

Abstract
Objectives/Hypothesis
In response to chronic cigarette smoke exposure, a subset of patients present with edematous vocal folds, characteristically referred to as Reinke's edema. This phenotype differs from the tissue changes associated with prolonged smoke exposure in the lower airway, and the mechanism underlying Reinke's edema remains poorly described. We hypothesize that the effects of smoke are diffuse and involve both the epithelium and mucosa.
Study Design
In vitro, ex vivo experiment.
Methods
Transepithelial resistance (RT) was quantified in an ex vivo, viable, porcine vocal fold model. Excised tissue was exposed to cigarette smoke condensate (CSC) and RT was computed at baseline and 1 and 4 hours after exposure. In vitro, human vocal fold fibroblasts were exposed to CSC. Cyclooxygenase 2 (COX-2), microsomal prostaglandin E synthase-1, and 15-hydroxyprostaglandin dehydrogenase mRNA expression were assessed at 4 hours. Prostaglandin E2 (PGE2) synthesis was quantified via immunoassay following 24 hours of CSC exposure.
Results
CSC had no effect on RT. CSC did, however, induce COX-2 mRNA expression as well as its downstream lipid mediator PGE2. PGE2 metabolism appears to be regulated via both synthetic and degradative enzymes in response to cigarette smoke.
Conclusions
In vitro, CSC initiates an inflammatory response in vocal fold fibroblasts. However, in isolation, the epithelial resistance is not altered by CSC, at least acutely. These data may suggest a role for the interaction between the inflammatory response in the mucosa and compromised epithelial barrier function, as has been shown in other tissues.
Key Words
Vocal folds, voice, Reinke's edema, cyclooxygenase-2, prostaglandin, transepithelial resistance, cigarette smoke.
Introduction
The majority of investigations regarding the effects of cigarette smoke have focused on the lower airway, despite the anatomic position of the vocal folds and their propensity for exposure to a myriad of airborne pathogens and pollutants, including smoke. Although numerous studies have eloquently described the biochemical sequelae of smoke exposure on lung tissue, including aberrant mesenchymal cell activity and compromised epithelial integrity, no single study has attempted to describe the underlying pathophysiology associated with the benign response to cigarette smoke exposure in the vocal folds (i.e., the lack of tumorigenesis and/or Reinke's edema); this information is particularly germane because the response in the vocal folds may vary significantly from the response in the lower airway. To address this issue, we seek to provide preliminary data in regard to the pathophysiologic response to cigarette smoke exposure in both mesenchymal and epithelial cells of the vocal folds. The pathophysiologic response of both cell types is critical, as aberrant mesenchymal cell activity is implicated in alterations to the vocal fold microarchitecture, and the overlying epithelium purportedly provides some level of protection to the underlying mesenchyme (i.e., lamina propria).
The association between cigarette smoke and lower airway disease is well documented. Cigarette smoking has been shown to play a causative role in 95% of all cases of emphysema.1 Emphysema, a component of chronic obstructive pulmonary disease, is characterized by inflammation and permanent destruction of the bronchioles and surrounding aveoli.2 The mechanism of this chronic and debilitating disease process is likely twofold. First, cigarette smoke increases the permeability of bronchial epithelia and thereby enhances the access of noxious agents into the underlying connective tissue.3–6 The stratified squamous epithelial cells and junctional complexes of the vocal folds provide a barrier to regulate the flow of solute and solvent into the underlying connective tissue.7 Previously, our group has shown that a transient hypertonic challenge compromised barrier function by rapidly decreasing the transepithelial resistance (RT) in native vocal folds.8 Given the frank toxicity of many of the components of cigarette smoke, we hypothesize that short-term exposure to cigarette smoke condensate (CSC) will disrupt vocal fold epithelial barrier function by reducing the resistance across epithelial tissue, thereby providing support for an emerging translational model where exposure to cigarette smoke increases tissue permeability and places vocal folds at increased risk for damage from both the toxic constituents of cigarette smoke itself and other environmental pollutants.
Second, cigarette smoke has been shown to elicit a significant inflammatory response in many tissues; this response has been investigated extensively given the relationship between inflammation and malignancy. The mesenchymal cell response with regard to cyclooxygenase -2 (COX-2) and its downstream lipid mediator product, prostaglandin E2 (PGE2), is particularly relevant; in models of colon cancer, for example, fibroblasts underlying the epithelial cells express COX-2 first.9,10 Prostaglandin synthesis by these fibroblasts is thought to lead to epithelial cell damage and the increased potential for malignant transformation. Little is known about the effects of cigarette smoke on mesenchymal cells in the vocal folds, and increased insight may prove useful in the development of novel therapeutics.
We hypothesize that Reinke's edema is likely mediated by aberrant extracellular matrix metabolism and chronic inflammatory mediator synthesis by fibroblasts within the lamina propria. In addition, there is likely a cyclical effect whereby smoke independently augments epithelial barrier function, but this effect is also exacerbated by the secretion of inflammatory mediators within the mucosa leading to the stimulation of epithelial cells, prolonged inflammation and matrix aberrations, and further compromised epithelial barrier integrity, as shown in the lung.11 We therefore sought to provide preliminary data in regard to the acute effects of cigarette smoke on vocal fold epithelial barrier integrity independent of the underlying mucosa, as well the regulatory effects of cigarette smoke on COX-2 and PGE2 metabolism in vocal fold fibroblasts.
Materials and Methods
Vocal Fold Tissue
Larynges (N = 6) were obtained from domestic pigs following sacrifice and transported to the laboratory in cold, isotonic saline. After removal of surrounding muscle tissue, each larynx was cut open on the dorsal surface. The vocal fold epithelium from each hemisected half of the larynx was dissected from the underlying lamina propria and muscle for the measurement of RT.
Electrophysiology Measurement
The apparatus for the measurement of RT included the Ussing system and associated voltage-current clamp (World Precision Instruments, Sarasota, FL). The dissected tissue sample was mounted on a 6-mm Lucite cell, placed in the Ussing apparatus, and bathed with warm, oxygenated Hanks Balanced Salt Solution (Invitrogen, Carlsbad, CA) on both the apical and serosal surfaces. Potential difference and short-circuit current were recorded with the aid of current and voltage electrodes placed on the apical and serosal surface of the tissue. Ohms law was applied offline to calculate RT from the ratio of clamp voltage to change in short-circuit current. The clamp voltage was of 2-mV magnitude. Vocal fold tissue was monitored until the short circuit current stabilized (between 45 and 60 minutes). CSC was then added to the apical surface of the tissue at one of the following concentrations (1, 5, and 10μg/mL; n = 3 for each). Three vocal fold epithelia served as vehicle controls (dimethyl sulfoxide [DMSO]). Tissue was monitored for 4 hours after the addition of CSC or vehicle. RT measures were computed at baseline, immediately following the addition of CSC or DMSO, and then at 1 hour and 4 hours following CSC or DMSO exposure.
CSC
CSC was purchased from Murty Pharmaceuticals (Lexington, KY). This formulation had been used by our laboratory previously12; it contains cigarette particulate suspended in DMSO. As such, DMSO/vehicle controls at the highest concentration were used for each experiment.
Cell model
The genetically immortalized human vocal fold fibroblast (HVOX) cell line established in our laboratory was used in the current series of experiments.13
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total cellular RNA was isolated from HVOX after 4 hours of CSC treatment and was then reverse-transcribed and amplified by using the OneStep RT-PCR Kit (Qiagen Inc., Santa Clara, CA) following the manufacturer's protocol. Briefly, 13.0 μL of OneStep PCR Mix was added to 10 ng RNA with 2.0 μL of each primer, incubated at 37°C for 1 hour, heated to 95°C for 6 minutes, and then subjected to 28 cycles of 30 seconds at 37°C, 30 seconds at 60°C, and 1 minute at 72°C. Polymerase chain reaction products were electrophoresed on a 1.5% agarose gel containing ethidium bromide. All experiments were performed in triplicate. Photographs of the resulting gel electrophoreses were subjected to image analysis using ImageJ (National Institutes of Health; http://rsb.info.nih.gov/ij/docs/faqs.html) and standardized to β-actin run with the same samples.
Enzyme-Linked Immunoassay
PGE2 synthesis was assayed via a commercially available enzyme-linked immunoassay kit (R&D Systems, Minneapolis, MN) following 24 hours of CSC treatment.
Results
CSC Did Not Alter RT
Although a trend toward increased RT was observed at both 1 and 4 hours of CSC treatment relative to vehicle controls, this effect did not achieve statistical significance (Fig. 1) (P = .075 and P =.167, respectively).
Fig. 1.
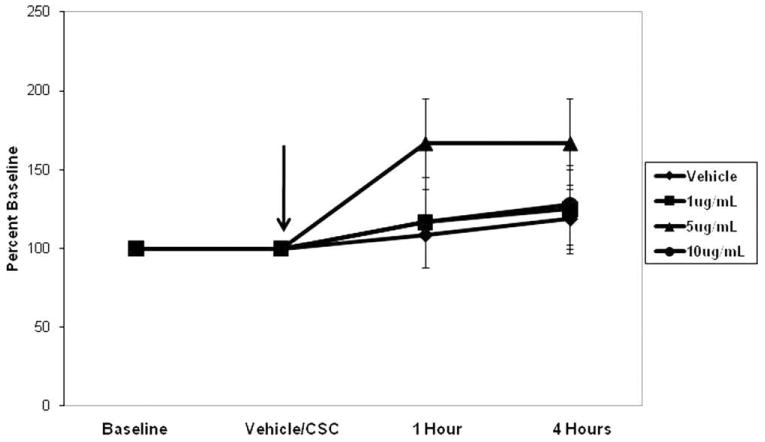
Cigarette smoke condensate (CSC) did not alter transepithelial resistance at either 1 or 4 hours, relative to the controls (P = .075 and P = .167, respectively). Data are plotted relative to baseline (100%).
CSC Increased COX-2 mRNA Expression
As shown in Figure 2A, 4 hours of CSC treatment increased COX-2 mRNA expression in HVOX cells, qualitatively, compared to vehicle controls. When subjected to densitometric analyses, this trend was confirmed. However, a statistically significant increase in COX-2 mRNA expression was only obtained at 10 μg/mL CSC (Fig. 2B) (main effect: P = .01; 10% CSC: P = .006). It is unclear whether this response is “permanent” or if the inflammatory phenotype resolves with the removal of CSC.
Fig. 2.
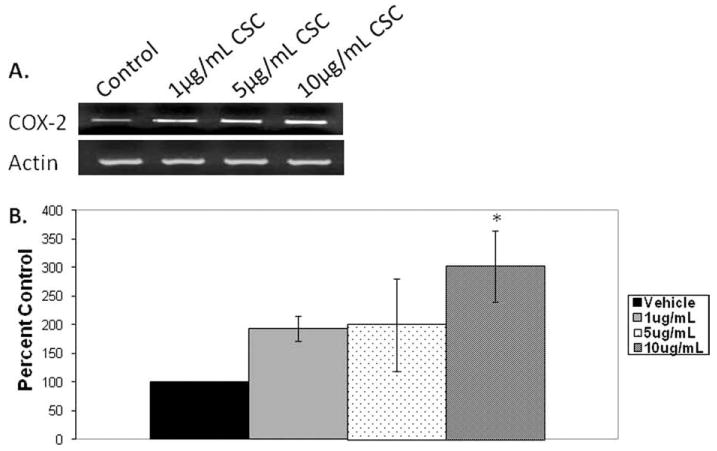
Cigarette smoke condensate (CSC) increased cyclooxygenase 2 (COX-2) mRNA expression at 4 hours in human vocal fold fibroblasts (P = .006). (A) Representative gel. (B) Densitometric analysis.
Fig. 4.
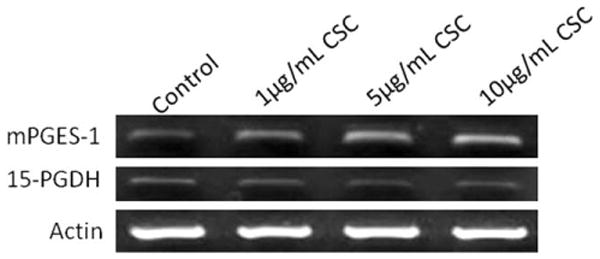
Cigarette smoke condensate (CSC) increased microsomal prostaglandin E synthase-1 (mPGES-1) and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) mRNA expression at 4 hours in human vocal fold fibroblasts (representative gels).
CSC Increased PGE2 Synthesis and Altered PGE2 Metabolizing Enzyme mRNA
PGE2 synthesis into the supernatant was quantified following 24 hours of CSC treatment in HVOX cells. CSC induced a statistically significant increase in PGE2 synthesis (Fig. 3) (P = .007). Although the lowest dose of CSC increased PGE2 synthesis by nearly 100%, post hoc analysis confirmed that this response was only significant at 5 and 10 μg/mL CSC (P = .011 and P = .013, respectively), compared to vehicle controls. Both of these higher concentrations increased PGE2 synthesis approximately three-fold. The increase in PGE2 synthesis correlated with increased steady state mRNA levels of the PGE2 biosynthetic enzyme, microsomal prostaglandin E synthase-1 (mPGES-1). CSC, however, had little effect on the biodegradation enzyme, 15-hydroxyprostaglandin dehydrogenase (Fig. 4).
Fig. 3.
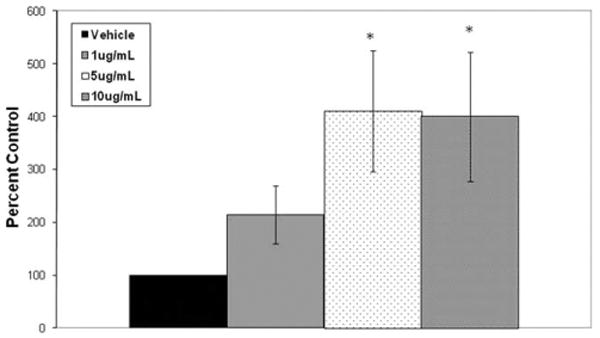
Cigarette smoke condensate increased prostaglandin E2 synthesis at 24 hours by human vocal fold fibroblasts (*P =.007).
Discussion
Our knowledge of the benign effects of cigarette smoke arises largely from investigations of the lower airway. In bronchial and lung tissue, smoke exposure induces fibrosis and is associated with reactive airway disease. The proposed link between tissue fibrosis and malignancy is also receiving increased attention, as fibroblasts have been shown to drive malignant tumorigenesis. In contrast, the tissue phenotype in the vocal folds appears quite different, and it has been hypothesized that Reinke's edema, the most common voice problem in smokers, appears to be mediated via both alterations in epithelial barrier function as well as an inflammatory response; edema may, in fact, be protective against malignant transformation.12 Changes to epithelia and fibroblast function were investigated in the current study, in the context of a diverse tissue response in the vocal folds as compared to other airway tissue.
The stratified squamous epithelia of the vocal folds have been posited to provide an effective barrier to noxious, inhaled agents. Our data suggest that even high concentrations of CSC for 4-hour exposure periods did not adversely affect RT; this finding supports a role for the robust protection provided by the epithelia. In native canine tracheal epithelia, the addition of cigarette smoke particulate to the apical surface actually increased RT.14 The reasons for the nonsignificant change in RT observed in the current study are unknown, but several explanations are plausible. First, it is possible that 4 hours of CSC exposure does not mimic the chronic exposure observed in vivo. Furthermore, the translational equivalent with regard to both the concentration and duration of CSC exposure in vitro and ex vivo conditions are largely unknown, but were modeled after previous investigation of the lower airway. Chronic exposure may be required to degrade the stratified squamous epithelial barrier over time. Second, as mentioned in the Introduction, we hypothesize that the underlying mucosa and the inherent inflammatory response may contribute to the degradation of epithelial barrier function in the vocal folds. This phenomenon warrants further investigation.
Based on these barrier function data, it is difficult to determine the extent to which the mesenchymal cells of the vocal fold are exposed to the components of cigarette smoke. However, our data suggest that CSC induces COX-2 expression in human vocal fold fibroblasts. This finding is consistent with data from human lung fibroblasts, and this upregulation has been hypothesized to play an integral role in inflammatory diseases such as chronic obstructive pulmonary disease as well as tumorigenesis.15 Our cells also secreted PGE2 in a temporal manner similar to that seen in previous investigations of cells in the lower airway. This response correlated to an increase in steady-state mPGES-1 expression with CSC exposure. mPGES-1 is the enzyme responsible for converting prostaglandin H2 to PGE2, and this lipid mediator has been implicated in a variety of pathologic processes, including activation of uncontrolled cell proliferation and tumorigenesis in the presence of DNA damage.15 Cumulatively, these data are the first to show the effects of cigarette smoke on both the epithelial and mesenchymal cells of the vocal folds. We hypothesize that increased insight into the biologic mechanisms underlying vocal fold tissue health may ultimately drive novel therapeutics. Furthermore, future studies should determine whether exposure to general irritants such as nitrogen components in smog or ammonia yield similar effects, as these irritants are likely much more prevalent that tobacco exposure.
Conclusion
CSC elicited a similar response with regard to the induction of COX-2 and its downstream lipid mediator, PGE2, when compared to the lower airway in studies by others. Acute CSC exposure did not alter epithelial resistance; this finding suggests that increased duration of CSC exposure may be required for alterations to epithelial barrier integrity. Furthermore, methods used in the current study isolated the vocal fold epithelium. The interactions between the mucosa and epithelium are nontrivial and warrant further investigation.
Acknowledgments
The authors wish to express their sincere gratitude to Elizabeth Erickson, Jennifer Montgomery, and Jenna Olson for their assistance at various stages of this study.
This work was funded by the NIH/NIDCD (RO3 DC010267), Hackers for Hope, The Langeloth Foundation, and the Garban Fund. The authors have no other funding, financial relationships, or conflicts of interest to disclose.
Footnotes
Experiments on excised larynges were completed according to regulations at Purdue University.
Bibliography
- 1.Pauwels RA, Buist AS, Ma P, Jenkins CR, Hurd SS GOLD Scientific Committee. Respir Care. Vol. 46. 2001. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: National Heart, Lung, and Blood Intsitute and World Health Organization Global Initiative for Chronic Obstructive Lung Disease (GOLD): executive summary; pp. 798–825. [PubMed] [Google Scholar]
- 2.Snider GL. Chronic obstructive pulmonary disease: risk factors, pathophysiology, and pathogenesis. Ann Rev Med. 1989;40:411–429. doi: 10.1146/annurev.me.40.020189.002211. [DOI] [PubMed] [Google Scholar]
- 3.Simani AS, Inoue S, Hogg JC. Penetration of the respiratory epithelium of guinea pigs following exposure to cigarette smoke. Lab Invest. 1974;31:75–81. [PubMed] [Google Scholar]
- 4.Burns AR, Hosford SP, Dunn LA, Walker DC, Hogg JC. Respiratory epithelial permeability after cigarette smoke exposure in guinea pigs. J Appl Physiol. 1989;66:2109–2116. doi: 10.1152/jappl.1989.66.5.2109. [DOI] [PubMed] [Google Scholar]
- 5.Jones JG, Minty BD, Lawler P, Hulands G, Crawley JC, Veall N. Increased alveolar epithelial permeability in cigarette smokers. Lancet. 1980;12:66–68. doi: 10.1016/s0140-6736(80)90493-6. [DOI] [PubMed] [Google Scholar]
- 6.Dusser DJ, Minty BD, Collignon MA, Hinge D, Barritault LG, Huchon GJ. Regional respiratory clearance of aerosolized 99mTc-DTPA: posture and smoking effects. J Appl Physiol. 1986;60:2000–2006. doi: 10.1152/jappl.1986.60.6.2000. [DOI] [PubMed] [Google Scholar]
- 7.Fisher K, Telser A, Phillips J, Yeates D. Regulation of vocal fold transepithelial water fluxes. J Appl Physiol. 2001;91:1401–1411. doi: 10.1152/jappl.2001.91.3.1401. [DOI] [PubMed] [Google Scholar]
- 8.Sivasankar M, Erickson E, Rosenblatt M, Branski RC. Hypertonic challenge to porcine vocal folds: Effects on epithelial barrier function. Otolaryngol Head Neck Surg. 2010;142:79–84. doi: 10.1016/j.otohns.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheehan KM, Sheahan K, O'Donoghue DP, et al. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA. 1999;282:1254–1257. doi: 10.1001/jama.282.13.1254. [DOI] [PubMed] [Google Scholar]
- 10.Sonoshita M, Takaku K, Oshima M, Suighara K, Taketo MM. Cyclooxygenase-2 expression in fibroblasts and endothelial cells of intestinal polyps. Cancer Res. 2002;62:6846–6849. [PubMed] [Google Scholar]
- 11.Gauldie J, Kolb M, Sime PJ. A new direction in the pathogenesis of idiopathic pulmonary fibrosis? Respir Res. 2002;3:1. doi: 10.1186/rr158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Branski RC, Saltman B, Sulica L, et al. Cigarette smoke and reactive oxygen species metabolism: Implications for the pathophysiology of Reinke's edema. Laryngoscope. 2009;119:2014–2018. doi: 10.1002/lary.20592. [DOI] [PubMed] [Google Scholar]
- 13.Branski RC, Barbieri SS, Weksler BB, et al. The effects of transforming growth factor-beta1 on human vocal fold fibroblasts. Ann Otol Rhinol Laryngol. 2009;118:218–226. doi: 10.1177/000348940911800310. [DOI] [PubMed] [Google Scholar]
- 14.Welsh M. Cigarette smoke inhibition of ion transport in canine tracheal epithelium. J Clin Invest. 1983;71:1614–1623. doi: 10.1172/JCI110917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martey CA, Pollock SJ, Turner CK. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287:L981–L991. doi: 10.1152/ajplung.00239.2003. [DOI] [PubMed] [Google Scholar]