

Abstract
The peroxisome-proliferator-activated receptor gamma coactivator 1 α (PGC1α) has been implicated in the pathogenesis of several neurodegenerative disorders, including Huntington's disease (HD). Recent data demonstrating white matter abnormalities in PGC1α knock-out (KO) mice prompted us to examine the role of PGC1α in CNS myelination and its relevance to HD pathogenesis. We found deficient postnatal myelination in the striatum of PGC1α KO mice, accompanied by a decrease in myelin basic protein (MBP). In addition, brain cholesterol, its precursors, and the rate-limiting enzymes for cholesterol synthesis, HMG CoA synthase (HMGCS1) and HMG CoA reductase (HMGCR), were also reduced in PGC1α KO mice. Moreover, knockdown of PGC1α in oligodendrocytes by lentiviral shRNA led to a decrease in MBP, HMGCS1, and Hmgcr mRNAs. Chromatin immunoprecipitations revealed the recruitment of PGC1α to MBP promoter in mouse brain, and PGC1α over-expression increased MBP and SREBP-2 promoter activity, suggesting that PGC1α regulates MBP and cholesterol synthesis at the transcriptional level. Importantly, expression of mutant huntingtin (Htt) in primary oligodendrocytes resulted in decreased expression of PGC1α and its targets HmgcS1, Hmgcr, and MBP. Decreased expression of MBP and deficient myelination were found postnatally and in adult R6/2 mouse model of HD. Diffusion tensor imaging detected white matter abnormalities in the corpus callosum of R6/2 mice, and electron microscopy revealed thinner myelin sheaths and increased myelin periodicity in BACHD [bacterial artificial chromosome (BAC)-mediated transgenic model for Huntington's disease] mice expressing full-length mutant Htt. Together, these data suggest that PGC1α plays a role in postnatal myelination and that deficient PGC1α activity in oligodendrocytes may contribute to abnormal myelination in HD.
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by the expansion of the CAG repeat within the huntingtin gene that leads to expanded polyglutamine tracts in the huntingtin protein (Zoghbi and Orr, 2000). Mutant huntingtin is expressed throughout the body, but HD principally affects various brain regions such as the striatum (STR), cortex, thalamus, and subthalamic nucleus. Although the striatal neurons are preferentially affected, HD is not simply a striatal disease as evidenced by degeneration of many other brain regions in later stages of the disease (Zuccato et al., 2010).
Abnormalities in oligodendrocytes and white matter have been detected by structural MRI or pathological examinations in HD brains (Myers et al., 1991; Gomez-Tortosa et al., 2001; Fennema-Notestine et al., 2004; Reading et al., 2005; Paulsen et al., 2008; Tabrizi et al., 2009; Weaver et al., 2009; Rosas et al., 2010; Nopoulos et al., 2011), even in subjects farthest from predicted disease onset (Gomez-Tortosa et al., 2001; Bartzokis et al., 2007; Tabrizi et al., 2009), suggesting that disruptions in white matter integrity may represent an early event in HD pathogenesis. Although the precise nature of these abnormalities remains unknown, postmortem and neuroimaging data support the hypothesis that myelin breakdown contributes to white matter atrophy in HD (Bartzokis et al., 2007).
In the CNS, oligodendrocytes (OLs) form myelin wrapping around axons to ensure normal neural signal conduction. Developmental myelination in rodents occurs during the first month postnatally with highly coordinated accumulation of key myelin components, such as myelin basic protein (MBP) and cholesterol (Dietschy and Turley, 2004). MBP is one of the major proteins of CNS myelin and constitutes as much as 30% of protein. Direct evidence that MBP plays a major role in myelin compaction in the CNS was provided from studies of the shiverer mutant mouse that exhibit a large deletion of the MBP gene (Baumann and Pham-Dinh, 2001). Since cholesterol supply in the brain relies mostly on local synthesis (Dietschy and Turley, 2004), interruption of its synthesis results in impaired myelination (Saher et al., 2005). Interestingly, cholesterol metabolism impairments have been associated with several neurodegenerative disorders, including HD (Valenza et al., 2005, 2007; Valenza and Cattaneo, 2006).
Peroxisome-proliferator-activated receptor gamma coactivator 1 α (PGC1α) plays a role in the transcriptional regulation of energy metabolism and has been implicated in several neurodegenerative disorders (Finck and Kelly, 2006), including HD (Cui et al., 2006; Weydt et al., 2006, 2009; Chaturvedi et al., 2009, 2010; McConoughey et al., 2010; Róna-Vörös and Weydt, 2010; Hathorn et al., 2011). PGC1α knock-out mice exhibited vacuolar abnormalities in the CNS that were primarily associated with the white matter (Lin et al., 2004; Leone et al., 2005). Moreover, PGC1α expression increases dramatically during the peak of postnatal myelination (Cowell et al., 2007), suggesting that PGC1α may play a role in CNS myelinogenesis. To directly examine whether PGC1α participates in postnatal myelinogenesis, we investigated cholesterol biosynthesis and MBP expression in PGC1α knock-out mice and primary oligodendrocytes. In addition, we examined whether mutant huntingtin affects PGC1α expression in oligodendrocytes and whether myelination is altered in HD mouse models.
Materials and Methods
Animals.
PGC1α knock-out mice (Leone et al., 2005), R6/2 HD mice (Mangiarini et al., 1996), and BACHD [bacterial artificial chromosome (BAC)-mediated transgenic model for Huntington's disease] mice (Gray et al., 2008) and their littermate wild-type (WT) controls of either sex were used. All animal use procedures were in accordance with the National Institutes of Health guidelines and were approved by Massachusetts General Hospital.
Immunohistochemistry.
Sections were permeabilized in PBS containing 0.1% Triton X-100 and 5% donkey serum and then incubated with primary antibodies (goat anti-MBP [D18], 1:500; Santa Cruz Biotechnology; rabbit anti-Olig2, 1:400, Millipore Bioscience Research Reagents; mouse anti-neurofilament, 1:1000; Sigma) followed by appropriate fluorescence-conjugated secondary antibodies. Fluorescence images were acquired with a CCD camera attached to a fluorescence microscope (Olympus; BX50) or a confocal microscope. All quantifications were performed with ImageJ software (National Institutes of Health). To quantify MBP immunostaining intensity, regions of interest (ROIs) were delineated by adjusting threshold, and fluorescence intensities were measured. To quantify MBP immunostaining intensity of confocal images, ROIs were delineated by adjusting threshold in the red channel (neurofilament [NF] staining); fluorescence intensities were measured in both red channel (NF) and green channel (MBP), and the degree of myelination expressed as the ratio of MBP intensity/NF intensity.
Western blotting.
Brain tissue samples were homogenized in a buffer containing 40 mm Tris-HCl, pH 7.4, 4 m urea, 5% (w/v) SDS, 2% β-mercaptoethanol, and 1% (v/v) protease inhibitor mixture. For SDS PAGE, 30–50 μg proteins were separated in an 8–16% gradient gel and transferred to a PVDF membrane. The membrane was blotted with goat anti-MBP and mouse anti-α-tubulin (Sigma) sequentially. After incubation with HRP-conjugated secondary antibodies, the bound antibodies were visualized by chemiluminescence.
Real-time RT-PCR.
Total RNA was extracted with Trizol reagent and reverse transcribed into cDNA using Oligo(dT) primer (Invitrogen). Real-time PCR was performed on iCycler (Bio-Rad) using SYBR green PCR mix (Invitrogen). The PCR parameters were 35 cycles with 94°C denaturation for 20 s, 60°C annealing for 30 s, and 72°C extension for 50 s. The primers used were as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH, rat and mouse), (5′-acccagaagactgtggatgg-3′), (5′-cacattgggggtaggaacac-3′); PGC1α (rat), (5′-atgtgtcgccttcttgctct-3′), (5′-atctactgcctggggacctt-3); PGC1α (mouse), (5′-ccgagaattcatggagcaat-3′), (5′-tttctgtgggtttggtgtga-3′); 3-hydroxy-3-methylglutaryl-coenzyme A synthase 1 (HMGCS1, rat and mouse), (5′-ctctgcctgactgtggttca-3′), (5′-cctcaaacagctgcatcaaa-3′); 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR, rat), (5′-tgctgctttggctgtatgtc-3′), (5′-tgagcgtgaacaagaaccag-3′); HMGCR (mouse), (5′-cacctctccgtgggttaaaa-3′), (5′-gaagaagtaggcccccaatc-3′); MBP (rat), (5′-gcttctttagcggtgacagg-3′), (5′-gggttttcatcttgggtcct-3′); MBP (mouse), (5′-cttcaaagacaggccctcag-3′), (5′-cctgtcaccgctaaagaagc-3′); α-tubulin (rat and mouse), (5′-agagatcaccaatgcctgct-3′), (5′-actggatggtacgcttggtc-3′); cyclooxygenase-1 (COX-1, rat), (5′-agagatcaccaatgccagct-3′), (5′-actggatggtacgcttggtc-3′). The relative expression for any given gene was normalized with GAPDH or β-actin and then expressed as fold changes as indicated.
Oligodendrocyte primary culture and lentiviral infections.
Oligodendrocyte primary culture was prepared from P1 Sprague Dawley rat pups as described previously (McCarthy and de Vellis, 1980) and cultured in serum-free medium (high glucose DMEM and N2 supplement) with 30 ng/ml triiodothyronine (T3) and 20 ng/ml ciliary neurotrophic factor (CNTF) (Wang et al., 2006). The purity of the culture was 80–90% oligodendrocytes and 10–20% astrocytes by positive immunostaining to oligodendrocyte transcription factor 2 (Olig2) and glial fibrillary acidic protein (GFAP), respectively. For lentiviral infections, lenti-Htt Ex1–25Q and Htt Ex1–72Q were received from Dr. E. Regulier (Novartis, Basel, Switzerland); shPGC1α plasmid was purchased from Sigma (TRCN0000095313); shRNA control vector was purchased from Addgene (#1864). Lentivirus was produced as previously described (Tiscornia et al., 2006), and virus titer was determined using HIV-1 p24 Antigen ELISA kit (Zeptometrix).
Oligodendrocyte precursor cell transfection assay.
Mouse oligodendrocyte precursor (mOP) cells (Lin et al., 2006) were transfected with luciferase expression plasmid that contains the −1323 to +30 of the mouse MBP 5′-flanking region upstream of the luciferase (Miskimins et al., 2002) and empty pcDNA3 vector or 40 ng of pcDNA3-PGC1α (Puigserver et al., 1998). Cell extracts were prepared 48 h after transfection for luciferase assay using the Dual-Glo luciferase assay system (Promega), normalized to activity from the cotransfected Renilla luciferase expression vector.
Chromatin immunoprecipitation assay.
Fresh P15 mouse brains were homogenized in DMEM, cross-linked with 1% formaldehyde, and lysed in SDS lysis buffer. Cell lysates were sonicated and immunoprecipitated with normal IgG or specific antibody against PGC1α (sc-13067; Santa Cruz Biotechnology) using Magna Chromatin immunoprecipitation (ChIP) Protein A Magnetic Beads (Millipore), according to the manufacturer's instructions. Normal rabbit IgG served as a negative control. Precipitated DNA fragments were analyzed by PCR amplification using primers directed against the indicated regions of the mouse MBP promoter. MBP intron region and β-actin promoter were used as negative controls.
Sterol measurement.
Brain homogenates were prepared in PBS. Thirty microliters of homogenates were added to a screw-capped vial sealed with a Teflon-lined septum, together with 200 ng of 5a-cholest-7-en-3b-ol-1,2,5a,6a-d4 (D4-lathosterol; CDN Isotopes), 200 ng of D324S-hydroxycholesterol, 50 ng of D627S-hydroxycholesterol, and 5 μg of (D6-cholesterol) as internal standards. To prevent auto-oxidation, 50 μl of butylated hydroxytoluene (5 g/L) and 50 μl of EDTA (10 g/L) were added to each vial, and argon was flushed through to remove air. Alkaline hydrolysis was allowed to proceed at room temperature (22°C) for 1 h in presence of 1 m ethanolic potassium hydroxide solution under magnetic stirring. After hydrolysis, the neutral sterols and oxysterols were extracted two times with 5 ml of cyclohexane. The organic solvents were evaporated under a gentle stream of argon and converted into trimethylsilyl ethers (pyridine:hexamethyldisilazane:trimethylchlorosilane, 3:2:1 [v/v/v]). Gas chromatography–mass spectrometry to quantify single components was performed as described previously (Valenza et al., 2007).
ST14A cell culture and sterol regulatory element luciferase assay.
ST14A cells were transfected by electroporation for sterol regulatory element (SRE) luciferase assay as described previously (Valenza et al., 2005). The constructs used were SRE-luciferase reporter construct (pSynSRE), pCDNA-PGC1α, pSV-PGC1α, and pCDNA3.1 (Invitrogen) as empty vector. The electroporated cells were grown for 12 h followed by 24 h in either DMEM plus 10% FBS or 5% delipidated serum (to stimulate SRE activity) before the luciferase assay.
Diffusion tensor imaging.
Diffusion tensor imaging (DTI) was performed on an 11.7 tesla nuclear magnetic resonance (NMR) spectrometer (Bruker Biospin). Bird-cage radiofrequency coils with diameters of 15 mm were used for transmission and reception. A 3D diffusion-weighted multiple spin echo sequence was used to acquire images with four spin echoes (six spin echoes for embryonic and neonatal brains), a repetition time of 0.7 s, an echo time of 35 ms, and two signal averages. The field of view was 20 mm × 14 mm × 10 mm. The native imaging resolution was 0.125 mm × 0.125 mm × 0.125 mm. Images reconstructed from each echo were added together to form one image to enhance the signal-to-noise ratio. For each sample, eight diffusion-weighted images were acquired with a diffusion gradient duration (δ) of 6 ms and a separation between the pair of diffusion gradients (Δ) of 14 ms. Two images were acquired with a minimum b value (50 s/mm2) and six images with a maximum b value (1,500 s/mm2). Diffusion sensitizing gradients were applied along six different orientations: [0.707, 0.707, 0], [0.707, 0, 0.707], [0, 0.707, 0.707], [−0.707, 0.707, 0], [0.707, 0, −0.707], and [0, −0.707, 0.707]. The total imaging time was ∼24 h. The ex vivo 3D diffusion-weighted images were reconstructed on an off-line PC workstation using IDL (ITT Visual Information Solutions). Diffusion tensor fitting was performed using DtiStudio (Jiang et al., 2006). The six elements of diffusion tensor were determined by log-linear fitting. The tensor was diagonalized to obtain three eigenvalues (λ1–3) and corresponding eigenvectors (v1–3). Fractional anisotropy (FA) (Basser and Pierpaoli, 1996) was calculated. FA is a measure reflecting directional organization of the brain which is greatly influenced by the magnitude and orientation of white matter tracts. In general, a higher FA value indicates a more directional organization of the tissue and thus is generally accepted as an indication of tissue integrity. ROIs were manually placed on the genu and splenium of the corpus callosum.
Electron microscopy.
To analyze the thickness of myelin sheath wrapping individual axons, BACHD, and littermate WT mice at P14 were anesthetized and perfused intracardially with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4, for 15 min at a pressure of 120 mm mercury. The brains were removed and postfixed in the same fixative for 24 h. Coronal slices (150 μm) at the level of bregma 0 mm were cut with a vibratome. Ultrathin sections (60 to 90 nm thickness) were mounted on grids and stained with uranyl acetate and lead citrate. Images were collected using the Tecnai electron microscope at the Harvard Medical School Electron Microscope facility and analyzed as reported (Ohno et al., 2009) using ImageJ software. G-ratios (the diameter of the axon/the outer diameter of the myelinated fiber) of at least 300 myelinated axons per genotype and area were measured. Data were displayed as a scatter plot against axon diameter.
Statistics.
Data expressed as mean ± SEM, unless indicated otherwise. Prism software (Graphpad) was used for statistical analysis of Student's t test and one-way ANOVA with Newman–Keuls posttest with significance level set at p < 0.05.
Results
PGC1α KO mice exhibit developmental myelination deficits
Our recent data demonstrating white matter abnormalities in PGC1α KO mice (Lin et al., 2004) suggested a role of PGC1α in CNS myelination. To more directly examine whether PGC1α plays a role in postnatal myelinogenesis, we first quantified myelinated fibers in the striatum of PGC1α KO mice. We focused on the striatum because it was the region most affected in PGC1α KO mice (Lin et al., 2004). Immunostaining was performed with anti-MBP and anti-NF antibodies on P10 mouse brain sections and the degree of colocalization of MBP and NF in fiber bundles assessed (Fig. 1). While there was no significant difference in the total number of neuronal fiber bundles (NF+), the number of unmyelinated fiber bundles (NF+/MBP−) and the percentage of unmyelinated fiber bundles (% of NF+/MBP−) were significantly increased in PGC1α KO mice as compared with littermate WT controls (Fig. 1B). In addition, Western blotting, which detected four MBP isoforms with respective molecular weights at 14, 17, 18.5, and 21.5 kDa (Baumann and Pham-Dinh, 2001), revealed a significant decrease in total MBP protein in the striatum (Fig. 2A,B) and in the cortex (data not shown) of PGC1α KO mice compared with WT littermates. Interestingly, a significant decrease in MBP mRNA (Fig. 2B) was also detected, suggesting that MBP expression was suppressed at the level of transcription.
Figure 1.
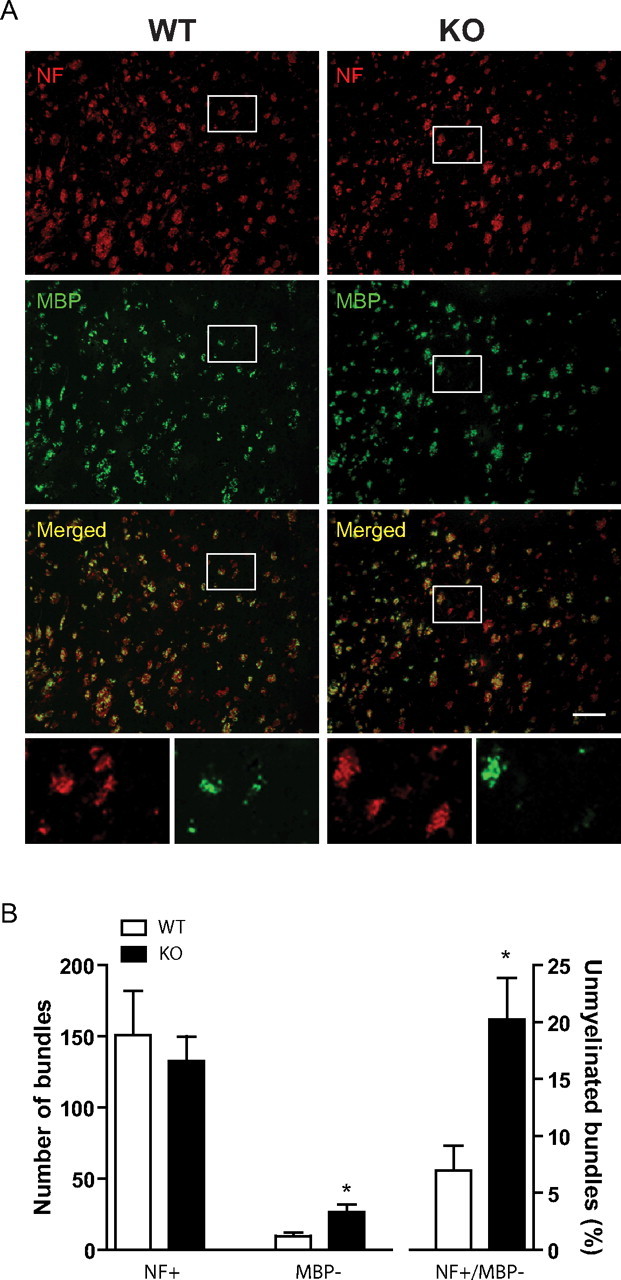
PGC1α KO mice displayed developmental myelination deficit in the striatum. A, Coronal brain sections from P10 PGC1α KO and WT mice (n = 5 each) were immunostained with NF (red) and MBP (green) antibodies. B, While there was no significant difference in the total number of NF+, the number of NF+/MBP− and the percentage of NF+/MBP− were significantly increased in PGC1α KO mice as compared with littermate WT controls. Data expressed as mean ± SEM. Student's t test; *p < 0.05. Scale bar, 100 μm.
Figure 2.
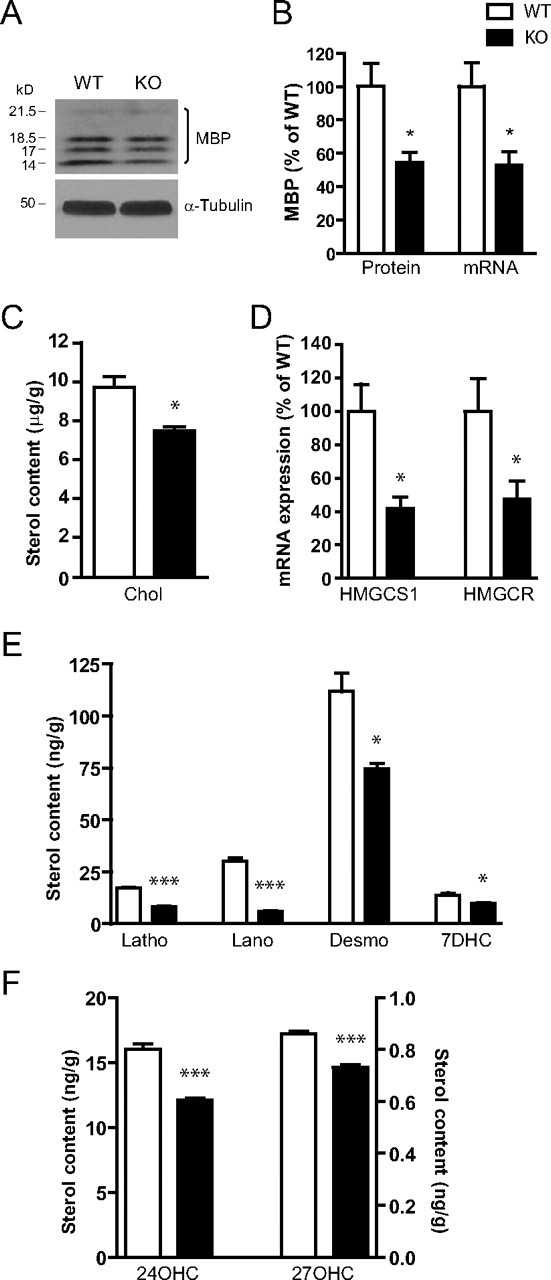
Decreased MBP expression and cholesterol metabolism in the brains of PGC1α KO mice. A, Western blotting of MBP expression in brain homogenates from P10 PGC1α KO mice (n = 5) and WT littermates (n = 4). B, Total MBP expression (quantitation of A, sum of all four isoforms normalized to α-tubulin) and MBP mRNA expression (P14 striatum, PGC1α KO mice [n = 3] and WT controls [n = 3]) were decreased in PGC1α KO mice compared with WT controls. The following was decreased in PGC1α KO brain at P14: cholesterol (Chol; C), HMGCS1 and HMGCR mRNA (D); cholesterol precursors lathosterol (Latho); lanosterol (Lano), desmosterol (Desmo), 7-dehydrocholesterol (7DHC) (E) and metabolites 24-hydroxycholesterol (24OHC) and 27-hydroxycholesterol (27OHC) (F). All sterol measurements were mean ± SD of weight over brain weight; n = 3 for both KO and WT mice. For B and D, data expressed as mean ± SEM. Student's t test; *p < 0.05, ***p < 0.001.
In addition to MBP, brain cholesterol represents another major component of myelin and cholesterol deficiency has been shown to significantly impact normal myelination (Saher et al., 2005). Therefore, we examined whether postnatal cholesterol synthesis was affected in PGC1α KO mice at P14 when myelination and cholesterol synthesis are most active (Dietschy and Turley, 2004). By isotope dilution mass spectrometry, we found significantly reduced brain cholesterol levels in PGC1α KO mice compared with WT littermates (Fig. 2C). In addition, the levels of cholesterol precursors lathosterol, lanosterol, desmosterol, and 7-dehydrocholesterol were all significantly reduced in the KO mice (Fig. 2E). To maintain homeostasis, excessive cholesterol is converted into 24S-hydroxycholesterol in neurons, and similarly converted into 27S-hydroxycholesterol in oligodendrocytes (Brown et al., 2004). We found that the levels of both 24S- and 27S-hydroxycholesterols were reduced in the PGC1α KO mice compared with WT mice (Fig. 2F). Since these results suggested that cholesterol biosynthesis was reduced in PGC1α KO mice, we examined expression of HMG CoA synthase 1 (HmgcS1) and HMG CoA reductase (Hmgcr), the rate-limiting enzymes for cholesterol synthesis. These experiments revealed that both HmgcS1 and Hmgcr mRNA were significantly decreased in the PGC1α KO mice (Fig. 2D), suggesting that a decrease in cholesterol, its precursors and metabolites was likely due to decreased expression of these enzymes that act upstream in the cholesterol biosynthesis pathway.
PGC1α regulates MBP and cholesterol gene expression in oligodendrocytes
Having found reduced cholesterol synthesis and myelination in PGC1α KO mice, we hypothesized that OLs represent the primary site of PGC1α action. Utilizing primary OL culture where OLs differentiate and produce MBP and other myelin components (Dubois-Dalcq et al., 1986), we found that upon differentiation, expression of MBP and PGC1α increased by ∼10 fold in these OLs (Fig. 3A).
Figure 3.
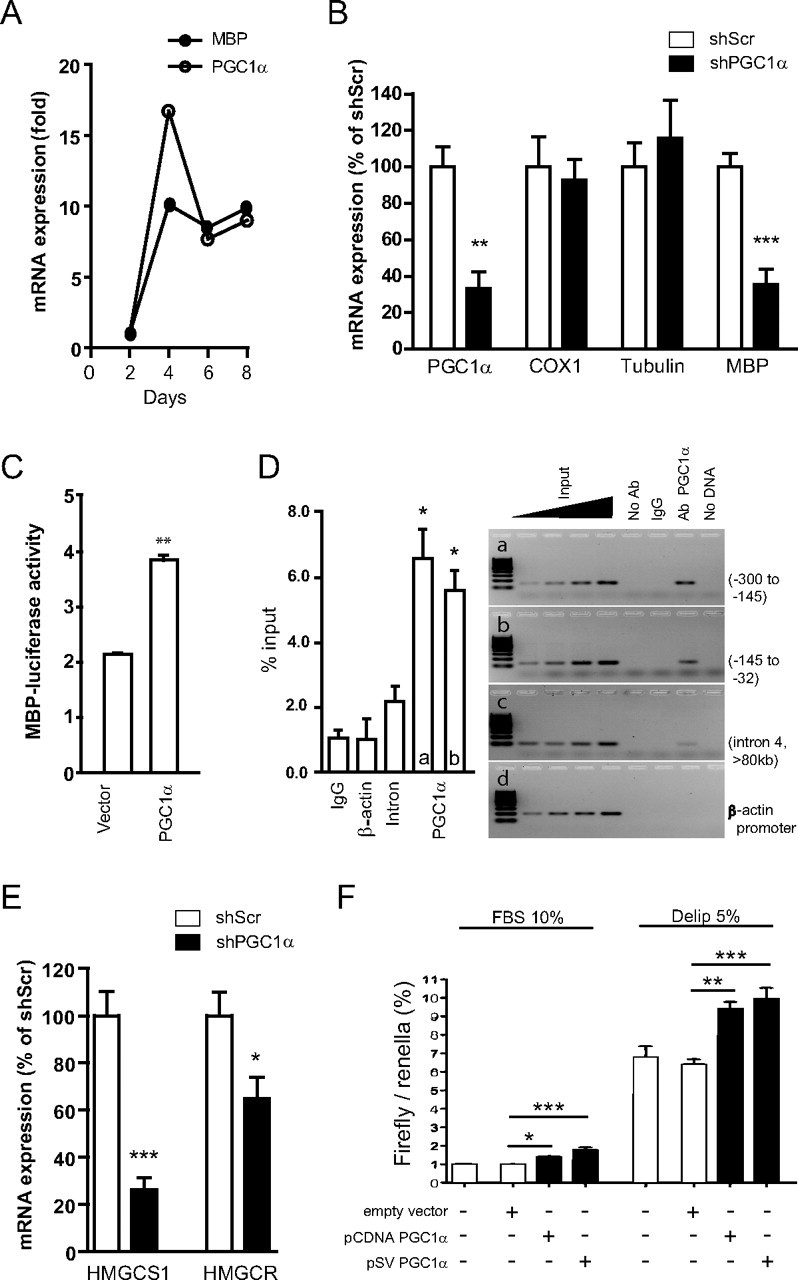
PGC1α regulates MBP expression and cholesterol synthesis. A, In primary oligodendrocyte culture, PGC1α and MBP mRNA expression increase in parallel upon differentiation. The x-axis indicates the number of days in serum-free medium containing triiodothyronine (T3) and CNTF. B, Lentiviral shRNA PGC1α (shPGC1α) knockdown decreased PGC1α and MBP gene expression but not cyclooxygenase-1 (COX1) or α-tubulin mRNA expression in primary OLs compared with scrambled shRNA (shScr). C, PGC1α over-expression increased MBP promoter activity in mOP cells. Data from three independent experiments are shown (means ± SEM; **p < 0.01 vs vector). D, ChIP revealed recruitment of PGC1α to MBP promoter. Chromatin from P15 mouse brain was immunoprecipitated with indicated antibodies, and the precipitated material amplified by PCR using primers for mouse MBP promoter (a) −300 to −145 bp; (b) −145 to −32 bp. MBP coding region (c) and β-actin promoter (d) were used as controls. Input lanes represent 0.5, 1, 2, and 4% of chromatin. Three independent experiments were performed and a representative blot is shown. *p < 0.05 compared with IgG or intron (left). E, PGC1α knockdown decreased expression of HmgcS1 and Hmgcr. Student's t test for panels B–E; *p < 0.05, **p < 0.01, and ***p < 0.001. F, PGC1α over-expression increases SRE activation in ST14 cells in lipid-containing (FBS 10%) or lipid-deprived (delip 5%) medium. One-way ANOVA; Newman–Keuls multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001.
Since our in vivo data demonstrated downregulation of MBP mRNA in PGC1α KO mouse brain (Fig. 2B), we examined whether PGC1α regulates MBP expression in OLs. To this end, we generated lentiviral shRNA construct that reduced endogenous PGC1α expression (Fig. 3B) in OLs by ∼60%. PGC1α knockdown led to a significant decrease in MBP mRNA, suggesting that PGC1α regulates MBP gene expression (Fig. 3B). We did not observe any toxicity or downregulation of control genes such as cyclooxygenase-1 or α-tubulin in OLs in the presence of PGC1α knockdown (Fig. 3B). To more directly examine whether PGC1α regulates MBP transcription, MBP promoter-reporter was expressed together with lenti-PGC1α in mouse OL precursor cells. These experiments showed that PGC1α over-expression significantly increased MBP promoter activity compared with vector control (Fig. 3C). Importantly, ChIP experiments revealed the recruitment of PGC1α to MBP proximal promoter in P15 mouse brain (Fig. 3D). Together, these results suggest that PGC1α gets recruited to MBP promoter to regulate its gene expression in OLs.
Since our in vivo data in PGC1α KO mice also implicated PGC1α in the regulation of cholesterol synthesis, we examined expression of HmgcS1 and Hmgcr in primary OLs. Knockdown of PGC1α led to significant downregulation of HmgcS1 and Hmgcr mRNA (Fig. 3E), further suggesting that PGC1α plays a role in the regulation of cholesterol genes. Intracellular cholesterol is tightly controlled by sterol regulatory element binding protein 2 (Srebp-2) which upon activation translocates to the nucleus and binds to SRE to activate transcription of cholesterol synthesis genes such as HmgcS1 and Hmgcr (Horton et al., 2002). We found that PGC1α over-expression significantly increased SRE activity in both lipid-containing (10% FBS) medium and in lipid-deprived (5% delip) medium (Fig. 3F), suggesting that PGC1α is involved in the regulation of expression of rate-limiting cholesterol synthesis genes.
Mutant huntingtin inhibits expression of PGC1α and cholesterol synthesis genes in oligodendrocytes
Our data so far suggest that PGC1α plays a role in postnatal myelinogenesis by regulating the expression of key components of myelin, MBP, and cholesterol. Impaired cholesterol metabolism has been reported in HD (Valenza and Cattaneo, 2006; Valenza et al., 2007, 2010; Leoni et al., 2008). In addition, white matter abnormalities were detected in the brain of HD patients (Gomez-Tortosa et al., 2001; Bartzokis et al., 2007). Since PGC1α function is repressed in HD (Cui et al., 2006; Weydt et al., 2006), we hypothesized that PGC1α repression may contribute to cholesterol deficiency and white matter abnormalities in HD. To examine this hypothesis, we first examined expression of PGC1α in OLs in the presence of mutant Htt. Our previous work showed that mutant Htt inhibits expression of PGC1α at the transcriptional level (Cui et al., 2006). While those studies were performed in primary neurons and brain homogenates, the role of mutant Htt in OLs was not investigated. To examine whether mutant Htt affects PGC1α in OLs, we transduced primary OLs with lenti-mutant Htt exon1 (72Q) or lenti-wt Htt (25Q) to achieve nontoxic levels of expression (data not shown). These experiments revealed that PGC1α expression was significantly decreased in the presence of mutant Htt when compared with control OLs transduced with lenti-wt Htt (Fig. 4A). The decrease of PGC1α was accompanied by a decrease in expression of MBP as well as cholesterol synthesis genes HmgcS1 and Hmgcr (Fig. 4A). These results suggest that mutant Htt inhibits expression of endogenous PGC1α in OLs. Moreover, our data suggest that the effect of mutant Htt on expression of HmgcS1, Hmgcr, and MBP in OLs at least in part occurs via the regulation of PGC1α.
Figure 4.
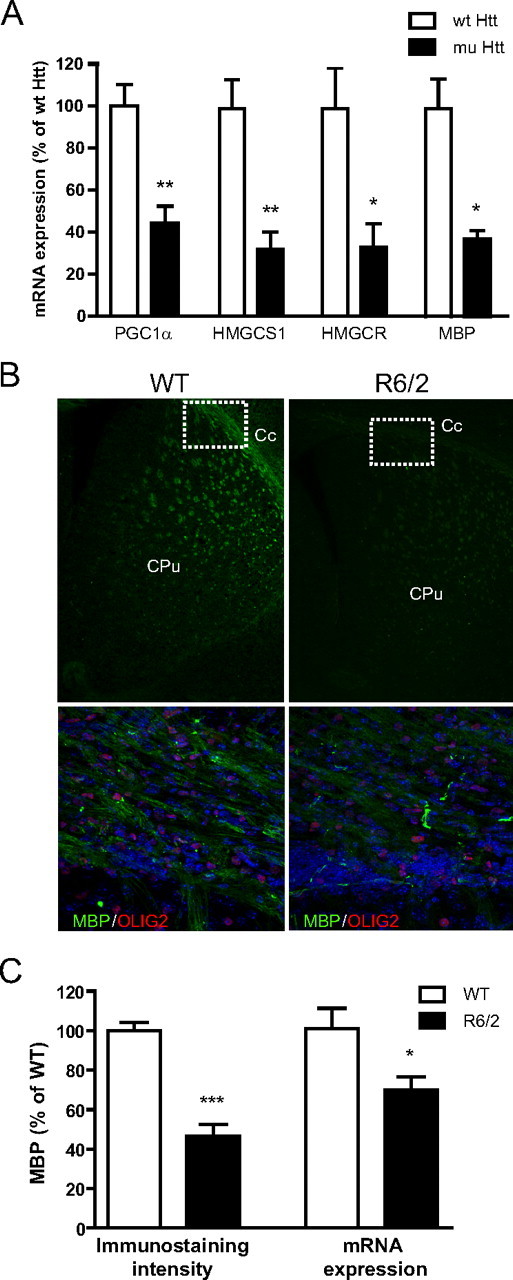
Inhibition of PGC1α by mutant Htt contributes to abnormal myelination in HD models. A, In primary OLs, over-expression of lenti-mutant Htt (72Q exon1) but not lenti-wt Htt (25Q) inhibited mRNA expression of PGC1α, cholesterol synthesis genes HmgcS1 and Hmgcr, and MBP. Data are mean ± SEM of three independent experiments. Student's t test; *p < 0.05, **p < 0.01. B, Representative images showing reduced MBP immunofluorescence intensity in R6/2 mice at P14 compared with WT brains (5 images/genotype). Higher magnification shows the distribution of MBP and Olig2 in corpus callosum (Cc; white boxes) of WT and R6/2 mice. The nuclei were stained with DAPI (blue). C, Reduced MBP immunofluorescence intensity (quantitation of B) and MBP mRNA expression by real-time RT PCR analysis in R6/2 mouse (n = 3) brains compared with WT mice (n = 4). Data are mean ± SEM. Student's t test; *p < 0.05, ***p < 0.001. CPu, Caudate putamen.
Abnormalities of CNS myelination in mouse models of HD
Having shown that PGC1α regulates expression of MBP and cholesterol synthesis genes (Figs. 2, 3) and that mutant Htt-mediated downregulation of PGC1α in OLs contributes to the downregulation of MBP and cholesterol synthesis (Fig. 4A), we next examined whether abnormalities of myelination exist in mouse models of HD. First, myelination was examined at P14 in the brain of R6/2 mouse model which expresses exon1 fragment of mutant Htt (Mangiarini et al., 1996). Immunohistochemistry experiments revealed a significant decrease in MBP staining intensity in the corpus callosum and also in the striatum of R6/2 mice (Fig. 4B,C). These changes in MBP staining were accompanied by decreased expression of MBP mRNA, further suggesting that postnatal myelination is abnormal in R6/2 HD mice (Fig. 4C).
To determine whether the deficiency in postnatal myelination persists in adult R6/2 mice, we performed MBP immunostaining in the brain sections from R6/2 HD mice at 6 and 12 weeks of age (Fig. 5A). We found a significant decrease in MBP staining intensity in the corpus callosum and striatum of R6/2 mice compared with WT controls at both 6 and 12 weeks (Fig. 5B). This decrease in MBP staining was consistent with decreased expression of MBP mRNA found at both time points (Fig. 5C).
Figure 5.
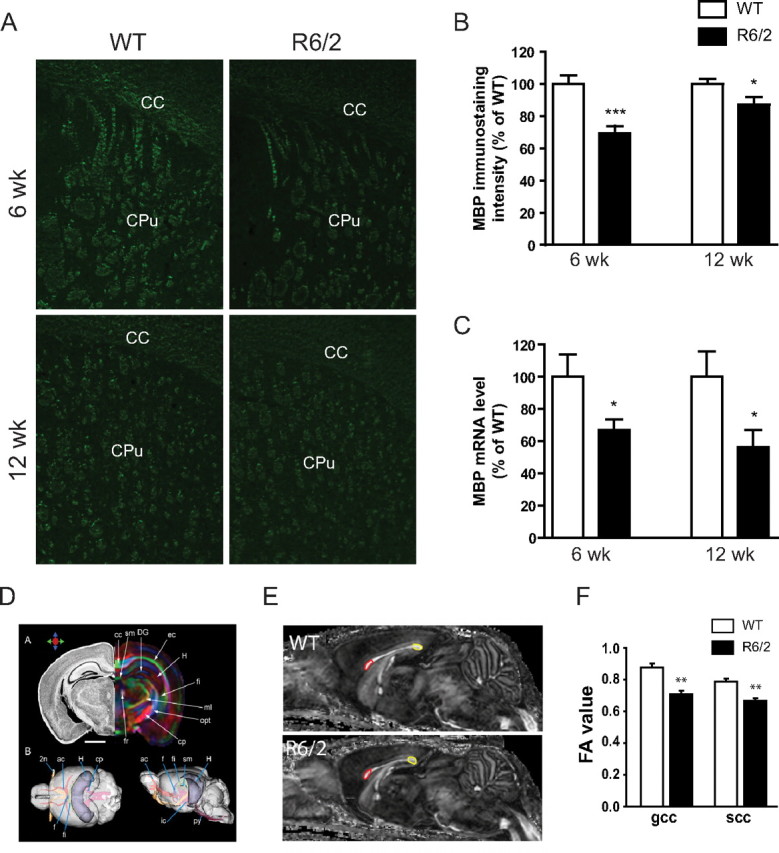
Deficiency in myelination persists in adult R6/2 HD mice. A, Brains from R6/2 mice at the age of 6 or 12 weeks were sectioned and MBP immunostaining performed. B, At both 6 and 12 weeks, MBP staining intensity was significantly decreased in the corpus callosum and striatum (CC+CPu) of R6/2 mice (n = 6 for both 6 and 12 weeks) compared with WT controls (n = 5 for 6 week, and 6 for 12 week). C, Brain MBP mRNA levels were decreased in both 6 week (n = 6) and 12 week (n = 3) R6/2 mice compared with their respective WT controls (n = 5 for 6 week, and 3 for 12 week). All data are mean ± SEM. Student's t test; *p < 0.05, ***p < 0.001. D, Nissl staining (left) and DTI (right) of a perfusion-fixed adult mouse brain were analyzed. Reconstructed white matter tracts from the DTI results are shown in color. 2n, Optic nerve; ac, anterior commissure; cc, corpus callosum; cp, cerebral peduncle; DG, dentate gyrus; ec, external capsule; f, fornix; fi, fimbria; H, hippocampus; ml, medial lemniscus; opt, optic tract; py, pyramidal tract; sm, stria medularis. Scale bar, 1 mm. The color arrows illustrate our color scheme. Red represents rostral–caudal, green for medial–lateral, and blue for dorsal–ventral. E, DTI detected deficits in white matter integrity in R6/2 mice. Mid-sagittal FA images of 12 week old WT and R6/2 mouse brains are shown. The red and yellow ROIs are defined for the genu and splenium of the corpus callosum (gcc and scc, respectively). F, FA values of the gcc and scc. Student's t test; **p < 0.01 between R6/2 mice and WT control mice (n = 3/genotype).
Next, high-resolution DTI with 11.7 tesla NMR spectrometer was used to examine FA in various brain regions of R6/2 mice (Fig. 5D). We found that the FA values were significantly decreased in 12 week old R6/2 mice as compared with WT littermate controls in genu and splenium of the corpus callosum (Fig. 5E,F). These results are in agreement with our findings by immunohistochemistry (Fig. 5B) and suggest that white matter abnormalities can be detected in R6/2 mice. Moreover, these studies are consistent with structural MRI studies in HD patients that demonstrated compromised white matter integrity in the corpus callosum in prodromal HD (Reading et al., 2005; Rosas et al., 2006; Weaver et al., 2009; Rosas et al., 2010).
To probe whether abnormalities in myelination were detected in another HD mouse model, we examined myelination in BACHD mice (Gray et al., 2008). The BACHD mice expressing full-length human mutant Htt with 97Q driven by an intact human Htt genomic locus exhibit multiple HD-like phenotypes, including progressive motor and psychiatric-like deficits and late-onset cortical and striatal neurodegenerative pathology (Gray et al., 2008). Compared with R6/2 mice, the BACHD model has more subtle and delayed phenotype, although behavioral deficits can be already detected at 2 months of age (Gray et al., 2008). Using confocal microscopy, we examined MBP immunostaining in the striata of 2 week old BACHD mice and found significantly decreased MBP immunostaining intensity in the BACHD mice compared with WT controls (Fig. 6A). To better characterize these changes, we used electron microscopy and examined myelin in the corpus callosum and striatum of BACHD mice at P14. We found that the percentages of myelinated axons in the corpus callosum and striatum were not significantly different compared with WT littermates (data not shown), suggesting there was no significant defect in the initiation of myelination. While the average axon diameters of myelinated fibers in both regions were comparable (corpus callosum [CC], WT = 801 ± 14 μm, BACHD = 773 ± 12 μm; STR, WT = 939 ± 15 μm, BACHD = 980 ± 19 μm; n>300; Student's t test, p > 0.05 for both), the ratios of axonal diameter and the outer diameter of myelinated fibers (G-ratios) were significantly larger in the corpus callosum (Fig. 6C) and striatum (Fig. 6D) of BACHD mice compared with WT littermate controls. These results indicated that the myelin sheaths were significantly thinner in BACHD mice. Myelin periodicity, measured as the distance between two major dense lines, is often used as an index of myelin compaction. We found that the periodicity was significantly larger in the corpus callosum but not in the striatum of BACHD mice compared with WT littermates (Fig. 6E,F). Together, the results suggested that myelin sheaths were thinner (larger G-ratio) and less compacted (larger periodicity) in BACHD mice compared with WT mice, suggesting abnormalities in the progression of postnatal myelination process in HD mice.
Figure 6.
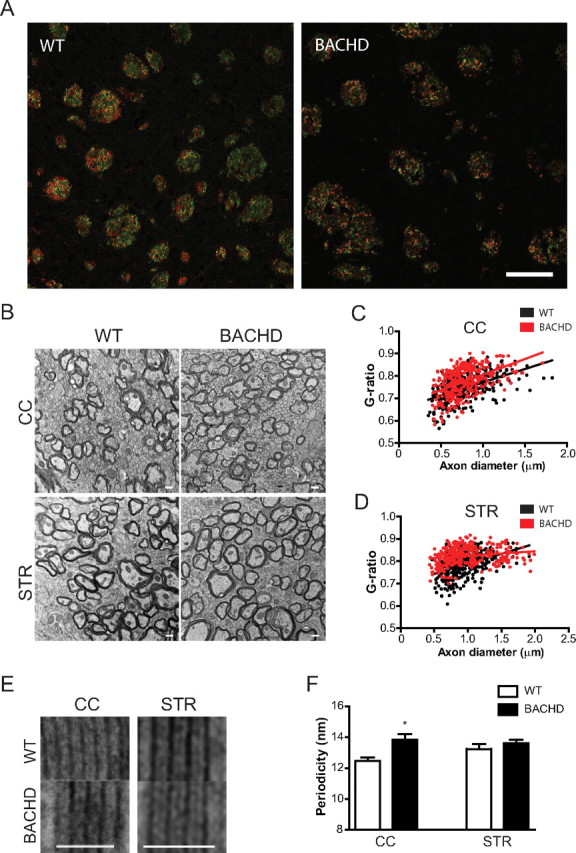
Myelination abnormalities in BACHD mouse model. A, P14 BACHD and WT mouse brains were sectioned coronally, immunostained with antibodies against NF (red) and MBP (green), and examined under confocal microscope. Merged images taken from the striatum are shown (yellow). MBP/NF is significantly decreased in BACHD mice (0.524 ± 0.027) compared with wild-type controls (0.597 ± 0.022). At least 200 fiber bundles were quantified in four animals per genotype. Student's t test; *p < 0.05. B, Electron microscopy reveals abnormalities in myelin in BACHD mice compared with WT mice. Representative images taken from the CC and STR are shown. In CC (C) and STR (D), G-ratios (the diameter of the axon/the outer diameter of the myelinated fiber) were determined, shown as scattered plot against axon diameter with linear regression. G-ratio is significantly increased in CC (0.774 ± 0.004) and STR (0.824 ± 0.002) of BACHD mice compared with wild-type controls (CC, 0.748 ± 0.003; STR, 0.783 ± 0.003; p < 0.001 for both). At least 300 axons per genotype and area were subjected to G-ratio analysis. E and F, Alignment of myelin sheath revealed differences in myelin periodicity. Periodicity was measured as the mean distance between two major dense lines from at least five randomly chosen myelin sheaths per genotype and area. All data are mean ± SEM. Student's t test; *p < 0.05, ***p < 0.001. Scale bars: 50 μm (A), 500 nm (B), and 50 nm in (E).
Discussion
Previous studies showed that PGC1α KO mice display white matter lesions in CNS (Lin et al. 2004) and that PGC1α levels dramatically increase during peak myelination (Cowell et al., 2007). In this study, we attempted to directly examine whether PGC1α plays a role in postnatal myelination. We found decreased MBP, cholesterol biosynthesis, and deficient myelination during the postnatal period in PGC1α KO mice. These findings were extended to primary oligodendrocytes where PGC1α expression was upregulated in parallel with expression of MBP upon differentiation of oligodendrocytes. Moreover, downregulation of endogenous PGC1α in oligodendrocytes resulted in decreased expression of MBP and the key cholesterol synthesis enzymes HmgcS1 and Hmgcr. Our finding that PGC1α activates MBP promoter in cultured cells and gets recruited to MBP promoter in vivo further suggests that PGC1α regulates MBP gene expression. Similarly, PGC1α can activate the SRE-containing promoter, suggesting that PGC1α regulates cholesterol synthesis by directly or indirectly regulating the SRE-binding protein, SREBP. PGC-1 coactivators have been previously reported to interact with nuclear receptors such as retinoid X receptor and liver X receptor, and non-nuclear receptor transcription factors involved in the regulation of cellular cholesterol metabolism (Oberkofler et al., 2003; Finck and Kelly, 2006). While the detailed mechanism remains to be elucidated, our results support the notion that PGC1α regulates MBP expression and cholesterol metabolism in oligodendrocytes at the transcriptional level.
Defective cholesterol biosynthesis results in deficient myelination (Saher et al., 2005; Verheijen et al., 2009); however, it is not clear how the production of cholesterol and other myelin components such as MBP is coordinated. In Schwann cells, the peripheral counterpart of OLs, the major myelin protein P0 requires cholesterol for exiting the ER and for peripheral myelin compaction (Saher et al., 2009). Similarly, decreased MBP gene expression was previously reported as a result of deficient cholesterol synthesis (Saher et al., 2005). Intriguingly, another recent study showed that MBP can also activate SREBP signaling by modifying the SRE-mediated gene expression, suggesting that MBP may directly participate in the regulation of cholesterol synthesis genes (Chatterjee et al., 2009). Although these studies suggest that the relationship between MBP and cholesterol synthesis is complex, our data that PGC1α regulates expression of both MBP and cholesterol synthesis genes suggested a coordinated regulation. It will be, therefore, of interest to further examine the molecular mechanism of PGC1α-mediated regulation of these key myelin components.
Reduced cholesterol biosynthesis and brain cholesterol levels have been reported in several HD mouse models, together with reduced brain cholesterol turnover in mice and humans (Valenza and Cattaneo, 2006; Valenza et al., 2007, 2010; Leoni et al., 2008). In addition, several recent reports have implicated dysfunction of PGC1α in the pathogenesis of HD (Cui et al., 2006; Weydt et al., 2006, 2009; Chaturvedi et al., 2009, 2010; McConoughey et al., 2010; Róna-Vörös and Weydt, 2010; Hathorn et al., 2011). While these studies suggest that PGC1α dysfunction in HD primarily affects neurons and muscle cells, our findings indicate that inhibition of PGC1α in oligodendrocytes by mutant Htt also contributes to the disease pathogenesis in HD. This conclusion is supported by our finding of abnormal myelination in mouse models of HD.
Structural MRI measurements of gray and white matter volumes, and cortical thickness, have shown widespread areas of atrophy in subjects with prodromal HD (Paulsen et al., 2008; Tabrizi et al., 2009; Nopoulos et al., 2011). In HD patients, there are also signs of myelin abnormalities such as increased number of oligodendrocytes (Myers et al., 1991; Gomez-Tortosa et al., 2001) and increased ferritin iron level (myelin breakdown product) (Bartzokis et al., 2007). An increased density of oligodendrocytes is observed in the head of the caudate nucleus for the lower grades (0 and 1) in postmortem HD brain, possibly suggesting a compensatory increase due to deficient myelination in early stages of HD (Myers et al., 1991). However, it remains unclear whether these white matter changes represent the initial event in HD neuropathology or occur as a result of neuronal damage. Recent MRI studies highlighted the possibility that white matter changes might not be due to simply the loss of cortical gray matter neurons (Stoffers et al., 2010). In support of this hypothesis, we were able to detect deficient myelination during the early postnatal period in R6/2 and BACHD mouse models of HD, suggesting that abnormalities in myelination occur very early in HD pathogenesis. Compared with R6/2 mice, BACHD mouse model expresses the full-length mutant Htt and has a milder phenotype (Gray et al., 2008). Unsurprisingly, less striking defects in postnatal myelination were found in these mice. Since no deficits in neuronal function have been reported during the early postnatal period in HD mouse models, these results suggest that oligodendrocytes may be more sensitive to mutant Htt expression than neurons and that perturbations of oligodendrocyte function may be an important early pathogenic event in HD. It remains to be established whether these alterations in myelination observed in HD mice correlate with white matter abnormalities detected in human HD. Our DTI studies in R6/2 mice detect compromised white matter integrity in the corpus callosum where we also observed deficient myelination suggesting that at least in HD mice a positive correlation exists between myelination and white matter abnormalities. These data are also consistent with the recent neuroimaging study that detected compromised white matter integrity of corpus callosum in premanifest HD (Rosas et al., 2006, 2010). Similarly, structural MRI studies in another fragment model of HD (171aa) revealed white mater atrophy that correlate with findings in human HD (Cheng et al., 2011). Although our data showed that myelin deficiency persists throughout the life of R6/2 mice, it remains to be determined how dysmyelination may contribute to neuronal dysfunction and subsequent neurodegeneration. It will be of interest to employ more sensitive neuroimaging methods to examine whether early white matter atrophy occurs in brain areas devoid of any gray matter pathology or vice versa.
In sum, our data suggest that PGC1α plays a role in postnatal myelination by regulating expression of MBP and cholesterol synthesis. In addition, we found deficient myelination in HD mouse models and decreased PGC1α activity in oligodendrocytes expressing mutant huntingtin suggesting that PGC1α may contribute to abnormal myelination in HD. These findings raise a possibility that upregulating PGC1α activity in oligodendrocytes may represent a novel strategy for early therapeutic interventions in HD.
Footnotes
This work was supported by National Institutes of Health Grants to D.K. (R01NS050352) and S.R. (R01 NS035996), by Fondazione Cariplo (Italy; 2008/2406) to M.V., by Ministero della Salute and Fondi per giovani Ricercatori (Italy; GR-2008-1145270) to L.V., by CHDI Foundation to W.D., and by Huntington's Disease Coalition for the Cure (USA) and CHDI (Cure Huntington's Disease Initiative) Foundation to E.C. We thank William Yang and Xiaofeng Gu for BACHD brain samples, and Marian Difiglia and Ellen Sapp for assistance with electron microscopy.
References
- Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S. Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications. Neurochem Res. 2007;32:1655–1664. doi: 10.1007/s11064-007-9352-7. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Brown J, 3rd, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, Lee JM, Yager D, Crowley J, Sambamurti K, Rahman MM, Reiss AB, Eckman CB, Wolozin B. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J Biol Chem. 2004;279:34674–34681. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Szustakowski JD, Nanguneri NR, Mickanin C, Labow MA, Nohturfft A, Dev KK, Sivasankaran R. Identification of novel genes and pathways regulating SREBP transcriptional activity. PLoS One. 2009;4:e5197. doi: 10.1371/journal.pone.0005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M, Cannella M, Sassone J, Ciammola A, Squitieri F, Beal MF. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum Mol Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF. Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum Mol Genet. 2010;19:3190–3205. doi: 10.1093/hmg/ddq229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Peng Q, Hou Z, Aggarwal M, Zhang J, Mori S, Ross CA, Duan W. Structural MRI detects progressive regional brain atrophy and neuroprotective effects in N171–82Q Huntington's disease mouse model. Neuroimage. 2011;56:1027–1034. doi: 10.1016/j.neuroimage.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1alpha to GABAergic neurons during maturation of the rat brain. J Comp Neurol. 2007;502:1–18. doi: 10.1002/cne.21211. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Dubois-Dalcq M, Behar T, Hudson L, Lazzarini RA. Emergence of three myelin proteins in oligodendrocytes cultured without neurons. J Cell Biol. 1986;102:384–392. doi: 10.1083/jcb.102.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennema-Notestine C, Archibald SL, Jacobson MW, Corey-Bloom J, Paulsen JS, Peavy GM, Gamst AC, Hamilton JM, Salmon DP, Jernigan TL. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology. 2004;63:989–995. doi: 10.1212/01.wnl.0000138434.68093.67. [DOI] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Tortosa E, MacDonald ME, Friend JC, Taylor SA, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel JP, Myers RH. Quantitative neuropathological changes in presymptomatic Huntington's disease. Ann Neurol. 2001;49:29–34. [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathorn T, Snyder-Keller A, Messer A. Nicotinamide improves motor deficits and upregulates PGC-1alpha and BDNF gene expression in a mouse model of Huntington's disease. Neurobiol Dis. 2011;41:43–50. doi: 10.1016/j.nbd.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, van Zijl PC, Kim J, Pearlson GD, Mori S. DtiStudio: resource program for diffusion tensor computation and fiber bundle tracking. Comput Methods Programs Biomed. 2006;81:106–116. doi: 10.1016/j.cmpb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni V, Mariotti C, Tabrizi SJ, Valenza M, Wild EJ, Henley SM, Hobbs NZ, Mandelli ML, Grisoli M, Björkhem I, Cattaneo E, Di Donato S. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington's disease. Brain. 2008;131:2851–2859. doi: 10.1093/brain/awn212. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin T, Xiang Z, Cui L, Stallcup W, Reeves SA. New mouse oligodendrocyte precursor (mOP) cells for studies on oligodendrocyte maturation and function. J Neurosci Methods. 2006;157:187–194. doi: 10.1016/j.jneumeth.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConoughey SJ, Basso M, Niatsetskaya ZV, Sleiman SF, Smirnova NA, Langley BC, Mahishi L, Cooper AJ, Antonyak MA, Cerione RA, Li B, Starkov A, Chaturvedi RK, Beal MF, Coppola G, Geschwind DH, Ryu H, Xia L, Iismaa SE, Pallos J. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med. 2010;2:349–370. doi: 10.1002/emmm.201000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskimins R, Srinivasan R, Marin-Husstege M, Miskimins WK, Casaccia-Bonnefil P. p27(Kip1) enhances myelin basic protein gene promoter activity. J Neurosci Res. 2002;67:100–105. doi: 10.1002/jnr.10080. [DOI] [PubMed] [Google Scholar]
- Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, Richardson EP, Jr, Bird ED. Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus. J Neuropathol Exp Neurol. 1991;50:729–742. doi: 10.1097/00005072-199111000-00005. [DOI] [PubMed] [Google Scholar]
- Nopoulos PC, Aylward EH, Ross CA, Mills JA, Langbehn DR, Johnson HJ, Magnotta VA, Pierson RK, Beglinger LJ, Nance MA, Barker RA, Paulsen JS. Smaller intracranial volume in prodromal Huntington's disease: evidence for abnormal neurodevelopment. Brain. 2011;134:137–142. doi: 10.1093/brain/awq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberkofler H, Schraml E, Krempler F, Patsch W. Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor gamma co-activator 1 alpha. Biochem J. 2003;371:89–96. doi: 10.1042/BJ20021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Hiraoka Y, Matsuoka T, Tomimoto H, Takao K, Miyakawa T, Oshima N, Kiyonari H, Kimura T, Kita T, Nishi E. Nardilysin regulates axonal maturation and myelination in the central and peripheral nervous system. Nat Neurosci. 2009;12:1506–1513. doi: 10.1038/nn.2438. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Reading SA, Yassa MA, Bakker A, Dziorny AC, Gourley LM, Yallapragada V, Rosenblatt A, Margolis RL, Aylward EH, Brandt J, Mori S, van Zijl P, Bassett SS, Ross CA. Regional white matter change in pre-symptomatic Huntington's disease: a diffusion tensor imaging study. Psychiatry Res. 2005;140:55–62. doi: 10.1016/j.pscychresns.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Róna-Vörös K, Weydt P. The role of PGC-1alpha in the pathogenesis of neurodegenerative disorders. Curr Drug Targets. 2010;11:1262–1269. doi: 10.2174/1389450111007011262. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Tuch DS, Hevelone ND, Zaleta AK, Vangel M, Hersch SM, Salat DH. Diffusion tensor imaging in presymptomatic and early Huntington's disease: Selective white matter pathology and its relationship to clinical measures. Mov Disord. 2006;21:1317–1325. doi: 10.1002/mds.20979. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Lee SY, Bender AC, Zaleta AK, Vangel M, Yu P, Fischl B, Pappu V, Onorato C, Cha JH, Salat DH, Hersch SM. Altered white matter microstructure in the corpus callosum in Huntington's disease: implications for cortical “disconnection”. Neuroimage. 2010;49:2995–3004. doi: 10.1016/j.neuroimage.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Saher G, Quintes S, Möbius W, Wehr MC, Krämer-Albers EM, Brügger B, Nave KA. Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J Neurosci. 2009;29:6094–6104. doi: 10.1523/JNEUROSCI.0686-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers D, Sheldon S, Kuperman JM, Goldstein J, Corey-Bloom J, Aron AR. Contrasting gray and white matter changes in preclinical Huntington disease: an MRI study. Neurology. 2010;74:1208–1216. doi: 10.1212/WNL.0b013e3181d8c20a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Valenza M, Cattaneo E. Cholesterol dysfunction in neurodegenerative diseases: is Huntington's disease in the list? Prog Neurobiol. 2006;80:165–176. doi: 10.1016/j.pneurobio.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, Strand A, Tarditi A, Woodman B, Racchi M, Mariotti C, Di Donato S, Corsini A, Bates G, Pruss R, Olson JM, Sipione S, Tartari M, Cattaneo E. Dysfunction of the cholesterol biosynthetic pathway in Huntington's disease. J Neurosci. 2005;25:9932–9939. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza M, Leoni V, Tarditi A, Mariotti C, Björkhem I, Di Donato S, Cattaneo E. Progressive dysfunction of the cholesterol biosynthesis pathway in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2007;28:133–142. doi: 10.1016/j.nbd.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Valenza M, Leoni V, Karasinska JM, Petricca L, Fan J, Carroll J, Pouladi MA, Fossale E, Nguyen HP, Riess O, MacDonald M, Wellington C, DiDonato S, Hayden M, Cattaneo E. Cholesterol defect is marked across multiple rodent models of Huntington's disease and is manifest in astrocytes. J Neurosci. 2010;30:10844–10850. doi: 10.1523/JNEUROSCI.0917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen MH, Camargo N, Verdier V, Nadra K, de Preux Charles AS, Meéard JJ, Luoma A, Crowther M, Inouye H, Shimano H, Chen S, Brouwers JF, Helms JB, Feltri ML, Wrabetz L, Kirschner D, Chrast R, Smit AB. SCAP is required for timely and proper myelin membrane synthesis. Proc Natl Acad Sci U S A. 2009;106:21383–21388. doi: 10.1073/pnas.0905633106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SZ, Dulin J, Wu H, Hurlock E, Lee SE, Jansson K, Lu QR. An oligodendrocyte-specific zinc-finger transcription regulator cooperates with Olig2 to promote oligodendrocyte differentiation. Development. 2006;133:3389–3398. doi: 10.1242/dev.02522. [DOI] [PubMed] [Google Scholar]
- Weaver KE, Richards TL, Liang O, Laurino MY, Samii A, Aylward EH. Longitudinal diffusion tensor imaging in Huntington's Disease. Exp Neurol. 2009;216:525–529. doi: 10.1016/j.expneurol.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Gellera C, Didonato S, Weidinger C, Oberkofler H, Landwehrmeyer GB, Patsch W. The gene coding for PGC-1alpha modifies age at onset in Huntington's Disease. Mol Neurodegener. 2009;4:3. doi: 10.1186/1750-1326-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]