aggregates in PBS or in culture media within several minutes. Therefore, PM in PBS was vortexed immediately prior to instillation [24]. After treatment, the gavage tube was removed gently and the animals were placed in their cages until harvesting of stomach, small bowel and colon at 24 and 48 hours. Permeability experiments were also performed at 48 hours.
Histology
At 24 or 48 hours after treatment with PM, mice were euthanized for collection of stomach, small bowel and colon tissue. After harvesting, the tissues were fixed and paraffin embedded and 5-μm sections were stained with hematoxylin/eosin.
TUNEL staining of colonic sections
End labeling of exposed 3'-OH ends of DNA fragments in paraffin-embedded tissue was done utilizing the TUNEL AP In Situ Cell Death Detection Kit (Roche Diagnostics Corp.) according to the manufacturer's directions. After staining, 20 fields of colonic sections (400x) were randomly chosen and nuclei and total nuclei were counted using an automated program (Image J) [25].
Assessment of intestinal permeability in mice
GI permeability was measured based on the intestinal permeability towards 4000Da-fluorescent-dextran- FITC (DX-4000-FITC) (FD4000 Sigma-Aldrich, St. Louis, Mo, USA) as described [26,27]. Briefly, mice were given DX-4000-FITC by gavage (500 mg/kg body-weight, 125 mg/ml). After 1 hour, 120 μl of blood were collected via right atrial puncture. The blood was centrifuged at 4°C, 12000×g for 3 min. Plasma was diluted in an equal volume of PBS (pH 7.4) and analyzed for DX-4000-FITC concentration with a fluorescence spectrophotometer at the excitation wavelength of 485 nm and the emission wavelength of 535 nm. Standard curves were obtained by diluting FITC-dextran in non-treated plasma diluted with PBS (1:3 v/v).
Cell culture
Caco-2 cells (a human colon adenocarcinoma cell line) (American Type Culture Collection, Manassas, VA) were chosen for our studies because they form monolayers that morphologically resemble intestinal cells with defined apical brush borders. Cells were cultured in Dulbecco's minimum essential medium at 37°C, 5% CO2, 100% relative humidity. Cells were exposed to PM diluted in media or control vehicle (media). To ensure equal dispersion, the PM solution was vortexed with the media before application to the cells. For barrier function experiments, Caco-2 cells were split at a ratio of 1:2 and seeded at a density of 200,000 cells/cm2 into 0.4 μM Biocoat Collagen I Cell Culture Inserts (0.3 cm2 growth surface; Becton Dickinson Labware, Bedford, MA) and experiments were performed at least 3 weeks post-confluence. The confluence of Caco-2 monolayers was confirmed with transepithelial electrical resistance (TEER) measurements as described below [28,29]. Only monolayers of cells with TEER values greater than 250 Ω × cm2 (after subtraction of the bare filter inserts [100-150 Ω × cm2]) were used for the experiments as previously described by Turner et al and other investigators [28,30-33]. The confluent monolayers were exposed to PM suspended in media or vehicle control (media alone). To ensure equal dispersion, the PM solution was vortexed with the media before application to the cells.
Measurement of transepithelial electrical resistance (TEER)
Transepithelial electrical resistance (TEER) was determined in polarized Caco-2 cell monolayers, cultured in 6-well plates, using a Millicell-ERS Resistance System (Millipore, Bedford, MA) that includes a dual electrode volt-ohm-meter. TEER was calculated as TEER = (Rm-Ri) × A, where Rm is transmembrane resistance; Ri, intrinsic resistance of a cell-free media; and A, the surface area of the membrane in cm2 [28,29,34]. The TEER measurements were performed in serum free media.
Measurement of the apparent permeability coefficient (Papp) of fluorescein sulfonic acid (FSA)
Barrier permeability was determined using Caco-2 cells grown to confluence on 6.5-mm/0.4 μm 24-well tissue culture plate inserts (Transwell, Corning) as we have previously described [35,36]. The measurements were performed in serum free media. Permeability of insert Caco-2 monolayers was evaluated by measurements of the apparent permeability coefficient (Papp) of the fluorescent marker fluorescein-5-(and-6)-sulfonic acid trisodium salt (FSA, 200 μg/ml, 478 Da) (Invitrogen) in the apical to basolateral direction [33]. Fluorescent signals from samples were measured using a fluorescence multiplate reader (FSA excitation, 485 nm; FSA emission, 530 nm). The Papp was calculated by the following formula: Papp (cm/sec) = (dQ/dt)/(Co × A), where dQ/dt is the permeability rate (mol/s); Co is the initial concentration in the donor compartment (mol/ml) and A is the surface area of the monolayer (0.33 cm2).
Assessment of cell death
Cell death was assessed using a commercially available photometric immunoassay that detects histone-associated DNA fragments (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's directions, as we have previously described [37].
Generation of Caco-2 cells stably expressing mitochondrially localized oxidant sensitive GFP (mito-Ro-GFP) probes
An adenoviral vector encoding Ro-GFP with a mitochondrial localization sequence (mito-Ro-GFP) was generated as described previously and commercially amplified (ViraQuest, Iowa City, IA) [37,38]. A lentiviral vector encoding the mito-Ro-GFP probe with a mitochondrial localization sequence was created using the ViraPower lentiviral transformation kit according to the manufacturer's directions (Invitrogen) as previously described [37]. Expression of the probe was confirmed by examining the cells using fluorescence microscopy. Appropriate localization of the probe to the mitochondria was confirmed by co-staining with the mitochondrial probe MitoTracker (10 μM) (Invitrogen, Carlsbad, CA).
Generation of Caco-2 cells lacking mitochondrial DNA (ρ0 Caco-2 cells)
The ρ0 Caco-2 cells were generated by incubating Caco-2 cells in medium containing ethidium bromide (100 ng/ml), sodium pyruvate (1 mm), and uridine (100 μg/ml) for 3-5 weeks. The lack of DNA encoding cytochrome oxidase subunit IV was confirmed by PCR using the primer sequences described previously [37,39,40]. Cells depleted of mitochondrial DNA, or ρ0 cells, lack 13 proteins critical for normal electron transport. Although ρ0 cells contain petite mitochondria, they cannot support normal oxidative phosphorylation and must survive and replicate using ATP derived solely from glycolysis. Without a functional electron transport chain, ρ0 cells cannot generate ATP from electron transport and their mitochondria are incapable of generating reactive oxygen species [40,41].
Measurement of Reactive Oxygen Species
We employed an oxidant-sensitive ratiometric probe (Ro-GFP) that was originally described by Hanson and co-workers who validated its responsiveness to a variety of intracellular oxidants both ex vivo and in living cells [42,43]. We generated Caco-2 cells stably expressing the mito-Ro-GFP probe and measured the oxidation state of the probe using flow cytometry as we have previously described [37]. Briefly, the cells were removed from the plate using trypsin, and equal aliquots of the resulting suspension were transferred to tubes containing medium alone or medium containing 1 mM dithiothreitol or 1 mM t-butyl hydroperoxide (t-BOOH). After 10 min, the ratio of fluorescence (emission of 535 nm) at excitations of 405 and 488 nm was measured in 5,000 cells/condition using a DakoCytomation CyAn high speed multilaser droplet cell sorter. The oxidation state of the cells was calculated as follows: (value after treatment - value after dithiothreitol (completely reduced state))/(value after t-BOOH (completely oxided state) - value after dithiothreitol (completely reduced state)) [37,38]. In some experiments, we also treated cells with EUK-134 (20 μM) (Cayman Chemicals, Ann Arbor, MI), which is a combined superoxide dismutase and catalase mimetic that has been shown to prolong the lifespan of the nematode Caenorhabditis elegans and to prevent alveolar epithelial cell death during injurious stimuli [44,45].
Immunoblotting determination of nitration and oxidation of tubulin to assess cytoskeleton
Oxidation and nitration of the tubulin backbone of microtubules were assessed by measuring protein carbonyl and nitrotyrosine formation, respectively [46]. Carbonylation and nitrotyrosination of tubulin were determined in a similar manner as the quantitative blotting of tubulin. To avoid unwanted oxidation of tubulin samples, all buffers contained 0.5 mM dithiothreitol and 20 mM 4,5-dihydroxy-1,3-benzene sulfonic acid (Sigma).
Immunofluorescent staining and high-resolution laser scanning confocal microscopy of the actin cytoskeleton
Integrity of actin cytoskeleton integrity in monolayers of Caco-2 cells was determined as previously described [47]. Cells were subsequently processed by incubation with fluorescein isothiocyanate (FITC)-conjugated phalloidin (specific for F-actin staining, Sigma, St. Louis, MO), 1:40 dilution for 1 h at 37°C. Desired areas of monolayers were processed using the image processing with Zeiss Axiovision and NIH Image J software.
Analysis of NF-κB activation
NF-κB (p65 and p50 subunit) activation was assessed by ELISA as we have previously described [35,48]. The NF-κB activity test is based on a validated ELISA principle in which NF-κB is captured by a double stranded oligonucleotide probe containing the consensus-binding sequence for either NF-κB p65 or p50 subunits [49]. The results were quantified by a chromogenic reaction, which was then read for absorbance at 450 nm by a Seivers NOA 280 microplate analyzer (Sievers, CO) [49].
Immunofluorescence staining of zonula occludens-1 (ZO-1) and occludin for tight junction morphology
Immunofluorescence staining for ZO-1 and occludin was performed in Caco-2 cells and fresh frozen mouse small bowel tissue sections using primary anti-ZO-1 antibody (Zymed) (1:200 dilution) or for anti-occludin antibody (Zymed) (1:100 dilution) for1 hour at 37°C followed by incubation with a secondary antibody (fluorescein isothiocyanate-conjugated goat anti-mouse; Sigma-Aldrich, St. Louis, MO; 1:50 dilution) for 1 hour at room temperature. Imaged areas of monolayers were processed using Zeiss Axiovision and NIH Image J image processing software.
Evaluation of tight junction disruption
Caco-2 cells were stained for ZO-1 and the slides were examined for their overall morphology, orientation, and disruption as we previously described [35,50]. To avoid bias, the slides were coded so that the examiner was blinded to the experimental protocol. After the scoring was complete, the slides were decoded for analysis. Briefly, the "ZO-1 integrity" in Caco-2 cells was considered abnormal or injured based on one or more of the following criteria: fragmentation, kinking, disruption of actin cortex, or detachment from membrane areas (areas of cell-to-cell contact). We examined 200 cells per slide (well) by LSCM microscopy, and the percentage (%) of cells displaying normal ZO-1 or abnormal ZO-1 was determined (e.g., 90 cells [displaying injured ZO-1] divided by 200 is equal to 45% abnormal ZO-1 or 55% normal ZO-1). Overall, at least 1,200 cells per group (200 × 6 slides) were examined in four different fields by LSCM and the percentage of cells displaying normal ZO-1 was then determined per treatment group.
Statistics
Differences between groups were explored using analysis of variance. When the analysis of variance indicated a significant difference, individual differences were explored using t tests with a Dunnett correction for multiple comparisons against control conditions. All of the analyses were performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA).
Results
Particulate matter induces a dose-dependent ROS generation and cell death in colonic epithelial cells
We previously reported that exposure of alveolar epithelial cells to PM10 collected from Düsseldorf, Germany induced the generation of ROS [51]. To determine whether PM can induce ROS generation in the GI tract epithelia, we treated Caco-2 cells stably expressing mitochondrial oxidant sensitive GFP (mito-Ro-GFP) with vehicle (media) or different concentrations of urban PM from Washington, D.C. Four hours later, we assessed PM-induced ROS production by measuring the oxidation of the probe. Treatment with t-butyl hydroperoxide was used as a positive control. Exposure of Caco-2 cells to PM caused a dose-dependent ROS generation from the mitochondria (Figure 1A). To determine whether PM can induce cell death in the GI tract epithelia, we treated Caco-2 cells with vehicle (media) or different concentrations of PM for 24 hours and measured apoptotic cell death (DNA fragmentation). Exposure of Caco-2 cells to PM caused a dose-dependent increase in the percentage of TUNEL positive cells (Figure 1B).
Figure 1.

Particulate matter induces dose-dependent ROS generation and cell death in colonic epithelial cells. (A) Caco-2 cells stably expressing an oxidant-sensitive GFP probe (mito-Ro-GFP, lentivirus) were treated with control vehicle (media) or different doses of PM. The percentage oxidation of the probe was measured using flow cytometry 4 hours later. The t-butyl hydroperoxide (t-BOOH) was used as a positive control. (B) Caco-2 cells were treated as described above and cell death (DNA fragmentation via enzyme-linked immunosorbent assay) was measured 24 hours later. (*P < 0.05 PM compared with PBS, n ≥ 4).
The generation of mitochondrial ROS is required for PM-induced cell death in colonic epithelial cells
We treated Caco-2 cells stably expressing mitochondrial oxidant sensitive GFP (mito-Ro-GFP) with vehicle (media) or different concentrations of PM from Washington, D.C in the absence or presence of the combined superoxide dismutase and catalase mimetic, EUK -134 (20 μM), which was added to the media 1 hour before exposure. We then measured oxidation of the mitochondrially localized probe 4 hours later and apoptotic cell death 24 hours later. Both PM-induced ROS generation and cell death were attenuated in cells treated with (EUK-134) therapy (Figure 2A,B).
Figure 2.

The generation of mitochondrial ROS is required for PM-induced cell death in colonic epithelial cells. (A) ROS production and (B) cell death were measured 4 and 24 hours, respectively, after treatment of Caco-2 cells stably expressing mitochondrial oxidant sensitive GFP (mito-Ro-GFP) with control (media) or PM in the absence or presence of the combined superoxide dismutase and catalase mimetic, EUK -134 (20 μM). (C) ROS production and (D) cell death were measured at 4 and 24 hours, respectively, after treatment of wild-type Caco-2 cells and Caco-2 cells lacking mitochondrial DNA (ρ0 CaCo-2 cells) with vehicle (media) or PM. The t-butyl hydroperoxide (t-BOOH) was used as a positive control. (P < 0.05 *PM compared with PBS, **EUK-134 compared with vehicle, n ≥ 4).
To determine whether mitochondrially generated ROS are required for PM-induced cell death, we generated Caco-2 cells lacking mitochondrial DNA (ρ0 Caco-2 cells). Wild type and ρ0 Caco-2 cells were infected with an adenovirus encoding the mito-Ro-GFP, and the oxidation of the probe was measured at 4 hours after treatment with PM (25 μg/cm2) suspended in media or control vehicle (media) using flow cytometry. Compared to wild-type cells, ρ0 Caco-2 cells produced minimal ROS in response to PM (Figure 2C). We treated wild-type and ρ0 Caco-2 cells with PM and measured cell death 24 hours later. Compared with wild-type cells, PM-induced cell death was attenuated in the ρ0 Caco-2 cells (Figure 2D).
Exposure to PM causes oxidant-dependent NF-κB activation and increased permeability in Caco-2 cell monolayers
As oxidant stress in the gut oxidation has been associated with activation of the nuclear transcription factor, NF-κB [52], we measured NF-κB in Caco-2 cells exposed to either PM or vehicle (PBS). Exposure to PM caused a dose-dependent increase in NF-κB activity, which was prevented by, EUK-134 (Figure 3A).
Figure 3.

Exposure of Caco-2 cells to PM causes oxidant-dependent NF-κB activation and increased permeability. (A) NF-κB activation was measured in Caco-2 cells treated with vehicle (PBS) or different doses of PM in the absence or the presence of the combined superoxide dismutase and catalase mimetic, EUK -134 (20 μM). NF-κB activation was presented as % activation compared to H2O2 (positive control). To evaluate the permeability of the Caco-2 monolayer, cells were treated with either vehicle (media) or different concentrations of PM and (B) transepithelial electrical resistance (TEER) was measured over time. (C) TEER values compared to control (as percent control) at 2 hours are shown. (D) We also measured the apparent permeability co-efficient (Papp) of fluorescein sulfonic acid (FSA) in the apical to basolateral direction across the Caco-2 cells. (E) Percent change in Papp values at 2 hours is shown. (P < 0.05 *PM compared with PBS, **EUK-134 compared with vehicle, n ≥ 4).
Intestinal barrier disruption has been implicated in several intestinal and systemic disorders and has been associated with ROS generation, NF-κB activation and cell death [50,53]. We treated Caco-2 cells with either vehicle (media) or different concentrations of PM and assessed the permeability of the monolayer by measuring the transepithelial electrical resistance (TEER) (Figure 3B) and the apparent permeability coefficient (Papp) of fluorescein sulfonic acid (FSA) in the apical to basolateral direction (Figure 3C and 3D). Exposure to PM caused an increase in permeability (Figure 3).
Exposure to particulate matter causes nitration and oxidation injury and disruption of the microtubule cytoskeleton of intestinal cell monolayers
We assessed the effect of PM induced ROS generation on the cytoskeleton by measuring the amount of nitration (nitrotyrosination) and oxidation (carbonylation) of tubulin in Caco-2 cells as previously described [46]. Oxidant (H2O2, 0.5 mM) alone resulted in substantial levels of nitration (nitro and oxidation of the tubulin cytoskeleton (Figure 4A and 4B)). Exposure to Caco-2 cells to PM caused nitration and oxidation of the tubulin cytoskeleton, which was evident even at the lowest dose of PM (0.5 μg/cm2). High-resolution laser scanning confocal microscopic evaluation of Caco-2 monolayers confirmed that intestinal cells exhibited abnormal architecture of the microtubule cytoskeleton after treatment with PM. The changes seen at highest dose of PM (50 μg/cm2) were similar to those seen from cell monolayers that are challenged with H2O2 (Figure 4C).
Figure 4.

Exposure to particulate matter causes nitration and oxidation of tubulin and disruption of tight junctions in intestinal cell monolayers. The effect of PM on tubulin (A) nitration (nitrotyrosination) and (B) oxidation (carbonylation) in Caco-2 cell monolayers are shown. H2O2 (0.5 mM) was used as a positive control. (C) Images from high-resolution laser scanning confocal microscopic evaluation of intestinal cell monolayers stained for F-actin are shown.
Exposure of Caco-2 cells to particulate matter causes disruption of tight junctions
To determine whether PM-induced increased permeability in monolayers of Caco-2 cells is associated with changes in their tight junctions, we stained monolayers of Caco-2 cells for ZO-1, a tight junction protein. Caco-2 cells exposed to PM had discontinuity and irregularity of ZO-1 distribution at the intercellular junctions suggesting the loss of tension in intestinal cells. ZO-1 was rearranged from an uninterrupted band along cell borders into radially oriented sporadic aggregates (Figure 5A). The severity of tight junction disruption estimated from blinded analysis of ZO-1 stained sections for their overall morphology, orientation, and disruption was increased in PM compared with control treated Caco-2 cells (Figure 5B).
Figure 5.

Exposure of monolayers of Caco-2 cells to PM causes increased permeability and disruption of tight junctions. Caco-2 cells were treated with either PBS (vehicle) or different concentrations of PM and we (A) stained the cell monolayers for ZO-1, a tight junction protein and (B) measured mean tight junction disruption Intestinal sections from the animals were subjected to a blinded analysis of tight junction disruption. Representative images from 4 independent experiments are shown and each bar represents the mean of 20 observations.
Exposure to PM causes increased gut permeability and inflammation and colonic epithelial cell death in mice
To determine whether PM exposure affects GI epithelia in vivo, we treated mice with PM administered via gavage and harvested the stomach and colon 48 hours later. Exposure to PM did not cause any detectable histologic changes in the GI tract (Figure 6). However, compared to PBS treated animals, mice exposed to PM exhibited an increase in IL-6 mRNA in the small bowel and colon suggesting that PM induces an inflammatory response in the gut (Figure 7A).
Figure 6.
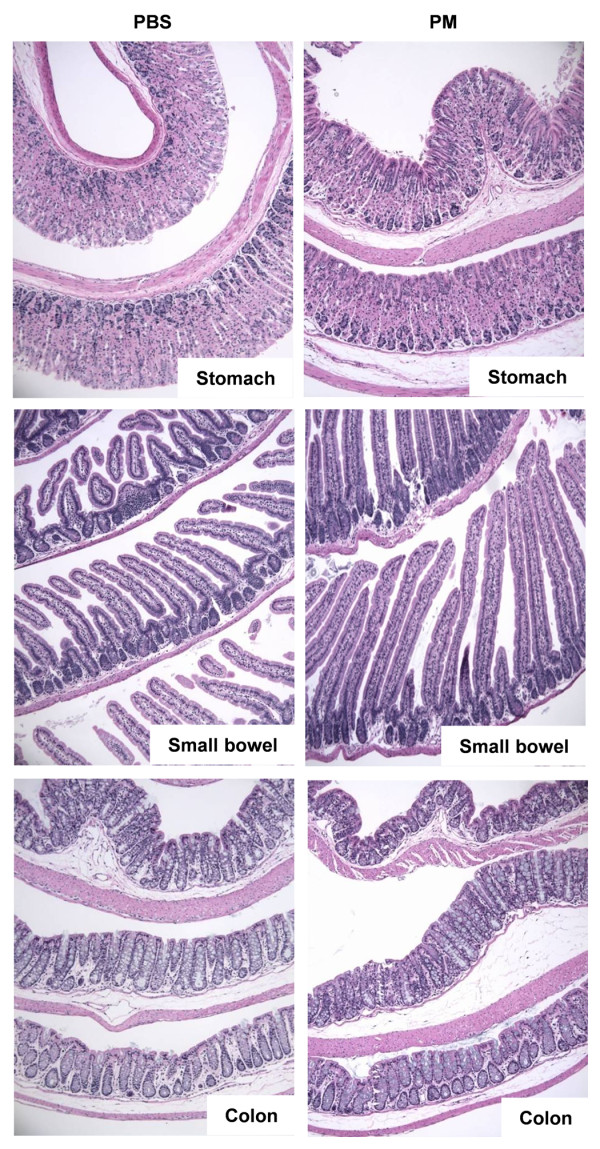
Exposure to PM does not cause histologic changes in gastric, small bowel and colonic mucosa at 24 or 48 hours. Wild-type cells were treated with either PBS (vehicle) or PM via gastric gavage and histology (H&E staining) of gastric, small bowel and colonic tissue was evaluated 48 hours after exposure. Representative photomicroographs from a total of 5 animals/treatment group are shown (x100 magnification).
Figure 7.

Exposure to PM causes increased gut permeability and inflammation and colonic epithelial cell death in mice. Wild-type mice were treated with either PBS (vehicle) or PM via gastric gavage for measurement of (A) IL-6 mRNA (qRT-PCR) in small bowel and colonic homogenates (24 and 48 hours after treatment) (n = 6/treatment group), (B) intestinal permeability to 4kD dextran (48 hours after treatment) (n = 5/treatment group), (C) cell death (TUNEL-positive nuclei) in the colon (48 hours after treatment) (n = 4/treatment group), (D) ZO-1 mRNA (qRT-PCR) in the small bowel and colon (n = 4/treatment group) and (E) ZO-1 protein expression in the small bowel (24 and 48 hours after treatment) (n = 4/treatment group) and (F) tight junction disruption (ZO-1 and occludin localization) in the small bowel by immunofluorescence (48 hours after treatment) (Representative images, n = 4/treatment/group). (P < 0.05 *PM compared with PBS).
We measured the permeability of the gut permeability to a 4kD dextran (Figure 7B) 48 hours after the administration of PM or PBS via gavage. PM exposure was associated with a significant increase in permeability. We then counted the number of TUNEL-positive nuclei observed in colonic sections obtained 48 hours after treatment with PM or PBS. Treatment with PM was associated with an increase in the number of TUNEL positive nuclei (Figure 7C). Forty-eight hours after treatment with PM, we observed a significant reduction in the mRNA encoding ZO-1 measured by qRT-PCR in the small bowel and colon (Figure 7D). The ZO-1 protein expression in the small bowel did not change significantly after PM treatment (Figure 7E). Examination of ZO-1 and occludin stained sections of small bowel tissue was altered in the PM compared with the PBS treated mice (Figure 7F).
Discussion
Exposure to air pollution is associated with well-known health effects on the cardiovascular and respiratory systems [54-56]. While less known, there also are several reports linking exposure to air pollution to adverse health effects in the GI tract [12-19]. Consistent with our findings in lung epithelial cells, we observed that Caco-2 cells exposed to PM generated ROS from the mitochondria, which were required for the activation of NF-κB and the development of apoptotic cell death in vitro. In confluent monolayers of Caco-2 cells, we found that PM reduced the TER of the monolayer and caused disruption of the architecture of the tight junctions. In mice exposed to PM by oral gavage, we found evidence of increased IL-6 transcription, disruption of small bowel tight junctions, increased numbers of TUNEL positive nuclei and increased gut permeability to FITC labeled dextran.
We found that colonic epithelial cells generate significant amounts of mitochondrially derived ROS and undergo apoptosis in response to PM exposure in vitro. Cells depleted of mitochondrial DNA (ρ0 Caco-2 cells) were resistant to PM induced apoptosis. ρ0 cells lack the 13 proteins encoded in the mitochondrial genome, which are required for normal electron transport. As a result, these cells are deficient in oxidative phosphorylation and cannot produce mitochondrial ROS. However, as most of the mitochondrial proteome is encoded by nuclear genes, these cells still contain mitochondria, maintain a mitochondrial membrane potential supported by glycolysis and undergo apoptosis normally. We reported a similar dependence of PM-induced signaling and apoptosis in alveolar epithelial cells and other investigators have highlighted the importance of mitochondrially derived ROS in the cellular response to PM [22,37,51]. Inflammation the GI tract during diseases such as inflammatory bowel disease is associated with increased local ROS generation and activation of NF-κB[57-61]. We observed that the administration of PM to Caco-2 cells resulted in the oxidant dependent activation of NF-κB. The importance of these findings is highlighted by our observation that the administration of PM by oral gavage increased the transcription of an NF-κB target gene IL-6 in the small bowel and colon.
We observed that the administration of PM to Caco-2 monolayers reduced the transepithelial resistance of the monolayers and caused a disruption in the organization of the tight junctions. In vivo, the administration of PM was associated with decreased transcription of the tight junction proteins ZO-1 and occludin and histologic evidence of alterations in tight junction structure. There was also an increase in the number of apoptotic cells in the small bowel from these animals. Our data do not allow us to determine whether PM-induced alterations in tight junctions, apoptosis or other mechanisms are responsible for the resulting increase in gut permeability. However, this finding is important as chances in gut permeability are thought to play a pathogenic role in many gastrointestinal disorders including alcoholic liver disease, inflammatory bowel disease and celiac disease [57,61,62].
While the lungs are the primary organs exposed to ambient PM; a significant portion of PM exposure can be deposited in the upper airway and also get access to the GI tract via direct entrance due to swallowing of air or indirectly via swallowing of the PM deposited in the upper airway. Furthermore, most of the inhaled PM is transported via mucociliary system from the lung to the upper airway and then swallowed. Our data support the recent reports, which showed an epidemiologic link between acute, short-term exposure to air pollution and inflammation in the gut. We speculate that PM-induced ROS generation, NF-κB activation and activation of inflammation may be responsible for exacerbations of inflammatory conditions in the gut such as inflammatory bowel disease.
Urban PM is a complex mixture of solid and particles that primarily are formed during the combustion of fossil fuels [63,62]. As the carbon core of the particle cools, organic components (including quinones) and transition metals (including iron and vanadium) condense on the surface of the particles. The resulting particles are composed of a carbonaceous core, organic components, sulfate, nitrate, ammonium, crustal material and transition metals such as iron or vanadium. Organic constituents in the particles, particularly quinones, can undergo single electron transfer reactions with intracellular electron acceptors, creating the opportunity for the transfer of an electron to molecular oxygen to form a superoxide radical. In the presence of a transition metal, the superoxide anion can be converted to a hydroxyl radical through Fenton chemistry [63-67]. In addition, sulfates and nitrates may combine with water to form acids which may modulate their toxicity.
An important limitation of our study is the dose of PM that we employed for our in vivo experiments. PM enters the lung via inhalation and larger particles are deposited in the oropharynx and large airways, while smaller particles gain access to the distal airspaces. A very small fraction of PM may gains access to the systemic circulation and affect systemic organs including the gut. Semmler-Benke followed the fate of iridium labeled carbon black nanoparticles administered via inhalation to rats for 6 months after the exposure. Most of the particles (> 90%) were taken up by alveolar macrophages, cleared by mucociliary mechanisms and excreted in the feces [21]. Because the GI tract is also exposed to PM deposited in the oropharynx, PM that has sedimented on unwashed fruits and vegetables and PM contained in swallowed air, the total dose of PM to which the gut is exposed is likely at least as large as that seen by the lung. However, the more rapid transit time of the mucous layer over the gut compared with the lung mucosa may decrease the concentration of PM to which the gut is exposed at any given time. Furthermore, the administration of PM may alter the transit time of material through the gut.
In 2002, the U.S. EPA reported a range of maximal city PM concentrations between 26 and 534 μg/m3 [54]. Many large cities in the world have much higher levels of PM with annual averages of 200 to 600 μg/m3 and peak concentrations frequently exceeding 1000 μg/m3 [68]. Using the highest value in U.S. and assuming a minute ventilation of 6 L/min (~8.6 m3 over 24 hours) for a healthy adult at rest, the total dose of PM inhaled over 24 hours would be 4,614 μg. However, in other "mega cities" of the world, the daily inhaled dose of particles may be as high as 20,000 μg [68,69]. Exposure of a mouse (with a minute ventilation of 35-50 ml/min) to a daily dose of 200 μg of PM corresponds to 24-30 mg of PM exposure for a human [70]. This is about 6-8 times higher than a US adult will be exposed in even the most polluted city. Further study is required to determine whether PM induces similar effects on GI function at lower levels of exposure.
Conclusions
Exposure to high doses of PM causes oxidant dependent GI epithelial cell death, disruption of tight junctional proteins, inflammation and increased permeability in the gut in vitro and in vivo. These PM-induced changes may be responsible for exacerbations of inflammatory disorders of the gut. More studies are warranted to better understand the health effects of air pollution in the GI tract.
Abbreviations
GFP: green fluorescent protein; GI: gastrointestinal; FITC: fluorescein isothiocyanate; PBS: phosphate buffered saline; PM: particulate matter; qRT-PCR: quantitative real-time PCR; ROS: reactive oxygen species; t-BOOH: t-butyl hydroperoxide; ZO-1: zonula occludens-1.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EAM, CF, AK, GRSB and GMM: Designed research, analyzed and interpreted data and wrote the manuscript. PE, SS, DU, CF, RN, SEC, KAR, AG and GMM performed research, analyzed data and reviewed the manuscript. SJ, EAM and AK reviewed histology slides.
All authors have read and approved the final manuscript.
Contributor Information
Ece A Mutlu, Email: Ece_Mutlu@rush.edu.
Phillip A Engen, Email: Phillip_A_Engen@rush.edu.
Saul Soberanes, Email: saulaxo@gmail.com.
Daniela Urich, Email: danielaurich@gmail.com.
Christopher B Forsyth, Email: Christopher_B_Forsyth@rush.edu.
Recep Nigdelioglu, Email: recepnigdelioglu@hotmail.com.
Sergio E Chiarella, Email: sergio-chiarella@northwestern.edu.
Kathryn A Radigan, Email: k-radigan@northwestern.edu.
Angel Gonzalez, Email: angel-gonzalez@northwestern.edu.
Shriram Jakate, Email: shriram_jakate@rush.edu.
Ali Keshavarzian, Email: Ali_Keshavarzian@rush.edu.
GR Scott Budinger, Email: s-buding@northwestern.edu.
Gökhan M Mutlu, Email: g-mutlu@northwestern.edu.
Acknowledgements
We thank Dr. Andrew J Ghio from the United States Environmental Protection Agency, Research Triangle Park, NC for providing us with PM from Washington, DC and his suggestions and careful review of our manuscript. This work was supported by NIH ES015024 (to GMM), ES 013995 (to GRSB) and 1P01HL071643 (to GMM and GRSB).
References
- Downs SH, Schindler C, Liu LJ, Keidel D, Bayer-Oglesby L, Brutsche MH, Gerbase MW, Keller R, Kunzli N, Leuenberger P, Probst-Hensch NM, Tschopp JM, Zellweger JP, Rochat T, Schwartz J, Ackermann-Liebrich U. SAPALDIA Team. Reduced exposure to PM10 and attenuated age-related decline in lung function. N Engl J Med. 2007;357:2338–2347. doi: 10.1056/NEJMoa073625. [DOI] [PubMed] [Google Scholar]
- Gauderman WJ, Avol E, Gilliland F, Vora H, Thomas D, Berhane K, McConnell R, Kuenzli N, Lurmann F, Rappaport E, Margolis H, Bates D, Peters J. The Effect of Air Pollution on Lung Development from 10 to 18 Years of Age. N Engl J Med. 2004;351:1057–1067. doi: 10.1056/NEJMoa040610. [DOI] [PubMed] [Google Scholar]
- Krewski D, Burnett RT, Goldberg MS, Hoover K, Siemiatycki J, Abrahamowicz M, White WH. Validation of the Harvard Six Cities Study of particulate air pollution and mortality. N Engl J Med. 2004;350:198–199. doi: 10.1056/NEJM200401083500225. [DOI] [PubMed] [Google Scholar]
- McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, Harrington R, Svartengren M, Han IK, Ohman-Strickland P, Chung KF, Zhang J. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007;357:2348–2358. doi: 10.1056/NEJMoa071535. [DOI] [PubMed] [Google Scholar]
- Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-Term Exposure to Air Pollution and Incidence of Cardiovascular Events in Women. N Engl J Med. 2007;356:447–458. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- Mills NL, Tornqvist H, Gonzalez MC, Vink E, Robinson SD, Soderberg S, Boon NA, Donaldson K, Sandstrom T, Blomberg A, Newby DE. Ischemic and Thrombotic Effects of Dilute Diesel-Exhaust Inhalation in Men with Coronary Heart Disease. N Engl J Med. 2007;357:1075–1082. doi: 10.1056/NEJMoa066314. [DOI] [PubMed] [Google Scholar]
- Pope CA III, Ezzati M, Dockery DW. Fine-Particulate Air Pollution and Life Expectancy in the United States. N Engl J Med. 2009;360:376–386. doi: 10.1056/NEJMsa0805646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane B, Jerrett M, Burnett RT, Marrie T, Arain A, Loeb M. Long-Term Exposure to Ambient Air Pollution and Risk of Hospitalization with Community-acquired Pneumonia in Older Adults. Am J Respir Crit Care Med. 2010;181:47–53. doi: 10.1164/rccm.200901-0160OC. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Martinelli I, Zanobetti A, Grillo P, Hou LF, Bertazzi PA, Mannucci PM, Schwartz J. Exposure to Particulate Air Pollution and Risk of Deep Vein Thrombosis. Arch Intern Med. 2008;168:920–927. doi: 10.1001/archinte.168.9.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A, Martinelli I, Pegoraro V, Melly S, Grillo P, Zanobetti A, Hou L, Bertazzi PA, Mannucci PM, Schwartz J. Living near major traffic roads and risk of deep vein thrombosis. Circulation. 2009;119:3118–3124. doi: 10.1161/CIRCULATIONAHA.108.836163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales RE, Cakmak S, Vidal CB. Air Pollution and hospitalization for venous thromboembolic disease in Chile. Journal of Thrombosis and Haemostasis. 2010. 9999. [DOI] [PubMed]
- Kaplan GG, Hubbard J, Korzenik J, Sands BE, Panaccione R, Ghosh S, Wheeler AJ, Villeneuve PJ. The inflammatory bowel diseases and ambient air pollution: a novel association. Am J Gastroenterol. 2010;105:2412–2419. doi: 10.1038/ajg.2010.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan AN, McGinley EL, G BD, Saeian K. Air pollution and hospitalizations for inflammatory bowel disease: An ecologic analysis. Gastroenterology. 2010;138:S17–S18. doi: 10.1002/ibd.21455. [DOI] [PubMed] [Google Scholar]
- Kaplan GG, Dixon E, Panaccione R, Fong A, Chen L, Szyszkowicz M, Wheeler A, Maclean A, Buie WD, Leung T, Heitman SJ, Villeneuve PJ. Effect of ambient air pollution on the incidence of appendicitis. CMAJ. 2009. [DOI] [PMC free article] [PubMed]
- Orazzo F, Nespoli L, Ito K, Tassinari D, Giardina D, Funis M, Cecchi A, Trapani C, Forgeschi G, Vignini M, Nosetti L, Pigna S, Zanobetti A. Air pollution, aeroallergens, and emergency room visits for acute respiratory diseases and gastroenteric disorders among young children in six Italian cities. Environ Health Perspect. 2009;117:1780–1785. doi: 10.1289/ehp.0900599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guberan E, Usel M, Raymond L, Bolay J, Fioretta G, Puissant J. Increased risk for lung cancer and for cancer of the gastrointestinal tract among Geneva professional drivers. Br J Ind Med. 1992;49:337–344. doi: 10.1136/oem.49.5.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardsson de Verdier M, Plato N, Steineck G, Peters JM. Occupational exposures and cancer of the colon and rectum. Am J Ind Med. 1992;22:291–303. doi: 10.1002/ajim.4700220303. [DOI] [PubMed] [Google Scholar]
- Andersen A, Barlow L, Engeland A, Kjaerheim K, Lynge E, Pukkala E. Work-related cancer in the Nordic countries. Scand J Work Environ Health. 1999;25(Suppl 2):1–116. [PubMed] [Google Scholar]
- Goldberg MS, Parent ME, Siemiatycki J, Desy M, Nadon L, Richardson L, Lakhani R, Latreille B, Valois MF. A case-control study of the relationship between the risk of colon cancer in men and exposures to occupational agents. Am J Ind Med. 2001;39:531–546. doi: 10.1002/ajim.1052. [DOI] [PubMed] [Google Scholar]
- Nel A. Atmosphere. Air pollution-related illness: effects of particles. Science. 2005;308:804–806. doi: 10.1126/science.1108752. [DOI] [PubMed] [Google Scholar]
- Semmler-Behnke M, Takenaka S, Fertsch S, Wenk A, Seitz J, Mayer P, Oberdorster G, Kreyling WG. Efficient elimination of inhaled nanoparticles from the alveolar region: evidence for interstitial uptake and subsequent reentrainment onto airways epithelium. Environ Health Perspect. 2007;115:728–733. doi: 10.1289/ehp.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich D, Soberanes S, Burgess Z, Chiarella SE, Ghio AJ, Ridge KM, Kamp DW, Chandel NS, Mutlu GM, Budinger GR. Proapoptotic Noxa is required for particulate matter-induced cell death and lung inflammation. FASEB J. 2009;23:2055–2064. doi: 10.1096/fj.08-114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y, Elias JA. Interleukin-6-induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol. 2000;22:535–542. doi: 10.1165/ajrcmb.22.5.3808. [DOI] [PubMed] [Google Scholar]
- Mutlu GM, Budinger GR, Green AA, Urich D, Soberanes S, Chiarella SE, Alheid GF, McCrimmon DR, Szleifer I, Hersam MC. Biocompatible nanoscale dispersion of single-walled carbon nanotubes minimizes in vivo pulmonary toxicity. Nano Lett. 2010;10:1664–1670. doi: 10.1021/nl9042483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budinger GR, Mutlu GM, Eisenbart J, Fuller AC, Bellmeyer AA, Baker CM, Wilson M, Ridge K, Barrett TA, Lee VY, Chandel NS. Proapoptotic Bid is required for pulmonary fibrosis. Proc Natl Acad Sci USA. 2006;103:4604–4609. doi: 10.1073/pnas.0507604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Erlejman AG, Fraga CG, Oteiza PI. Procyanidins protect Caco-2 cells from bile acid- and oxidant-induced damage. Free Radic Biol Med. 2006;41:1247–1256. doi: 10.1016/j.freeradbiomed.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Buzza MS, Netzel-Arnett S, Shea-Donohue T, Zhao A, Lin CY, List K, Szabo R, Fasano A, Bugge TH, Antalis TM. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci USA. 2010;107:4200–4205. doi: 10.1073/pnas.0903923107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narai A, Arai S, Shimizu M. Rapid decrease in transepithelial electrical resistance of human intestinal Caco-2 cell monolayers by cytotoxic membrane perturbents. Toxicol In Vitro. 1997;11:347–354. doi: 10.1016/S0887-2333(97)00026-X. [DOI] [PubMed] [Google Scholar]
- Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC kinase. Am J Physiol. 1999;277:C554–562. doi: 10.1152/ajpcell.1999.277.3.C554. [DOI] [PubMed] [Google Scholar]
- Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- Hubatsch I, Ragnarsson EG, Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc. 2007;2:2111–2119. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- Biganzoli E, Cavenaghi LA, Rossi R, Brunati MC, Nolli ML. Use of a Caco-2 cell culture model for the characterization of intestinal absorption of antibiotics. Farmaco. 1999;54:594–599. doi: 10.1016/S0014-827X(99)00069-5. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Key role of PLC-gamma in EGF protection of epithelial barrier against iNOS upregulation and F-actin nitration and disassembly. Am J Physiol Cell Physiol. 2003;285:C977–993. doi: 10.1152/ajpcell.00121.2003. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang Y, Losurdo J, Keshavarzian A. Carbonylation and disassembly of the F-actin cytoskeleton in oxidant induced barrier dysfunction and its prevention by epidermal growth factor and transforming growth factor alpha in a human colonic cell line. Gut. 2000;46:830–837. doi: 10.1136/gut.46.6.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soberanes S, Urich D, Baker CM, Burgess Z, Chiarella SE, Bell EL, Ghio AJ, De Vizcaya-Ruiz A, Liu J, Ridge KM, Kamp DW, Chandel NS, Schumacker PT, Mutlu GM, Budinger GR. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J Biol Chem. 2009;284:2176–2186. doi: 10.1074/jbc.M808844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budinger GR, Tso M, McClintock DS, Dean DA, Sznajder JI, Chandel NS. Hyperoxia-induced apoptosis does not require mitochondrial reactive oxygen species and is regulated by Bcl-2 proteins. J Biol Chem. 2002;277:15654–15660. doi: 10.1074/jbc.M109317200. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett. 1999;454:173–176. doi: 10.1016/S0014-5793(99)00783-8. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Buccellato LJ, Tso M, Akinci OI, Chandel NS, Budinger GR. Reactive oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem. 2004;279:6753–6760. doi: 10.1074/jbc.M310145200. [DOI] [PubMed] [Google Scholar]
- Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A. Novel effect of NF-kappaB activation: carbonylation and nitration injury to cytoskeleton and disruption of monolayer barrier in intestinal epithelium. Am J Physiol Cell Physiol. 2004;287:C1139–1151. doi: 10.1152/ajpcell.00146.2004. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Farhadi A, Fields JZ, Shaikh M, Forsyth CB, Choudhary S, Keshavarzian A. Critical role of the atypical {lambda} isoform of protein kinase C (PKC-{lambda}) in oxidant-induced disruption of the microtubule cytoskeleton and barrier function of intestinal epithelium. J Pharmacol Exp Ther. 2005;312:458–471. doi: 10.1124/jpet.104.074591. [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Farhadi A, Fields JZ, Shaikh M, Keshavarzian A. PKC-beta1 isoform activation is required for EGF-induced NF-kappaB inactivation and IkappaBalpha stabilization and protection of F-actin assembly and barrier function in enterocyte monolayers. Am J Physiol Cell Physiol. 2004;286:C723–738. doi: 10.1152/ajpcell.00329.2003. [DOI] [PubMed] [Google Scholar]
- Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Ethanol-induced barrier dysfunction and its prevention by growth factors in human intestinal monolayers: evidence for oxidative and cytoskeletal mechanisms. J Pharmacol Exp Ther. 1999;291:1075–1085. [PubMed] [Google Scholar]
- Mutlu GM, Snyder C, Bellmeyer A, Wang H, Hawkins K, Soberanes S, Welch LC, Ghio AJ, Chandel NS, Kamp D, Sznajder JI, Budinger GR. Airborne particulate matter inhibits alveolar fluid reabsorption in mice via oxidant generation. Am J Respir Cell Mol Biol. 2006;34:670–676. doi: 10.1165/rcmb.2005-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33:1836–1846. doi: 10.1111/j.1530-0277.2009.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC Jr, Tager I. Air pollution and cardiovascular disease: a statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation. 2004;109:2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, Foiles N, Kamp DW, Ghio AJ, Chandel NS, Dean DA, Sznajder JI, Budinger GR. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest. 2007;117:2952–2961. doi: 10.1172/JCI30639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, von Klot S, Heier M, Trentinaglia I, Hormann A, Wichmann HE, Lowel H. Exposure to traffic and the onset of myocardial infarction. N Engl J Med. 2004;351:1721–1730. doi: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- McKenzie SJ, Baker MS, Buffinton GD, Doe WF. Evidence of oxidant-induced injury to epithelial cells during inflammatory bowel disease. J Clin Invest. 1996;98:136–141. doi: 10.1172/JCI118757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banan A, Farhadi A, Fields JZ, Mutlu E, Zhang L, Keshavarzian A. Evidence that nuclear factor-kappa B activation is critical in oxidant-induced disruption of the microtubule cytoskeleton and barrier integrity and that its inactivation is essential in epidermal growth factor-mediated protection of the monolayers of intestinal epithelia. J Pharmacol Exp Ther. 2003;306:13–28. doi: 10.1124/jpet.102.047415. [DOI] [PubMed] [Google Scholar]
- Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Scholmerich J, Gross V. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–369. doi: 10.1016/S0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut. 1998;42:477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavarzian A, Banan A, Farhadi A, Komanduri S, Mutlu E, Zhang Y, Fields JZ. Increases in free radicals and cytoskeletal protein oxidation and nitration in the colon of patients with inflammatory bowel disease. Gut. 2003;52:720–728. doi: 10.1136/gut.52.5.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, Rallabhandi P, Shea-Donohue T, Tamiz A, Alkan S, Netzel-Arnett S, Antalis T, Vogel SN, Fasano A. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–204. doi: 10.1053/j.gastro.2008.03.023. e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y, Arimoto T, Shinyashiki M, Shimojo N, Nakai Y, Yoshikawa T, Sagai M. Generation of reactive oxygen species during interaction of diesel exhaust particle components with NADPH-cytochrome P450 reductase and involvement of the bioactivation in the DNA damage. Free Radic Biol Med. 1997;22:479–487. doi: 10.1016/S0891-5849(96)00341-3. [DOI] [PubMed] [Google Scholar]
- Xia T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C, Nel A. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity. Environ Health Perspect. 2004;112:1347–1358. doi: 10.1289/ehp.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiura TS, Kaszubowski MP, Li N, Nel AE. Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J Immunol. 1999;163:5582–5591. [PubMed] [Google Scholar]
- Karczewski JM, Peters JG, Noordhoek J. Prevention of oxidant-induced cell death in Caco-2 colon carcinoma cells after inhibition of poly(ADP-ribose) polymerase and Ca2+ chelation: involvement of a common mechanism. Biochem Pharmacol. 1999;57:19–26. doi: 10.1016/S0006-2952(98)00286-X. [DOI] [PubMed] [Google Scholar]
- Ferruzza S, Scacchi M, Scarino ML, Sambuy Y. Iron and copper alter tight junction permeability in human intestinal Caco-2 cells by distinct mechanisms. Toxicol In Vitro. 2002;16:399–404. doi: 10.1016/S0887-2333(02)00020-6. [DOI] [PubMed] [Google Scholar]
- Mage D, Ozolins G, Peterson P, Webster A, Orthofer RVV, Gwynne M. Urban air pollution in megacities of the world. Atmospheric Environment. 1996;30:681–686. doi: 10.1016/1352-2310(95)00219-7. [DOI] [Google Scholar]
- Benchmarking Urban Air Quality Management and Practice in Major and Mega Cities of Asia. http://www.unep.org/PDF/APMA_Benchmarking_report.pdf
- de Hennezel L, Debarre S, Ramisse F, Delamanche S, Harf A, Alonso JM, Calvet JH. Plethysmography for the assessment of pneumococcal pneumonia and passive immunotherapy in a mouse model. Eur Respir J. 2001;17:94–99. doi: 10.1183/09031936.01.17100940. [DOI] [PubMed] [Google Scholar]