

Abstract
Objectives/Hypothesis
Inflammation and its role in a coordinated fibroplastic response, which disrupts the structure of the vocal folds following injury, is critical. Cyclooxygenase-2 (COX-2) is an important enzyme involved in both inflammation and fibrosis; in addition, it is a prime target for therapeutic intervention. We sought to study this pathway in vocal fold fibroblasts to provide a foundation for future interventional studies.
Study Design
In vitro.
Methods
Human vocal fold fibroblasts were incubated with IL-1β to determine the effects on COX-2 signaling, along with upstream regulatory mechanisms and downstream mediators of wound healing. In vitro methods to assess mRNA expression, as well as intracellular and secreted protein (sodium dodecyl sulfate polyacrylamide gel electrophoresis and enzyme-linked immunosorbent assay) were employed.
Results
IL-1β regulation of COX-2 mRNA and protein levels was dose and time dependent and IL-1β altered PGE2 metabolism, via regulation of both synthetic and degradative enzymes. IL-1β increased nuclear factor (NF)-κB activation and nuclear translocation. Inhibition of the p50 and p65 subunits of NF-κB decreased IL-1β-induced COX-2 transcription. IL-1β also altered mRNA expression of four cell-surface prostaglandin receptors.
Conclusions
Inflammation and fibrosis are important in the vocal fold pathophysiologic response to injury. Our data suggest that COX-2 and PGE2 are inducible in human vocal fold fibroblasts, and this response appears to be NF-κB-dependent. We purport this fundamental investigation will lead to increased insight regarding injury and repair in the vocal folds, with the ultimate goal of developing novel clinical care paradigms.
Keywords: Vocal fold, inflammation, cyclooxygenase, prostaglandin, NF-κB
INTRODUCTION
Inflammation and its putative role in the coordination of a fibrotic response following injury is important in many organ systems. Given that disruption to the structure of the vocal folds is accompanied by potentially significant dysphonia,1 insight into the pathophysiology underlying fibrosis might be particularly relevant. However, understanding of the specific biochemical and regulatory mechanisms underlying vocal fold injury and repair are only recently evolving. Cyclooxygenase (COX)-2 is an interesting enzyme given that its main downstream product, prostaglandin E2 (PGE2) has been shown to regulate both inflammatory and fibroplastic processes during tissue repair.2,3 This pathway is activated by various inflammatory mediators including interleukin (IL)-1β, and both COX-2 and IL-1β have been implicated in vocal fold injury. Our laboratory and others have previously described increased IL-1β in both surgical and phonotraumatic injury in human and animal models.4,5 In addition, increased COX-2 expression has been documented in phonotraumatic vocal fold lesions.6
In the skin, COX-2 expression and PGE2 production in the wound bed increase in the transition from scarless to a fibrotic healing phenotype,3 and in cases of impaired PGE2 signaling, wound healing is altered.7-14 In contrast, during early embryonic development, tissue repair is characterized by a limited inflammatory response due to the lack of inflammatory lineages. In humans, injury prior to the onset of the third trimester typically results in scarless healing.15 In addition, the oral mucosa is less prone to scar formation, which correlates with a diminished inflammatory profile,16 and anecdotal evidence suggests that the inflammatory response decreases with advancing age accompanied by reduced scarring.17 These phenomena have been confirmed in various animal models. For example, the temporal dynamics of healing in the PU.1 knockout model, missing several leukocytic lineages including mast cells, neutrophils, and macrophages, are similar to wild-type. However, these animals heal with significantly reduced scarring.18 Given that vocal fold fibrosis continues to perplex clinicians, perhaps the ideal therapeutic strategy is to avoid it through modulation of the inflammatory response.
Also of particular relevance to COX-2 and PGE2 signaling is that the regulatory effects of PGE2 appear to be organ specific. Baseline PGE2 concentrations are substantially higher in the lung compared to other tissues, and it is hypothesized that PGE2 acts to limit the immune-inflammatory response and facilitate a more regenerative model of healing.19 Furthermore, increased bleomycin-induced pulmonary fibrosis was observed in COX-2 deficient mice,20 suggesting the potential that the lung is a privileged site for PGE2.19 Given the anatomical proximity of the larynx to the lung, insight regarding COX-2 and PGE2 is critically important in the development of novel clinical care paradigms. Our laboratory previously observed PGE2 to have a bimodal expression pattern with peak expression in the acute inflammatory phase and then increased concentrations approximately 1 to 2 weeks following injury.4 This pattern of expression is likely related to the complex nature of the PGE2 interactions with mesenchymal cells.
With regard to PGE2 metabolism, PGE2 synthases (PGES) convert PGH2 into PGE2.21 PGE2 acts through four E-prostanoid receptors coupled to G-proteins and calcium and cyclic adenosine monophosphate cAMP signaling.22-27 PGE2 inactivation occurs primarily via enzymatic oxidation by 15-hydroxy prostaglandin dehydrogenase (15-PGDH).28 Upstream, regulation of COX-2 appears to be nuclear factor (NF)-κB-dependent in many cases. Mammalian NF-κB is made up of multiple subunits, including p65 (RelA) and p50. These complexes remain inactive and sequestered in the cytoplasm through binding to the inhibitor of NF-κB (IκB) family of proteins. Activation of this pathway is mediated via IκB kinases that induce phosphorylation and degradation of IκB proteins. This activated complex then translocates to the nucleus where it binds to specific DNA sequences (κB sites) to regulate transcription of multiple genes, including COX-2.29
We hypothesize that increased insight into the inflammatory response in vocal fold fibroblasts, and furthermore the relationship between inflammatory events and the fibrotic fibroblast phenotype, will eventually lead to improved therapeutic strategies for this challenging patient population. We sought to describe this signaling pathway, acknowledging that altering various components of the cascade might lead to less favorable outcomes, similar to the lower airway. In the current study, we sought to describe the regulatory effects of IL-1β on COX-2 signaling in an immortalized human vocal fold fibroblast cell line, with the goal providing a foundation for future, intervention studies.
MATERIALS AND METHODS
Cell Model and Reagents
The HVOX human vocal fold fibroblast cell line was used in the current series of experiments.30 IL-1β was purchased from Sigma-Aldrich (St. Louis, MO). Specific inhibitors to the p50 (NK-κB p50 NLS inhibitory peptide) and p65 (NF-κB p65 Ser529/536 inhibitory peptide) subunits of NF-κB were purchased from Imgenex (San Diego, CA).
Reverse Transcriptase-Polymerase Chain Reaction
Total cellular RNA was isolated from HVOX, reverse transcribed, and amplified utilizing the OneStep RT-PCR Kit (Qiagen Inc., Santa Clara, CA) following the manufacturer’s protocol. Briefly, 13.0 μL of OneStep PCR Mix was added to 10 ng RNA with 2.0 μL of each primer, incubated at 37°C for 1 hour, heated to 95°C for 6 minutes, and then subjected to 28 cycles of 30 seconds at 37°C, 30 seconds at 60°C, and 1 minute at 72°C. Polymerase chain reaction products were electrophoresed on a 1.5% agarose gel containing ethidium bromide. All experiments were performed in triplicate. Photographs of the resulting gel electrophoreses were subjected to image analysis using ImageJ (National Institutes of Health, Bethesda, MD) and standardized to β-actin, run concurrently with the same samples.
Western Blots
Cells were lysed with M-PER (Mammalian Protein Extraction Solution; Pierce, Rockford, IL). Equivalent amounts of proteins were separated by 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel and transferred to nitrocellulose membranes (Whatman, Kent, United Kingdom). The membranes were blocked overnight in 5% nonfat milk, and after rinsing, incubated at room temperature for 1 hour with the primary antibodies against COX-2 (1:10,000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). After washing, blots were subsequently incubated for 1 hour at room temperature with horseradish peroxidase-labelled secondary antibody (Bio-Rad Western Blot Kit; Bio-Rad Laboratories, Hercules, CA). The Bio-Rad Western Blot Kit was then utilized according to the manufacturer’s recommended protocol to detect specifically labeled bands.
Enzyme-Linked Immunoassay
PGE2 synthesis was assayed via a commercially-available enzyme-linked immunoassay (ELISA) kit (R&D Systems, Minneapolis, MN). Concentrations were standardized to total cellular protein.
NF-κB Activation and Nuclear Translocation
Activated nuclear NF-κB in HVOX cells 1 hour following treatment with IL-1β was assayed using commercially available ELISAs for both the p50 and p65 subunits (Active Motif, Carlsbad, CA). Nuclear/cytoplasm fractionation was performed via a commercially available kit (Active Motif).
Statistical Analyses
One-way analyses of variance were performed using SPSS version 12.0 (SPSS Inc., Chicago, IL). Pending a significant main effect at P = .05, post hoc comparisons were employed using the Tukey method.
RESULTS
IL-1β Induced COX-2 Transcription, Steady State Protein Levels, and PGE2 Synthesis
As shown in Figure 1A and 1B, IL-1β increased COX-2 mRNA expression as early as 1 hour (P < .001) and expression continued to increase through 4 hours in a dose-dependent fashion (P = .002). Steady state COX-2 protein levels increased as early as 6 hours following IL-1β treatment (Fig. 1C, representative gel). Following 24 hours of IL-1β treatment, PGE2 secretion into the supernatant increased in a dose-dependent manner (Fig. 1D; P < .001).
Fig. 1.
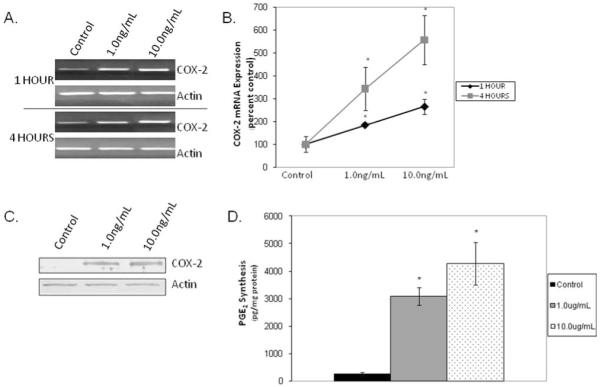
Interleukin (IL)-1β-induced cyclooxygenase-2 (COX-2) mRNA expression at 1 and 4 hours was dose and time dependent; (A) representative gel and (B) densitometric analyses; n = 3. IL-1β increased steady state COX-2 protein levels as early as 6 hours; (C) representative gel; as well as (D) prostaglandin E2 (PGE2) synthesis at 24 hours (n = 3; *P < .05).
IL-1β Regulated P50 and P65 Transcription, Activation, and Nuclear Translocation
One hour of IL-1β treatment increased both p50 and p65 subunit mRNA expression modestly (Fig. 2A). The ratio of nuclear to cytoplasmic-activated p50 and p65 subunits was calculated based on ELISA data to quantify NF-κB activation. At baseline, the nuclear to cytoplasmic ratios of both p50 and p65 were <1 (i.e., increased concentrations in the cytoplasm compared to the nucleus). As shown in Figure 2B, within 1 hour of IL-1β treatment, nuclear translocation of both subunits was observed with ratios of approximately 1.5 to 2.5 for both subunits (P < .001; control versus IL-1β treatment). No significant dose response was observed for either subunit.
Fig. 2.
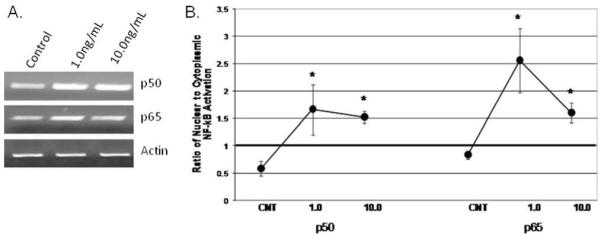
Interleukin (IL)-1β treatment for 1 hour increased transcription of the p50 and p65 subunits of nuclear factor (NF)-κB (A) (representative gel) and (B) increased nuclear translocation of both subunits (n = 3; *P < .05) at 1 hour.
IL-1β-Induced COX-2 Expression is NF-κB Dependent
We sought to determine the effects of inhibiting the p50 and p65 subunits both separately and in combination to confirm the regulatory role of these proteins in IL-1β-induced COX-2 mRNA expression. As proof of principle, we investigated the effect of these inhibitors on NF-κB activation as determined by ELISA of the cytoplasmic and nuclear fractions. As shown in Figure 3A, these inhibitors diminished the inherent NF-κB response to IL-1β, back to baseline. Interestingly, as shown in Figure 3B, the p50 inhibitor alone increased baseline COX-2 mRNA expression, and when the cells were cotreated with the p50 inhibitor and IL-1β, COX-2 mRNA expression increased when compared to IL-1β treatment alone. In contrast, the p65 inhibitor had no effect on basal COX-2 mRNA expression and yielded a moderate decrease in IL-1β-induced COX-2 expression. However, when used in combination, the p50 and p65 inhibitors completely abrogated IL-1β-induced COX-2 mRNA expression.
Fig. 3.
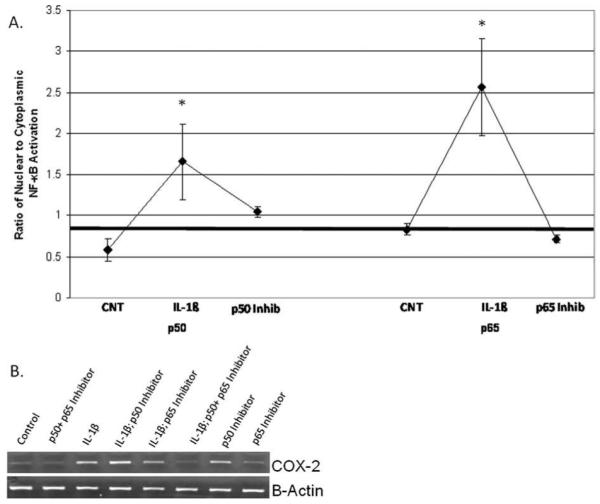
Pretreatment (2 hours) with inhibitors of the p50 and p65 subunits of nuclear factor (NF)-κB decreased interleukin (IL)-1β-induced nuclear translocation of each subunit (A) (n = 3). Inhibition of the p50 and p65 subunits individually had no effect on IL-1β-induced cyclooxygenase-2 (COX-2) mRNA expression. (B) Combined inhibition of both subunits abrogated this effect (representative gel).
IL-1β Regulates the PGE2 Synthetic and Degradative Enzymes
Next, we sought to investigate the effects of IL-1β on the synthetic and degradative enzymes associated with PGE2 metabolism. As shown in Figure 4, IL-1β increased mPGES-1 mRNA in a time- and dose-dependent manner at 1 hour and 4 hours (Fig. 4A; P = .017 and P = .001, respectively). IL-1β decreased 15-PGDH mRNA expression in HVOX cells (Fig. 4B, representative gel), but this effect was not observed until 24 hours.
Fig. 4.

Interleukin (IL)-1β increased mPGES-1 mRNA expression in a dose- and time-dependent manner (A) (densitometric analysis; n = 3; *P < .05). IL-1β decreased 15-PGDH mRNA expression in a similar dose- and time-dependent manner (B).
Vocal Fold Fibroblasts Express the Four EP Receptors, and mRNA Expression of These Receptors Is Regulated by IL-1β
Given that PGE2 signaling is likely both paracrine and autocrine, we sought to describe the EP receptor response to IL-1β. As shown in Figure 5 (representative gel), HVOX express all four EP receptors. Receptor mRNA levels increased in a dose-dependent manner in response to IL-1β. This response was consistent across all four receptors.
Fig. 5.
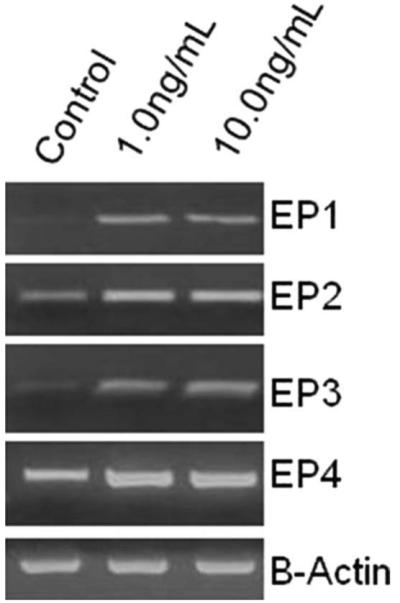
Interleukin-1β increased transcription of the four cell surface prostaglandin receptors (1 hour; representative gel).
DISCUSSION
During early embryonic development, tissue repair is rapid and results in recapitulation of the uninjured tissue. This response is characterized by a limited inflammatory response due to the lack of inflammatory lineages.15 To date, this link between inflammation and fibrosis has not been exploited therapeutically for vocal fold injury. We hypothesize that modulation of the acute, inflammatory response following vocal fold injury might lead to improved long-term histological and functional outcomes. Specifically, we hypothesize that COX-2 expression and PGE2 production in the wound bed increase in the transition from scarless to a fibrotic healing phenotype as has been described in the skin.3 Our laboratory and others have provided cursory information regarding the acute response to surgical injury implicating both COX-2 and PGE2 in vocal fold injury and repair.6,32 Systemic COX-2 inhibition has been shown to effectively limit fibrosis in multiple tissues, including the kidney, ureter, and heart.32-35 In addition, topical COX-2 inhibition has been shown to diminish cutaneous fibrosis.36 However, as mentioned previously, COX-2 inhibition in the lung is in fact profibrotic, and therefore a suboptimal therapeutic option.
As such, we sought to describe the fundamental COX-2 and PGE2 signaling machinery in our human vocal fold fibroblast cell line. IL-1β, a prototypic proinflammatory mediator implicated in vocal fold injury,4,38,39 induced COX-2 mRNA and protein levels in a dose- and time-dependent manner. Transcription of COX-2 appears to be NF-κB dependent, as inhibition of both the p50 and p65 subunits of NF-κB attenuates IL-1β-induced COX-2 transcription. However, our finding that the inhibition of the p50 subunit in isolation does not in fact limit transcription is confounding, as these subunits are known to both homo- and heterodimerize,39 and inhibition of either of the subunits should effectively inhibit transcription. One might speculate the COX-2 transcription is therefore highly dependent on the p65 subunit as has been previously described in endothelial cells, for example.40 This phenomena warrants future investigation. In addition, this observation might also suggest the potential for alternative signaling pathways mediating COX-2 metabolism in vocal fold fibroblasts as described in synoviocytes, for example. In these cells, stress proteins were actually shown to block NF-κB activation, and transcription of multiple proinflammatory genes was mediated via map kinase.41
In our cells, IL-1β regulated PGE2 metabolism via increased transcription of the major synthetic enzyme (mPGES-1) as well as decreased expression of the degradative enzyme 15-PGDH. Similar to other cell lines, PGE2 signaling in vocal fold fibroblasts appears to be both autocrine and paracrine as IL-1β increased PGE2 production, limited PGE2 degradation, and altered the expression profile of the four cell surface prostaglandin receptors. Beyond insight regarding fundamental signaling pathways in human vocal fold fibroblasts, it is hypothesized that this type of investigation might eventually provide a platform for advances in clinical care paradigms.
CONCLUSION
Fundamental investigation regarding cell behavior in the context of vocal fold injury and repair might hold the potential to ultimately lead to physiologically sound pharmacological therapies for many patients with voice disorders. Our data suggest that COX-2 and its lipid mediator byproduct, PGE2 are inducible in human vocal fold fibroblasts, and induction of this enzyme appears to be NF-κB dependent. COX-2 has been shown to be a viable therapeutic target for several models of injury and repair; pharmacological agents with a direct impact on COX-2 signaling are readily available. We hypothesize that this pathway might prove useful as a means to limit the inflammatory response following vocal fold injury, and therefore facilitate a more regenerative model of vocal fold repair.
Acknowledgments
This work was funded by the NIH/NIDCD (RO3 DC010267), Hackers for Hope, The Langeloth Foundation, and the Garban Fund. The authors have no other funding, financial relationships, or conflicts of interest to disclose.
Footnotes
Portions of this manuscript were presented at the 2010 American Laryngological Association Combined Otolaryngology Spring Meetings, Las Vegas, Nevada, U.S.A., April 28–May 2, 2010.
BIBLIOGRAPHY
- 1.Benninger MS, Alessi D, Archer S, et al. Vocal fold scarring: current concepts and management. Otolaryngol Head Neck Surg. 1996;115:474–482. doi: 10.1177/019459989611500521. [DOI] [PubMed] [Google Scholar]
- 2.Futagami A, Ishizaki M, Fukuda Y, Kawana S, Yamanaka N. Wound healing involves induction of cyclooxygenase-2 expression in rat skin. Lab Invest. 2002;82:1503–1513. doi: 10.1097/01.lab.0000035024.75914.39. [DOI] [PubMed] [Google Scholar]
- 3.Wilgus TA, Bergdall VK, Tober KL, et al. The impact of cyclooxygenase-2 mediated inflammation on scarless wound healing. Am J Pathol. 2004;165:753–761. doi: 10.1016/S0002-9440(10)63338-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branski R, Rosen C, Verdolini K, Hebda P. Biochemical markers associated with acute vocal fold wound healing: a rabbit model. J Voice. 2005;19:283–289. doi: 10.1016/j.jvoice.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Verdolini K, Rosen C, Branski R, Hebda P. Shifts in biochemical markers associated with wound healing in laryngeal secretions following phonotrauma: a preliminary study. Ann Otol Rhinol Laryngol. 2003;112:1021–1025. doi: 10.1177/000348940311201205. [DOI] [PubMed] [Google Scholar]
- 6.Karahan N, Baspinar S, Yariktas M, Kapucuoglu N. Matrix metalloproteinases (MMP-2 and MMP-9) and cyclooxygenase-2 (COX-2) expressions in vocal cord polyps. J Voice. 2009;23:29–33. doi: 10.1016/j.jvoice.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Noguchi K, Iwasaki K, Shitashige M, et al. Downregulation of lipopolysaccharide-induced intercellular adhesion molecule-1 expression via EP2/EP4 receptors by prostaglandin E2 in human fibroblasts. Inflammation. 2001;25:75–81. doi: 10.1023/a:1007110304044. [DOI] [PubMed] [Google Scholar]
- 8.Noguchi K, Iwasaki K, Shitashige M, Ishikawa I. Prostaglandin E2 receptors of the EP2 and EP4 subtypes downregulate tumor necrosis factor alpha-induced intercellular adhesion molecule-1 expression in human gingival fibroblasts. J Periodontal Res. 1999;34:478–485. doi: 10.1111/j.1600-0765.1999.tb02284.x. [DOI] [PubMed] [Google Scholar]
- 9.Kohyama T, Ertl RF, Valenti V, et al. Prostaglandin E(2) inhibits fibroblast chemotaxis. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1257–L1263. doi: 10.1152/ajplung.2001.281.5.L1257. [DOI] [PubMed] [Google Scholar]
- 10.Choung J, Taylor L. Role of EP2 receptors and cAMP in prostaglandin E2 regulated expression of type 1 collagen alpha 1, lysyl hydroxylase, and cyclooxygenase-1 genes in human embryo lung fibroblasts. J Cell Biochem. 1998;71:254–263. [PubMed] [Google Scholar]
- 11.Schmitz T, Dallot E, Leroy MJ, Breuiller-Fouche M, Ferre F, Cabrol D. EP(4) receptors mediate prostaglandin E(2)-stimulated glycosaminoglycan synthesis in human cervical fibroblasts in culture. Mol Hum Reprod. 2001;7:397–402. doi: 10.1093/molehr/7.4.397. [DOI] [PubMed] [Google Scholar]
- 12.Kanekura T, Higashi Y, Kazaki T. Cyclooxygenase-2 expression and prostaglandin E2 biosynthesis are enhanced in scleroderma fibroblasts and inhibited by UVA irradiation. J Rheumatol. 2001;28:1568–1572. [PubMed] [Google Scholar]
- 13.Vancheri C, Sortino MA, Tomaselli V, et al. Different expression of TNF-alpha receptors and prostaglandin E(2) production in normal and fibrotic lung fibroblasts: potential implications for the evolution of the inflammatory process. Am J Respir Cell Mol Biol. 2000;22:628–634. doi: 10.1165/ajrcmb.22.5.3948. [DOI] [PubMed] [Google Scholar]
- 14.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest. 1995;95:1861–1865. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzick NS, Longaker MT. Scarless fetal healing. Therapeutic implications. Ann Surg. 1992;215:3–7. doi: 10.1097/00000658-199201000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szpaderska AM, Zuckerman JD, DiPietro LA. Differential injury responses in oral mucosal and cutaneous wounds. J Dent Res. 2003;82:621–626. doi: 10.1177/154405910308200810. [DOI] [PubMed] [Google Scholar]
- 17.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. 2007;127:1009–1017. doi: 10.1038/sj.jid.5700811. [DOI] [PubMed] [Google Scholar]
- 18.Martin P, D’Souza D, Martin J, et al. Wound healing in the PU.1 null mouse—tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;1:1122–1128. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- 19.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004;24:40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Keerthisingam CB, Jenkins RG, Harrison NK, et al. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol. 2001;158:1411–1422. doi: 10.1016/s0002-9440(10)64092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naraba H, Yokoyama C, Tago N, et al. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J Biol Chem. 2002;277:28601–28608. doi: 10.1074/jbc.M203618200. [DOI] [PubMed] [Google Scholar]
- 22.Abramovitz M, Adam M, Boie Y, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochem Biophys Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 23.Desai S, April H, Nwaneshiudu C, Ashby B. Comparison of agonist-induced internalization of the human EP2 and EP4 prostaglandin receptors: role of the carboxyl terminus in EP4 receptor sequestration. Mol Pharmacol. 2000;58:1279–1286. doi: 10.1124/mol.58.6.1279. [DOI] [PubMed] [Google Scholar]
- 24.Bhattacharya M, Peri K, Ribeiro-da-Silva A, et al. Localization of functional prostaglandin E2 receptors EP3 and EP4 in the nuclear envelope. J Biol Chem. 1999;274:15719–15724. doi: 10.1074/jbc.274.22.15719. [DOI] [PubMed] [Google Scholar]
- 25.Breyer MD, Breyer RM. Prostaglandin receptors: their role in regulating renal function. Curr Opin Nephrol Hypertens. 2000;9:23–29. doi: 10.1097/00041552-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 27.Nasrallah R, Zimpelmann J, Singh S, Hebert RL. Molecular and biochemical characterization of prostacyclin receptors in rat kidney. Am J Physiol Renal Physiol. 2001;280:F266–F277. doi: 10.1152/ajprenal.2001.280.2.F266. [DOI] [PubMed] [Google Scholar]
- 28.Ivano AI, Scheck AC, Romanovsky AA. Expression of genes controlling transport and catabolism of prostaglandin E2 in lipopolysaccharid fever. Am J Physiol Regul Integr Comp Physiol. 2003;284:R698–R706. doi: 10.1152/ajpregu.00570.2002. [DOI] [PubMed] [Google Scholar]
- 29.Wan F, Lendardo MJ. The nuclear signaling of NF-kappaB: current knowledge, new insights, and future perspectives. Cell Res. 2010;20:24–33. doi: 10.1038/cr.2009.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Branski RC, Barbieri SS, Weksler BB, et al. Effects of transforming growth factor-beta1 on human vocal fold fibroblasts. Ann Otol Rhinol Laryngol. 2009;118:218–226. doi: 10.1177/000348940911800310. [DOI] [PubMed] [Google Scholar]
- 31.Welham NV, Lim X, Tateya I, Bless DM. Inflammatory factor profiles one hour following vocal fold injury. Ann Otol Rhinol Laryngol. 2008;117:145–152. doi: 10.1177/000348940811700213. [DOI] [PubMed] [Google Scholar]
- 32.Planaguma A, Claria J, Miquel R, et al. The selective cyclooxygenase-2 inhibitor SC-236 reduces liver fibrosis by mechanisms involving non-parenchymal cell apoptosis and PPAR-gamma activation. FASEB J. 2005;19:1120–1122. doi: 10.1096/fj.04-2753fje. [DOI] [PubMed] [Google Scholar]
- 33.Kucuk HF, Bingul SM, Kurt N, et al. Effect of a selective cyclooxygenase-2 inhibitor on renal scarring. Eur Surg Res. 2006;38:451–457. doi: 10.1159/000095088. [DOI] [PubMed] [Google Scholar]
- 34.Sciarra A, Salciccia S, Albanesi L, Cardi A, D’Eramo G, Di Silverio F. Use of cyclooxygenase-2 inhibitor for prevention of urethral strictures secondary to transurethral resection of the prostate. Urology. 2005;66:1218–1222. doi: 10.1016/j.urology.2005.06.090. [DOI] [PubMed] [Google Scholar]
- 35.LaPointe MC, Mendez M, Leung A, Tao Z, Yang XP. Inhibition of cyclooxygenase-2 improves cardiac function after myocardial infarction in the mouse. Am J Physiol Heart Circ Physiol. 2004;286:H1416–H1424. doi: 10.1152/ajpheart.00136.2003. [DOI] [PubMed] [Google Scholar]
- 36.Wilgus TA, Vodovotz Y, Vittadini E, Clubbs EA, Oberyszyn TM. Reduction of scar formation in full-thickness wounds with topical celecoxib treatment. Wound Rep Reg. 2003;11:25–34. doi: 10.1046/j.1524-475x.2003.11106.x. [DOI] [PubMed] [Google Scholar]
- 37.Branski RC, Rosen CA, Verdolini K, Hebda PA. Acute vocal fold wound healing in a rabbit model. Ann Otol Rhinol Laryngol. 2005;114:19–24. doi: 10.1177/000348940511400105. [DOI] [PubMed] [Google Scholar]
- 38.Branski RC, Verdolin K, Rosen CA, Hebda PA. Markers of wound healing in vocal fold secretions from patients with laryngeal pathology. Ann Otol Rhinol Laryngol. 2004;113:23–29. doi: 10.1177/000348940411300105. [DOI] [PubMed] [Google Scholar]
- 39.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 40.Schmedtje JF, Ji YS, Liu WL, Dubois RN, Runge MS. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J Biol Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- 41.Stuhlmeier KM, Broll J, Iliev B. NF-kappaB independent activation of a series of proinflammatory genes by hydrogen sulfide. Exp Biol Med. 2009;234:1327–1338. doi: 10.3181/0904-RM-137. [DOI] [PubMed] [Google Scholar]