

Abstract
We describe a novel approach, selectively amplified microsatellite (SAM) analysis, for the targeted development of informative simple sequence repeat (SSR) markers. A modified selectively amplified microsatellite polymorphic loci assay is used to generate multi-locus SSR fingerprints that provide a source of polymorphic DNA markers (SAMs) for use in genetic studies. These polymorphisms capture the repeat length variation associated with SSRs and allow their chromosomal location to be determined prior to the expense of isolating and characterising individual loci. SAMs can then be converted to locus-specific SSR markers with the design and synthesis of a single primer specific to the conserved region flanking the repeat. This approach offers a cost-efficient and rapid method for developing SSR markers for predetermined chromosomal locations and of potential informativeness. The high recovery rate of useful SSR markers makes this strategy a valuable tool for population and genetic mapping studies. The utility of SAM analysis was demonstrated by the development of SSR markers in bread wheat.
INTRODUCTION
Microsatellites, or simple sequence repeats (SSRs), are highly polymorphic and abundant sequences dispersed throughout most eukaryotic genomes (1). As co-dominant, locus-specific markers they are widely used for DNA fingerprinting, paternity testing, linkage map construction and population genetic studies (2–4). However, high development cost is a major impediment to the routine application of SSRs in the genetic study of non-commercial species and for identifying markers located in chromosomal regions of interest. The development of locus-specific SSR markers requires the isolation and characterisation of individual loci, a process involving the construction and screening of a DNA library with microsatellite-specific probes, followed by DNA sequencing of positive clones and subsequent PCR primer synthesis and testing (5). The recovery rate of useful SSRs is generally low due to non-specific amplification and monomorphic loci. In addition, the random selection of clones prevents predetermination of the chromosomal location and copy number of microsatellite loci.
Several techniques that generate multi-locus SSR fingerprints are able to utilise SSRs as molecular markers without the expense of single locus isolation. One approach is to employ oligonucleotide primers containing microsatellite repeats, either individually in PCR to amplify the region between two inverted SSR sequences (6) or in combination with arbitrary sequence primers to amplify the conserved region between a SSR and a primer-binding site located in the region flanking the repeat (7,8). A second approach, selectively amplified microsatellite polymorphic loci (SAMPL), employs PCR primers targeting compound SSR repeats in combination with primers specific to adapter sequences attached to DNA restriction fragments (9). The SAMPL technique allows the complexity of the multi-locus SSR fingerprint to be tailored and is therefore preferred in the analysis of large genomes. Briefly, SAMPLs are generated in a two-step process. First, adapter molecules of known sequence are ligated to doubly digested DNA restriction fragments, followed by amplification of subsets of these fragments using primers homologous to the adapter sequences. Next, subsets of the amplified DNA fragments containing SSR sequences are selectively amplified, then visualised following separation on sequencing gels. The complexity of SAMPL fingerprints is tailored by an affinity capture step using streptavidin-coated magnetic beads to remove one of the populations of restriction fragments and inclusion of additional ‘selective’ nucleotides on the 3′-ends of the adapter primers that extend into the DNA fragments.
A disadvantage of multi-locus SSR profiling is the capture of only some of the polymorphism associated with microsatellites due to the prevalence of dominant markers and difficulty in identifying allelic fragments in complex DNA fingerprints. The conversion of dominant markers into locus-specific, co-dominant formats by developing primer pairs specific to individual loci is restricted, as these methods amplify DNA from only one side of the repeat. Additional steps are therefore required to isolate the DNA flanking the other side of the microsatellite. In our experience the conversion of SAMPLs into locus-specific SSR markers is challenging in bread wheat, despite the availability of a range of techniques for this purpose (9–12). This difficulty results from the extremely large size and hexaploid nature of the bread wheat genome and the availability of only one primer determining PCR specificity. In this report we describe a simple approach using a modified SAMPL assay to develop locus-specific SSR markers with the design and synthesis of just one primer specific to the conserved region flanking the repeat. This approach, selectively amplified microsatellite (SAM) analysis, offers cost-efficient and rapid development of SSR markers, whose chromosomal location and potential informativeness can be determined prior to the expense of characterising the microsatellite locus.
MATERIALS AND METHODS
Preparation of template DNA
One microgram of genomic DNA isolated from hexaploid wheat (Triticum aestivum L.) was digested for 3 h at 37°C with 5 U each of MseI and PstI in 50 µl of 1× New England Biotechnologies (NEB) buffer II and 100 ng/µl BSA. Next, 5 and 50 pmol PstI and MseI adapters, respectively, were added and the mixture was heated at 45°C for 5 min to melt any cohesive adapter termini that had annealed. This step prevented self-ligation of the phosphorylated adapters before attachment to the restriction fragments. A solution containing 1× NEB buffer II, 100 ng/µl BSA, 6 mM dATP and 1 U T4 ligase (NEB) was added to give a total volume of 60 µl and the incubation was continued for a further 1 h at 37°C, then at room temperature for 2 h. The adapters were prepared by incubating equimolar amounts of two synthetic oligomers in annealing buffer (10 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA) at 65°C for 10 min, followed by slow cooling to room temperature over a 2 h period. The sequence of the PstI adapter sense strand was CTC GGA AGC CTC AGT CCC AGA CTG CGT ACA TGC A-OH and of the antisense strand was phos-TGT ACG CAG TCT GGG ACT GAG GCT TCC GAG A-OH. The sequence of the MseI adapter sense strand was GAG CAA GGC TCT CAC AAG GAC GAC CGA CGA G-OH and of the antisense strand was phos-TAC TCG TCG GTC GTC CTT GTG AGA GCC TTG CT-OH. Each adapter oligomer was purified by PAGE to enrich for full-length product. The sequences of PCR primers used in subsequent steps are given in Table 1.
Table 1. Sequences of the suppressor, adapter and 5′-anchored SSR primers.
| Primer name |
Sequence (5′→3′) |
| MseI suppressor | GAG CAA GGC TCT CAC A |
| PstI suppressor | CTC GGA AGC CTC AGT C |
| MseI adaptera | GAC GAC CGA CGA GTA ANN N |
| PstI adaptera | AGA CTG CGT ACA TGC AGN NNN |
| PAC6b | KKR YRY YAC ACA CAC ACA C |
| KKY RYR YCA CAC ACA CAC A | |
| PCT6b | KKV RVR VCT CTC TCT CTC T |
| KKR VRV RTC TCT CTC TCT C | |
| PGA6b | KKY HYH YAG AGA GAG AGA G |
| KKH YHY HGA GAG AGA GAG A | |
| PGT6b | KKH VHV HTG TGT GTG TGT G |
| KKV HVH VGT GTG TGT GTG T |
aN represents ‘selective’ nucleotides determining which subsets of DNA fragments are amplified (15).
bThe 5′-anchored SSR primers consist of equimolar amounts of the two listed oligonucleotides.
Amplification of the PstI–MseI restriction fragments was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer (Advanced Biotechnologies), 1.5 mM MgCl2, 5 pmol each MseI and PstI suppressor primer, 2 µl digested/ligated template DNA and 1 U Taq polymerase (Advanced Biotechnologies). PCR amplification was performed for 20 cycles (60 s at 92°C, 60 s at 56°C and 60 s at 72°C) in a Hybaid 96-well thermocycler. The reaction mixture was then diluted 1:20 with TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA).
Modified SAMPL assay
The SAMPL assay was performed essentially as described by Witsenboer et al. (9), with the following modifications. Preamplification was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 5 pmol each MseI and PstI adapter primer with selective nucleotides (one for the MseI adapter primer and one or two for the PstI adapter primer), 2 µl of the diluted pool of amplified PstI–MseI fragments and 1 U Taq polymerase. PCR was performed as described above and the reaction products were diluted 1:20 with TE. SAM amplification was performed in a similar 20 µl reaction mix, with 5 pmol MseI (or PstI) adapter primer with three or four selective nucleotides, 20 pmol SSR primer and 2 µl of diluted preamplification products as template DNA. The SSR primers were designed to faithfully anchor to the 5′-end of microsatellite repeats (13), and consisted of a mixture of two synthetic oligomers to maximise the degeneracy of the 5′-anchor sequence of the SSR primer. PCR was performed for 37 cycles with the profile 60 s at 92°C, 60 s annealing (see below) and 30 s at 72°C. The annealing temperature for the first cycle was 65°C, reducing by 1°C/cycle for the next seven cycles. The next five cycles were with annealing at 59°C. Following the twelfth cycle the denaturation and annealing steps were reduced to 30 s and the remaining 25 cycles were continued using annealing at 57°C. Following amplification, samples were mixed with an equal volume of gel loading buffer (98% formamide, 10 mM EDTA, 0.25% xylene cyanol as tracking dye), heated for 3 min at 95°C, chilled quickly on ice and run on 5% sequencing gels under standard conditions (14). SAMs were visualised with silver staining.
Sequencing of SAMs
Selected fragments were excised from dried polyacrylamide gels, reamplified and sequenced, either directly or after cloning into vector pGEM-T (Promega), by dye deoxy terminator chemistry using appropriate adapter primers. Primers specific to the sequence flanking the repeat array were designed with NetPrimer (Premier Biosoft International) using the parameters: primer length 16–24 nt, 3′-end stability –5.5 to –9.0 kcal/mol, oligomer Tm 55–70°C, GC content 30–70% and primer rating >90.
Amplification of SSR markers
Amplification of the target SSR loci from genomic DNA was performed using a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 5 pmol sequence-specific primer, 5 pmol SSR primer, 100 ng genomic DNA and 1 U Taq polymerase. The SSR primer corresponded to that used to amplify the fragment from which the sequence-specific primer was derived. PCR was performed using the SAM PCR profile except that 20–35 cycles were performed with the final 57°C annealing.
RESULTS AND DISCUSSION
Overview of the approach
The technical simplicity and flexibility of the SAMPL assay was improved through two significant modifications. First, a modified adapter ligation step was used to simplify the process for reducing the complexity of the template DNA prior to amplification. This was achieved by ligating a special type of adapter to the ends of the restriction fragments generated by the digestion of genomic DNA with two endonucleases, followed by fragment amplification using suppression PCR technology (discussed below). This modification eliminated the need for the affinity capture step using streptavidin-coated magnetic beads (9) and improved compatibility of the assay with silver staining of the products. The latter advantage arose because affinity capture removes only one of the two populations of restriction fragments with identical adapter sequences at both ends, whereas suppression PCR simultaneously removes both. The absence of both fragment populations avoids amplification arising from the adapter primer alone during the SAM PCR step and therefore ensures that only DNA fragments containing SSR sequences are visualised. Moreover, suppression PCR amplification further reduces the complexity of the template DNA for SAMPL analysis compared to the original protocol, thereby minimising the total number of selective nucleotides required on the adapter primers to generate a scorable SSR fingerprint (data not shown). Minimal use of selective nucleotides is desirable, as they reduce the chance of amplifying both alleles from a SSR locus (15). The second modification introduced 5′-anchored SSR primers to permit amplification of DNA fragments containing any type of repeat structure. These primers increase the proportion of SSR sequences in a DNA sample that can be assayed compared to the original protocol, which is restricted to compound repeats. These primers also overcome problems reported for generating clear and reproducible multi-locus SSR profiles with these types of primers (9) and can be extended to different types of tri-, tetra- and pentanucleotide repeats (13).
The preparation of template DNA for SAM analysis relies on PCR suppression effects generated when adapter-ligated restriction fragments are amplified with primers that are shorter in length than the adapters and capable of hybridising only with the outer regions of the adapter sequences (16). Upon denaturation, DNA fragments with identical adapter sequences at each end form single-stranded molecules with inverted terminal repeats. These molecules undergo intrastrand annealing to form pan-handled structures, which are more stable than primer–template hybrids, therefore suppressing exponential amplification (Fig. 1). Moreover, the small number of amplification products that arise from the successful extension of fragments with inverted terminal repeats are themselves subject to PCR suppression effects in subsequent cycles. In contrast, DNA fragments with different adapter sequences are efficiently and exponentially amplified because they cannot form the pan-handled structure (Fig. 1). Suppression PCR therefore generates an amplicon pool of reduced complexity, which is highly enriched for restriction fragments with different adapter sequences at each end.
Figure 1.
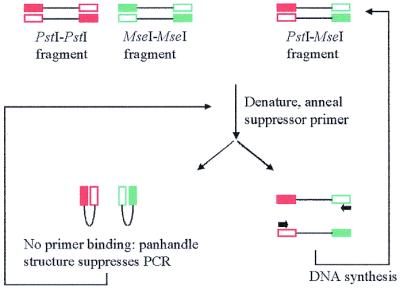
Illustration of the PCR suppression effect.
Preamplification was performed using adapter primers with selective nucleotides on their 3′-ends to further reduce the complexity of the template DNA. This step was essential to minimise the chance of mispriming and interference between fragment amplification from different loci (9,15). Selected subsets of SSR-containing DNA fragments were then amplified using a 5′-anchored SSR primer in combination with an adapter primer possessing additional selective nucleotides and the preamplification products as template DNA. Following separation and visualisation by silver staining on sequencing gels, SAMs were used directly as DNA markers in genetic studies to determine the chromosomal location and polymorphism of the amplified SSR loci.
The conversion of SAMs into locus-specific SSR markers was achieved with the design and synthesis of just one primer specific to the conserved region flanking the repeat. Following the strategy described by Fisher et al. (13) the corresponding SSR locus was amplified from genomic DNA using the sequence-specific primer in combination with the 5′-anchored SSR primer used to amplify the SAM marker. This PCR produces an amplicon with the entire microsatellite sequence by faithful anchoring of the SSR primer to the 5′-end of the repeat array (13). Therefore, the cost of developing a SSR marker of predetermined informativeness and chromosomal location was reduced to the synthesis of just one sequence-specific primer.
Development of SSR markers in bread wheat
To demonstrate the utility of this approach, SAM analysis was used to develop SSR markers in bread wheat, an allohexaploid with an extremely large (1C = 17.3 pg) genome composed of >80% repetitive DNA (17). A total of four or five selective nucleotides on the adapter primers were typically required during SAM amplification to generate reproducible SSR fingerprints when scoring was restricted to strongly amplified fragments (Fig. 2). The genetic mapping of 72 polymorphisms on an established linkage map for a recombinant inbred population confirmed the Mendelian inheritance of SAMs and their distribution throughout the genome (data not shown). While the chromosomal location of segregating loci could be determined, polymorphisms associated with repeat length variation were observed for only 13% of the SAMs due to marker dominance. However, the prevalence of dominant markers does not limit the utility of the technique. SAMs will typically be converted to locus-specific, co-dominant SSR markers when in chromosomal regions of interest, such as those containing economically important genes. These markers could be used to follow the chromosomal regions of interest during breeding, a process aided by the multi-allelic nature of SSRs. Hence, the informativeness of the SSR markers in the population from which the SAMs originated is unimportant. Similarly, initial knowledge of the informativeness of SSRs is less important when the markers are intended for use in natural or different populations than the one from which the SAMs were derived.
Figure 2.
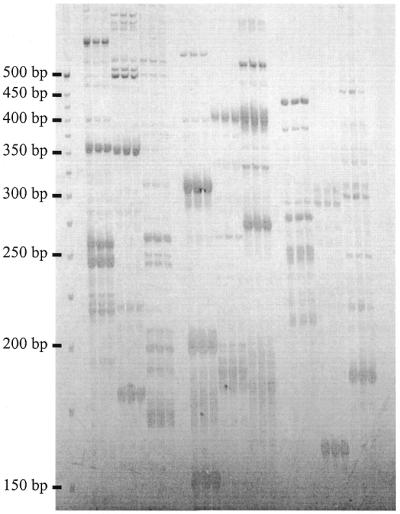
Multi-locus SSR profiles of bread wheat amplified using different combinations of PstI adapter primers with four selective nucleotides and 5′-anchored SSR primers. Reactions for each primer combination were prepared in triplicate. Lanes 1–3, PstI + GACG and PCT6; lanes 4–6, PstI + GACC and PCT6; lanes 7–9, PstI + GAGC and PCT6; lanes 10–12, PstI + GACG and PAC6; lanes 13–15, PstI + GACC and PAC6; lanes 16–18, PstI + GAGC and PAC6; lanes 19–21, PstI + GACG and PGT6; lanes 22–24, PstI + GACC and PGT6; lanes 25–27, PstI + GAGC and PGT6.
The sequences of 20 SAMs segregating in the mapping population were used to develop locus-specific SSR markers. Using a standard set of PCR conditions, 12 (60%) of the primers (Table 2) amplified the corresponding SSR sequence from genomic DNA. This was indicated by amplification of a SSR fragment of expected size from the parental cultivar from which the SAM originated and co-segregation of polymorphic SSRs with the SAM from which they were derived (Fig. 3). The latter also confirmed the Mendelian behaviour and reproducibility of the SSR markers. The presence of allelic variation at half of the loci confirmed faithful anchoring of the SSR primers to the 5′-ends of the repeats, while monomorphic markers probably reflected the absence of allelic variation rather than primer slippage (Fig. 4). Noteworthy was the minimal amplification of ISSR fragments, which were confined to the high molecular weight regions of the gel (Fig. 4). It is believed that the parameters used to design the primers specific to the conserved region flanking the repeats ensured their participation first in PCR to give preferential amplification of the target microsatellite fragment over amplification of ISSR sequences by the SSR primer alone. The amplification of additional SSR loci by some primer sets (Fig. 4) was an anticipated outcome for SSR markers developed in bread wheat due to the hexaploid nature and large size of the genome (5,18) and use of only one primer determining PCR specificity. Genetic mapping and DNA sequence analysis revealed that the additional SSRs were amplified from closely related non-homologous loci and homeoloci (data not shown). It is expected that the recovery rate and specificity of SSR markers would improve significantly in the analysis of smaller, less complex genomes.
Table 2. Sequence of specific primers, accompanying 5′-anchored SSR primers, expected size of target SSR fragment and number of PCR cycles used at final 57°C annealing to amplify the marker.
| Primer name |
Sequence (5′→3′) |
5′ SSR
primer mix |
Allele size (bp) |
PCR cycles |
| Sun4 | TCC AGT GCA GCA TGA CAG AAC A | PAC | 262 | 30 |
| Sun5 | TGC CCT TTT GGT TGA TCA CCA C | PCT | 150 | 30 |
| Sun6 | TCT CTT AGC TCT TGC GCC CAC C | PAC | 208 | 35 |
| Sun7 | CGA TGC GCT TCA GTC ACT GAC C | PAC | 158 | 30 |
| Sun8 | CTT AGT CGT GAG GCC AAC GCA T | PAG | 243 | 30 |
| Sun9 | CGG ACA CGG TCA CGA CGG A | PCT | 126 | 30 |
| Sun10 | CAG GGA ACT AGC AGG CCA A | PAC | 221 | 30 |
| Sun11 | TCC GGA TGA ACT GAA TGA TGT GG | PAC | 132 | 30 |
| Sun12 | TCC AGT GCA TCA TGA CAG AAC A | PAC | 254 | 30 |
| Sun13 | ACC AGC AGC AGC TTA AAC CG | PAC | 132 | 25 |
| Sun14 | TCT AGG AAG GCA CGT AGG CTG C | PCT | 128 | 30 |
| Sun15 | GAA CAA TTT CAC GGC TGA TGC G | PAC | 111 | 30 |
Figure 3.
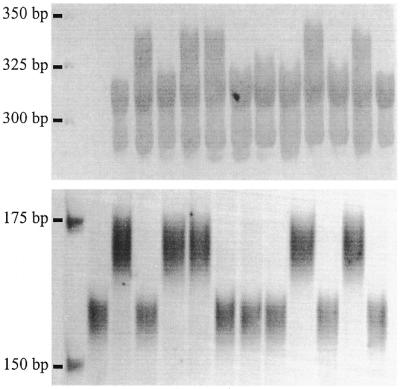
Co-segregation of a SSR (Sun7) with the dominant SAM from which it was derived. Shown from left to right are the parental cultivars, then 10 recombinant inbred progeny.
Figure 4.
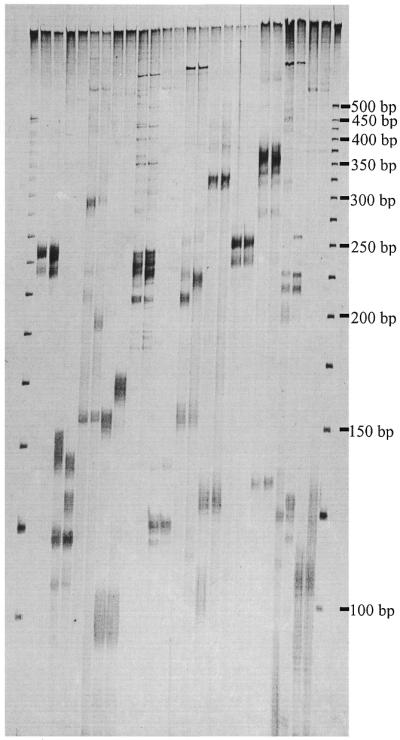
Amplification of SSR markers corresponding to specific SAM loci from genomic DNA of bread wheat. Each pair of lanes consists of the parental cultivars. Shown from left to right are Sun4, Sun5, Sun6, Sun7, Sun8, Sun9, Sun10, Sun11, Sun12, Sun13, Sun14 and Sun15.
Applications of the approach
SAM analysis provides a useful alternative to existing techniques for developing SSR markers. The capacity to determine the chromosomal location and capture some of the length polymorphism associated with SSR loci, prior to the expense of DNA sequencing and primer synthesis, will be particularly useful when the cost and resources required to develop SSR markers by traditional methods cannot be justified. This situation often arises in population genetics studies, where only small numbers of informative markers are usually required, and during the final stages of linkage map construction, when markers are sought to bridge gaps between linkage groups. For population genetic studies SAM analysis avoids the need to construct and screen a DNA library for SSR-containing clones and provides a good recovery rate of usable SSR markers. These advantages also apply to the development of SSR markers for chromosomal regions of interest, which can be achieved in several ways. SAM analysis can be used to screen chromosome substitution lines and aneuploid and microdeletion stocks (when available) to identify SSRs in specific chromosomal segments. Similarly, SAMs can be mapped on an existing genetic linkage map and those markers located in regions of interest converted to locus-specific SSR markers. Our approach can also be coupled with bulked segregant analysis (19) to discover SSRs linked to genes or chromosomal regions of interest. The application of SAM analysis to achieve the latter two objectives is described elsewhere (20).
References
- 1.Weber J.L. and May,P.E. (1989) Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am. J. Hum. Genet., 44, 388–396. [PMC free article] [PubMed] [Google Scholar]
- 2.Schlotterer C. and Pemberton,J. (1998) The use of microsatellites for genetic analysis of natural populations—a critical review. In DeSalle,R. and Schierwater,B. (eds), Molecular Approaches to Ecology and Evolution. Birkhauser Verlag, Basel, Switzerland, pp. 71–86.
- 3.Powell W., Morgante,M., Andre,C., Hanafey,M., Vogel,J., Tingey,S. and Rafalsky,A. (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed., 2, 225–238. [Google Scholar]
- 4.Gupta P.K., Balyan,H.S., Sharma,P.C. and Ramesh,B. (1996) Microsatellites in plants: a new class of molecular markers. Curr. Sci., 70, 45–54. [Google Scholar]
- 5.Roder M.S., Korzun,V., Wendehake,K., Plaschke,J., Tixier,M.-H., Leroy,P. and Ganal,M.W. (1998) A microsatellite map of wheat. Genetics, 149, 2007–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zietkiewicz E., Rafalski,A. and Labuda,D. (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics, 20, 176–183. [DOI] [PubMed] [Google Scholar]
- 7.Wu K., Jones,R., Danneberger,L. and Scolnik,P.A. (1994) Detection of microsatellite polymorphisms without cloning. Nucleic Acids Res., 22, 3257–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Provan J., Thomas,W.T.B., Forster,B.P. and Powell,W. (1999) Copia-SSR: a simple marker technique which can be used on total genomic DNA. Genome, 42, 363–366. [Google Scholar]
- 9.Witsenboer H., Vogel,J. and Michelmore,R.W. (1997) Identification, genetic localisation and allelic diversity of selectively amplified microsatellite polymorphic loci in lettuce and wild relatives (Lactuca spp.). Genome, 40, 923–936. [DOI] [PubMed] [Google Scholar]
- 10.Padegimas L.S. and Reichert,N.A. (1998) Adapter ligation-based polymerase chain reaction-mediated walking. Anal. Biochem., 260, 149–153. [DOI] [PubMed] [Google Scholar]
- 11.Chen X. and Wu,R. (1997) Direct amplification of unknown genes and fragments by uneven polymerase chain reaction. Gene, 185, 195–199. [DOI] [PubMed] [Google Scholar]
- 12.Roux K.H. and Dhanarajan,P. (1990) A strategy for single site PCR amplification of dsDNA: priming digested cloned or genomic DNA from an anchored-modified restriction site and a short internal sequence. Biotechniques, 8, 48–57. [PubMed] [Google Scholar]
- 13.Fisher P.J., Gardner,R.C. and Richardson,T.E. (1996) Single locus microsatellites isolated using 5′ anchored PCR. Nucleic Acids Res., 24, 4369–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 15.Vos P., Hogers,R., Bleeker,M., Reijans,M., van de Lee,T., Hornes,M., Frijters,A., Pot,J., Peleman,J., Kuiper,M. and Zabeau,M. (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res., 23, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukianov S., Gurskaya,N.K., Luklanov,V.S., Tarabukin,V.S. and Sverdlov,E.D. (1994) Highly efficient subtractive hybridisation of cDNA. Bioinorg. Chem., 20, 701–704. [Google Scholar]
- 17.Bennet M.D. and Smith,J.B. (1976) Nuclear DNA amounts in angiosperms. Phil. Trans. R. Soc. Lond. B Biol. Sci., 274, 227–274. [DOI] [PubMed] [Google Scholar]
- 18.Bryan G.J., Collins,A.J., Stephenson,P., Orry,A., Smith,J.B. and Gale,M.D. (1997) Isolation and characterisation of microsatellites from hexaploid bread wheat. Theor. Appl. Genet., 94, 557–563. [Google Scholar]
- 19.Michelmore R.W., Paran,I. and Kesseli,R.V. (1991) Identification of markers linked to disease resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl Acad. Sci. USA, 88, 9828–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayden M.J. and Sharp P.J. (2001) Targeting microsatellites (SSRs) in genetic linkage maps of bread wheat. Aust. J. Agric. Res., in press. [Google Scholar]