

Abstract
During peripheral nervous system development, successful communication between axons and Schwann cells is required for proper function of both myelinated and nonmyelinated nerve fibers. Alternatively spliced proteins belonging to the neuregulin1 (NRG1) gene family of growth and differentiation factors are essential for Schwann cell survival and peripheral nerve development. Although recent studies have strongly implicated membrane-bound NRG1 forms (type III) in the myelination at late stages, little is known about the role of soluble, heparin-binding forms of NRG1 (type I/II) in regulating early Schwann cell development in vivo. These forms are rapidly released from axons in vitro by Schwann-cell-secreted neurotrophic factors and, unlike membrane-bound forms, have a unique ability to diffuse and adhere to heparan sulfate-rich cell surfaces. Here, we show that axon-derived soluble NRG1 translocates from axonal to Schwann cell surfaces in the embryonic chick between days 5 and 7, corresponding to the critical period of Schwann cell survival. Downregulating endogenous type I/II NRG1 signaling either with a targeted NRG1 antagonist or by shRNA blocks their differentiation from precursors into immature Schwann cells and increases programmed cell death, whereas upregulating NRG1 rescues Schwann cells. Exogenous BDNF also promotes Schwann cell survival through promoting the local release of axonal NRG1. Consistently, increased Schwann cell death occurs both in trkB knock-out mice and after knocking down axonal trkB in chick embryos, which can then be rescued with soluble NRG1. These findings suggest a localized, axoglial feedback loop through soluble NRG1 and BDNF critical for early Schwann cell survival and differentiation.
Introduction
Precisely orchestrated communication between axons and Schwann cells is critical for normal peripheral nervous system development. Part of this communication comes from a family of alternatively spliced, neuron-derived growth and differentiation factors produced by the neuregulin1 (NRG1) gene. NRG1 has been shown to have multiple important functions in the development and maintenance of the peripheral nervous system with most attention on type III, membrane-bound forms that are critical for myelination (Fischbach and Rosen, 1997; Nave and Salzer, 2006; Mei and Xiong, 2008). NRG1 isoforms are produced as transmembrane precursors (proNRG) that are subsequently proteolytically cleaved to both soluble and membrane-bound proteins (Falls, 2003). All isoforms have an EGF-like domain that is essential for erbB receptor activation. Although type III (CRD–NRG1) forms remain tethered to the axonal membrane through a hydrophobic N-terminal cysteine-rich domain (CRD), types I and II forms (HBD–NRG1) are secreted (Loeb and Fischbach, 1995). These soluble forms have a unique N-terminal, positively charged heparin-binding domain (HBD) that targets them to cell surfaces rich in developmentally expressed heparan sulfate proteoglycans (HSPGs) (Loeb et al., 1999, 2002; Li et al., 2004a; Pankonin et al., 2005).
Evidence from both in vitro and in vivo studies has suggested important roles of NRG1 in Schwann cell development (Jessen and Mirsky, 2005; Birchmeier and Nave, 2008). Although soluble NRG1 can rescue Schwann cells from both normal occurring and nerve injury-induced apoptosis in vivo (Kopp et al., 1997; Winseck et al., 2002; Winseck and Oppenheim, 2006), the precise, developmentally specific roles of endogenous soluble NRG1 in regulating early Schwann cell development are still not clear.
Knocking out all NRG1 isoforms or their erbB2/3 receptors results in the almost complete loss of Schwann cells, followed by the death of motor and sensory neurons, suggesting that Schwann cells provide critical trophic support to neurons (Meyer and Birchmeier, 1995; Riethmacher et al., 1997; Woldeyesus et al., 1999). Because neuronal survival is mediated in part by Schwann-cell-secreted neurotrophic factors, including brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) (Jessen and Mirsky, 1999; Garratt et al., 2000), it seems reasonable to postulate a bidirectional signaling network between NRG1 and neurotrophic factors at the axon–Schwann cell interface. We have found that Schwann-cell-derived neurotrophic factors, including BDNF and GDNF, promote the rapid release of soluble NRG1 from both motor and sensory axons (Esper and Loeb, 2004). We have also found that neurotrophic factors produced by postsynaptic muscles at neuromuscular junctions promote activity-dependent soluble NRG1 release (Loeb et al., 2002). Here we provide additional in vivo evidence that endogenous BDNF signaling through axonal trkB receptors promotes a stage-dependent release of soluble NRG1 from axons to Schwann cells. We demonstrate that the release of soluble NRG1 is critical for the survival of Schwann cell precursors (SCPs) as well as their differentiation into immature Schwann cells. This localized, regulatory feedback loop between soluble NRG1 and neurotrophins may not only be important for axoglial communication but may also be helpful in understanding nervous system diseases that involve the axoglial interface.
Materials and Methods
Chick eggs and in ovo treatment.
Fertilized chicken eggs were obtained from Michigan State University Poultry Farms and incubated in a Kuhl rocking incubator at 50% humidity. Daily treatments of recombinant human NRG1-β1 extracellular domain (amino acids 2–246; catalog #377-HB; R & D Systems), recombinant human BDNF (amino acids 129–247; catalog #248-BD; R & D Systems), or the recombinant NRG1 antagonist (HBD–S-H4) on chick embryos were performed as described previously (Loeb et al., 2002; Winseck et al., 2002; Ma et al., 2009). In brief, 5 μg of NRG1, 1 μg of BDNF, and 10 or 20 μg of HBD–S-H4 were each prepared in saline containing 0.2% BSA and added onto the chorioallantoic membrane through a small hole in the air sac without damaging underlying blood vessels for 2 consecutive days. Staging of chick embryos was determined according to Hamburger–Hamilton (HH) stage series (Hamburger and Hamilton, 1951): E4 (stage 24); E5 (stage 26–27); E6 (stage 28–29); and E7 (stage 30–31).
trkB mice.
trkB heterozygous mice were generously provided by M. Barbacid (Bristol-Myers Squibb, New York, NY) (Klein et al., 1993). Homozygous timed-pregnant mutant embryos of either sex were obtained by heterozygous mating and harvested at E12.5 or E13.5 (the day when the vaginal plug was observed is designated as embryonic day 0). Four to five separate litters were collected at each stage. Mouse embryo experiments were performed with approval of Institutional Animal Care and Use Committee at Wayne State University.
Chick in ovo electroporation and shRNA knockdown.
Type I proNRGβ1a cDNA with a myc tag at the C terminus was subcloned into the pMES vector downstream from the chick β-actin promoter with IRES–EGFP (Krull, 2004). shRNA for the heparin-binding domain of chick NRG1 and chick trkB were designed and cloned into the pSilencer 1.0-U6 expression vector (Ambion) according to the instructions of the manufacturer. Three shRNAs against different regions of each cDNA were tested, and the shRNA with the best efficacy in vitro was selected for in ovo electroporation. The target sequence selected for chick HBD of soluble NRG1 was AAGCTAGTGCTAAGGTGTGAA and for chick trkB was AAGGAGCTATATTGAATGAGT. The pCAX vector expressing EGFP was used for coelectroporation with other plasmids to visualize electroporated cells. The final concentration of each plasmid was 3 μg/μl (George et al., 2007). The plasmid DNA was electroporated unilaterally into the ventral part of the neural tube at the lumbar level at E2.5 (HH stage 15–16) as described previously (Eberhart et al., 2002). Electrodes were placed ventrodorsal across the neural tube and pulsed for five times at 35 V for 50 ms with a square-wave pulse generator (Intracept TSS10; Intracel). Embryos were collected from E5 to E7, and only those with strong GFP expression were processed for additional analysis.
Immunostaining, TUNEL, and BrdU.
Chicken and mouse embryos were fixed with fresh 4% paraformaldehyde overnight. Embryos were then equilibrated in 30% sucrose after rinsing quickly with PBS and mounted in OCT (Tissue-Tek). Frozen sections were cut transversely at 14 μm and placed on Superfrost slides (Thermo Fisher Scientific). Immunofluorescence was performed at the lumbar level as described previously (Loeb et al., 1999; Ma et al., 2009). Sections were incubated with antibodies at the following dilutions: chicken soluble NRG1 ectodomain (1:200; 183N, rabbit polyclonal) (Loeb et al., 1999); P0 (1:5; 1E8), AP2α (1:10; 3B5), BrdU (1:100; G3G4), and neurofilament (1:10; 3A10) (Developmental Studies Hybridoma Bank, University of Iowa); GFP (1:100; ab6662) and p75 (1:100; ab70481) (Abcam); myc tag (1:100; 2272; Cell Signaling Technology); and trkB (1:200; sc-12; Santa Cruz Biotechnology). Sections were incubated with antibodies in blocking solution (10% normal goat serum, 0.2% Triton X-100 in PBS) overnight at 4°C, followed by incubation with the corresponding goat anti-mouse or anti-rabbit IgG Alexa Fluor antibodies (1:250; Invitrogen) for visualization. O4 (1:100; MAB345; Millipore) and the trkB antibody were prepared in a blocking solution containing 5% fetal bovine serum in PBS, and, after overnight incubation, goat anti-mouse IgM secondary antibody was used for O4 detection. For BrdU staining, chick embryos were given 10 μg of BrdU through the air sac for 3 h before harvest. Sections were pretreated with 2N HCl to denature DNA for the exposure of BrdU antigen, followed by incubation with blocking solution for 1 h. TUNEL assays were performed with the in situ cell death TMR kit (Roche) following the instructions of the manufacturer (Ma et al., 2009). Some sections were first treated with 1 m NaCl/PBS at 37°C for 90 min as described previously to disrupt ionic interactions between NRG1 and HSPGs (Loeb et al., 1999).
shRNA testing.
105 COS7 cells were seeded in each well of eight-well chamber slides (BD Biosciences) and cotransfected with type I proNRG1 and EGFP cloned in the pTriex vector (Novagen) together with a given shRNA in pSilencer vector using Lipofectamine 2000 (Invitrogen). The next day, cells were fixed in 4% paraformaldehyde for 30 min and stained for NRG1 (1:100; sc-348; Santa Cruz Biotechnology) as described above. Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI).
Immunoblotting and quantitative real-time PCR.
Spinal cords were harvested from BDNF/saline-treated chick embryos or electroporated embryos at E5.5 (HH stage 27) and then processed for protein or RNA extraction. A 5- to 6-somite-long segment at the lumbar level with high levels of GFP expression was isolated from each embryo as described previously (Liu, 2006), and total protein was extracted separately from the electroporated and control sides, using RIPA lysis and extraction buffer containing 25 mm Tris, pH 7.6, 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitors (Thermo Fisher Scientific). Protein samples from three animals were used for immunoblotting using antibodies at the following dilutions: NRG1 (1:500; sc-348; Santa Cruz Biotechnology), β-actin (1:1000; A5441; Sigma), neurofilament (1:2000; AB1987; Millipore), and GFP (1:2500; ab6556; Abcam). SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) was used for signal detection, and each blot was stripped and reprobed with different antibodies. COS7 cells after transfection were harvested by passive lysis buffer (Promega), and sc-348 antibody was used to detect the expression of exogenous NRG1 (this will detect all forms of NRG1 with a cytoplasmic tail). Quantification of band intensity was performed using MetaMorph image analysis software (Universal Imaging Corporation) as described previously (Li and Loeb, 2001; Esper and Loeb, 2004).
For quantitative RT-PCR, total RNA from chick spinal cords after in ovo treatment or electroporation was collected using the RNeasy kit (Qiagen). SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) was used for reverse transcription. Chick HBD–NRG1 transcripts (types I and II) were detected by using the following oligonucleotides directed at the HBD: forward, 5′-GACGGACGTCAACAGCAGTTAC; reverse, 5′-CAACCTCTTGGTTTTTCATTTCCT; and taqman probe, 6FAM-ACACAGTGCCTCCC. For detecting CRD-NRG1 (type III) transcripts, the following primers were used: forward, 5′-ACGGCATCTCAGGCACAAG; reverse, 5′-AAGTGGAAAGTTTTGGAGCAGTTT; and taqman probe, 6FAM-AACAGAAACCAATCTC (Applied Biosystems). Chicken glyceraldehyde-3-phosphate dehydrogenase (Gg03346990_g1; Applied Biosystems) was used for normalization. Quantitative PCR data were collected from five biological replicates, and ΔΔCt was used for calculations.
Imaging and quantitative analysis.
Digital fluorescent images were obtained with a Nikon Eclipse 600 epifluorescence microscope. Confocal stacks of images were acquired from 14-μm-thick sections with a z-step of 1.05 μm using a D-Eclipse C1 confocal system (Nikon), and all representative images are shown as single confocal planes for more precise determination of colocalization. At least five sections for each animal (total five animals) were used for each embryonic stage. MetaMorph image analysis software (Molecular Devices) was used to quantify colocalization. Quantification of TUNEL-positive Schwann cell nuclei was performed as described previously (Ma et al., 2009). Twelve to 20 sections (∼1500 Schwann cell nuclei) at the lumbar level were used to analyze each condition in each animal. shRNA knockdown of trkB was quantified by measuring the intensity of trkB staining divided by the total cell number in the ventral horn that were counted by DAPI staining. At this stage of development, the ventral horn consists almost entirely of motor neurons. Statistical significance was defined as p < 0.05 using either a one-way ANOVA or a two-tailed Student's t test. All data are presented as the mean ± SEM.
Results
Soluble, matrix-bound NRG1 transitions from axons to Schwann cells
Once released from neurons, soluble forms of NRG1 adhere to heparan sulfate-rich surfaces through the developmental expression of HSPGs (Loeb et al., 1999). To examine the spatial distribution of soluble NRG1 during early chick axon–glial interactions in the ventral nerve root, we performed confocal microscopy using specific antibodies against the extracellular domain of soluble type I/II NRG1 together with either Schwann cell (P0 protein) (Bhattacharyya et al., 1991) or axonal (neurofilament) markers from E5 to E7 (Fig. 1). Although at E5 soluble NRG1 is concentrated along motor axons and is not associated with Schwann cells, between E6 and E7, NRG1 becomes progressively localized to Schwann cells (Fig. 1A,B). Quantitative analysis shows that colocalization of NRG1 immunoreactivity on Schwann cells significantly increases from 25 to 75% between E5 and E7 (Fig. 1C), with a corresponding decrease in NRG1 axonal localization (Fig. 1D). To show that the NRG1 immunoreactivity seen is indeed attributable to soluble forms of NRG1 bound to the ECM, we used high-salt treatment to disrupt NRG–HSPGs ionic interactions (Fig. 1E) (Loeb et al., 1999). This treatment significantly reduced soluble NRG1 immunoreactivity, suggesting that the protein is bound to developmentally expressed heparan sulfates along both axons and Schwann cells.
Figure 1.
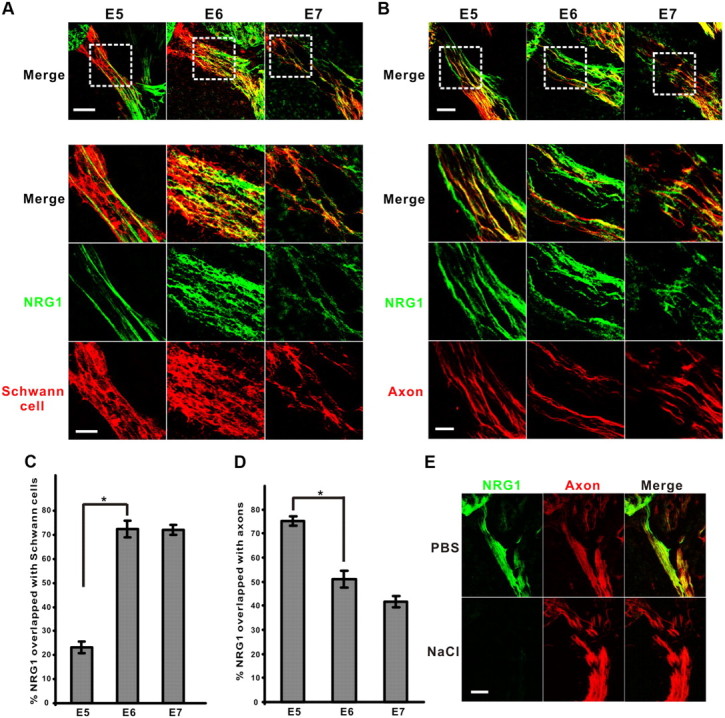
Transition of matrix-bound NRG1 from axons to Schwann cells. Representative confocal images show the colocalization of soluble NRG1 (green) and Schwann cells (P0 protein, red) (A) or axons (neurofilament, red) (B) between E5 and E7. Scale bar: top, 50 μm. Bottom shows high-power images of area from the dashed-line square; scale bar, 20 μm. The percentage of pixel overlapping between NRG1 immunoreactivity and Schwann cells (C) (n = 9, 9, 5 for each stage, respectively) or axons (D) (n = 7, 4, 4 for each stage, respectively) was measured in the ventral root (*p < 0.0001 for E5 vs E6). E, Matrix-bound soluble NRG1 signal (green) was dramatically reduced after high-salt (1 m NaCl) treatment compared with a PBS control at E6, whereas axon immunoreactivity (red) was intact. Scale bar, 50 μm.
Soluble NRG1 regulates Schwann cell survival in vivo
During early peripheral nerve development, an excess number of Schwann cells are born than are ultimately needed, and their survival has been shown to depend on axon-derived NRG1 signaling both in vitro and in vivo during normal development and after axon denervation (Dong et al., 1995; Wolpowitz et al., 2000; Winseck et al., 2002). To investigate additional soluble NRG1 function on Schwann cell survival during this transition of NRG1 from axonal to Schwann cell surfaces, we used a novel NRG1 antagonist (HBD–S-H4) that specifically targets the heparan sulfate-rich surfaces that NRG1 binds (Ma et al., 2009) (Fig. 2A,B). This is achieved by fusing the heparin-binding domain of NRG1 to a soluble “decoy” erbB4 receptor with high affinity for the EGF-like domain of NRG1. Treatment with this antagonist results in a dose-dependent increase in Schwann cell death at E7 along both motor and sensory axons. Consistent with previous studies (Winseck et al., 2002), exogenous soluble NRG1 significantly rescues normal-occurring Schwann cell death at both E5 and E6 (Fig. 2C).
Figure 2.
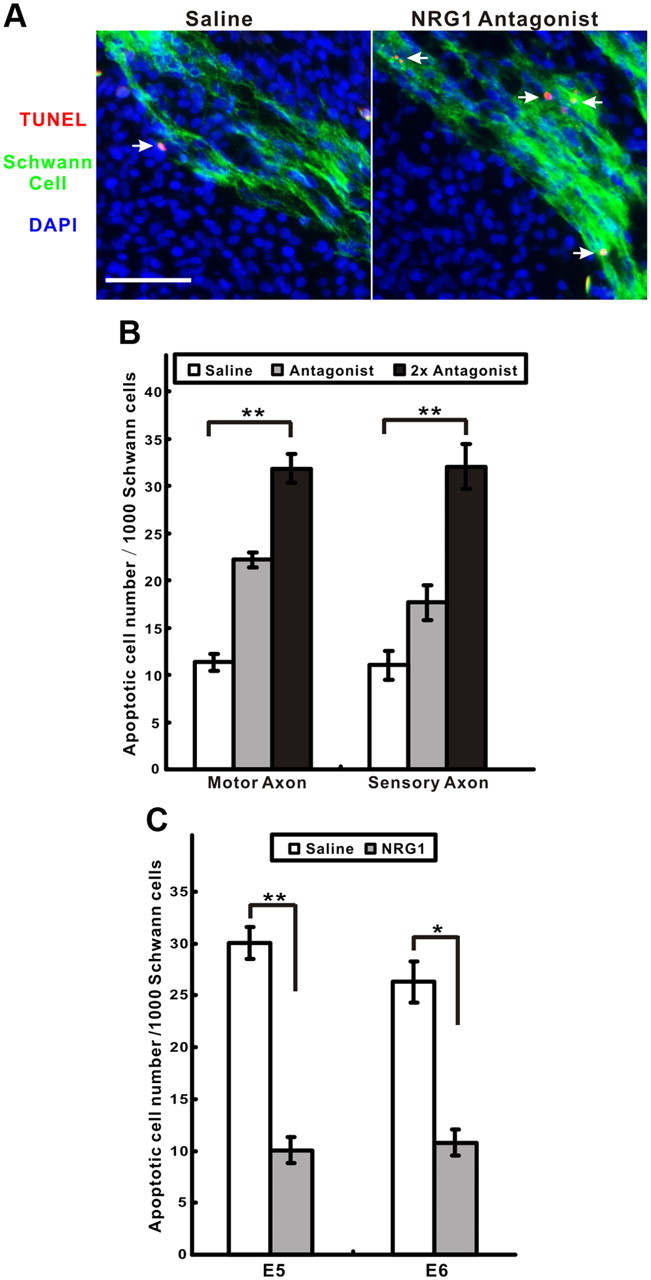
NRG1 signaling promotes Schwann cell survival in vivo. A, A significant increase in TUNEL-positive cells (red, arrows) were detected in motor axons after treatment with a targeted NRG1 antagonist compared with a saline control (Schwann cells, green; DAPI-stained nuclei, blue). Scale bar, 50 μm. B, Dose-dependent increase in apoptotic Schwann cells treated with the antagonist in both motor and sensory axons (n = 7, 6, 7 for each condition, respectively; **p < 0.0001 for both axon regions using one-way ANOVA). C, Addition of soluble type I NRG1 rescues Schwann cells from cell death at both E5 and E6 (n = 4 for each stage; *p < 0.001, **p < 0.0001, for saline vs NRG1 treatment).
To be certain that these survival effects are attributable to endogenous type I/II NRG1 isoforms, we used chick in ovo electroporation to downregulate only soluble NRG1 isoforms in motor neurons using an HBD-specific shRNA that effectively reduces NRG1 expression at protein level by >95% in vitro (Fig. 3A,B). Lumbar ventral spinal cord regions electroporated with this shRNA, together with a plasmid expressing GFP for localization, were compared with the contralateral side without GFP expression (Fig. 3C). Knocking down endogenous soluble NRG1 significantly increased Schwann cell death at E7 (Fig. 3E). In contrast, overexpression of full-length type I NRG1 with a C-terminal myc tag in motor neurons rescued Schwann cells from apoptosis at E6 but not E5 (Fig. 3F). Overexpression of NRG1 was confirmed by myc-tag staining at the electroporated side (Fig. 3D). These results demonstrate that axon-derived, soluble NRG1 isoforms mediate Schwann cell survival in vivo in a stage-dependent manner that parallels the expression of developmentally timed NRG1 deposition on Schwann cells.
Figure 3.

Implication of soluble NRG1 isoforms for Schwann cell survival. A, Efficacy of shRNA in downregulating soluble NRG1 in COS7 cells was measured by cotransfection of full-length NRG1 type I (proNRG1) together with an shRNA–HBD specific for the heparin-binding domain of NRG1. Both no shRNA and a scrambled shRNA (shRNA-con) were used as controls. Transfection efficiency was tracked by GFP (green) expression from a third plasmid. DAPI stains cell nuclei (blue). Scale bar, 50 μm. B, proNRG1 expression in COS7 cell lysates was significantly reduced, with shRNA–HBD transfection showing both the uncleaved (∼75 kDa) and the cleaved (cytoplasmic tail) fragment (∼50 kDa) of proNRG1 by immunoblot analysis. C, In ovo electroporation shows selective expression of GFP (green) in the ventral spinal cord of the electroporated side (EP) as well as in the exiting motor axons surrounded by Schwann cells (red) that are also shown at the contralateral side (CON). DAPI stains cell nuclei (blue). Scale bar, 50 μm. D, Overexpression of NRG1 in ventral spinal cord in vivo using a C-terminal myc-tagged proNRG1 shows myc expression in GFP-positive cells. Scale bar, 20 μm. E, Increased number of apoptotic Schwann cells was seen at E7 after electroporation with shRNA–HBD compared with shRNA-con or GFP alone (n = 7, 4, 6 for each condition, respectively; **p < 0.0005, for shRNA–HBD vs control or shRNA-con). F, Overexpression of proNRG1 in motor axons significantly promotes Schwann cell survival at E6 but not at E5 or control (pMES empty vector alone) (n = 4, 6, 4 for each condition, respectively; *p < 0.005, for proNRG1 at E6 vs vector control; **p < 0.0005, for proNRG1 at E6 vs E5).
Soluble NRG1 promotes differentiation of Schwann cell precursors to immature Schwann cells
SCPs differentiate into immature Schwann cells that elongate along axons and can then further differentiate into myelinating or nonmyelinating Schwann cells, depending on instructions provided by the axon (Taveggia et al., 2005). Although type III NRG1 forms contribute to Schwann cell development in vivo (Wolpowitz et al., 2000), they are not essential for Schwann cell survival and early differentiation, suggesting a role for other types of NRG1. Previous in vitro studies have shown that SCP survival is more critically dependent on soluble NRG1 signaling than are immature Schwann cells (Jessen and Mirsky, 2005). Given that peak Schwann cell death occurs at E5–E6 in the chick (Ciutat et al., 1996), it seems likely that the in vivo survival effects we and others have observed with NRG1 are on SCPs rather than on immature Schwann cells. To confirm this, we measured the transition of SCPs to immature Schwann cells from E4 to E7. Although AP2α, a transcription factor marker for SCPs (Jessen and Mirsky, 2005), was downregulated in motor axon Schwann cells between E4 and E7, it remained highly expressed in the DRG Schwann cells that mature more slowly (Fig. 4A). S100β is often used as a marker of immature Schwann cells in other species, but in the chick embryo, it is not expressed until E13 when the myelination process is initiated (Bhattacharyya et al., 1992). We therefore used an antibody against the lipid antigen O4 as a marker for immature Schwann cell differentiation (Dong et al., 1999). Figure 4A shows that O4 expression turns on rather abruptly in Schwann cells of motor axons at E7, suggesting that the normal transition of SCPs to Schwann cells occurs between E6 and E7. When we blocked NRG1 activity using the targeted NRG1 antagonist between E5 and E6, O4 expression at E7 was prevented in both motor and sensory axons, whereas AP2α expression was not affected (Fig. 4B). These findings suggest that soluble NRG1 signaling is not only critical for SCP survival but also for their differentiation into immature Schwann cells.
Figure 4.
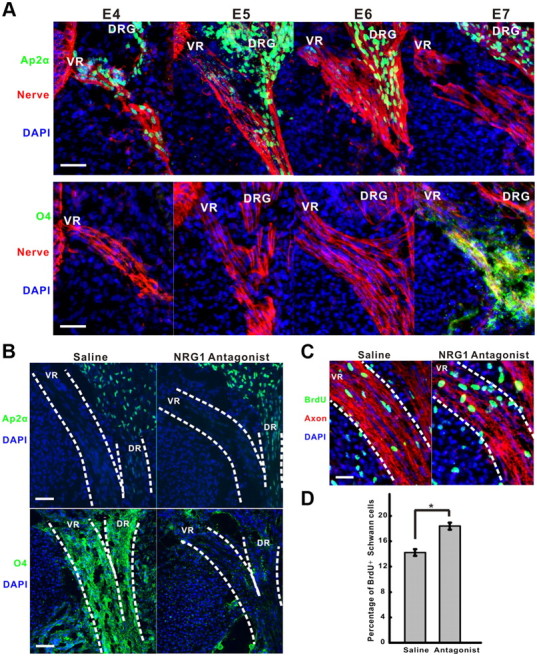
NRG1 signaling is required for Schwann cell differentiation but not proliferation. A, Schwann cell precursors differentiate into immature Schwann cells between E6 and E7 as evidenced by the downregulation of AP2α and upregulation of O4 antigens. AP2α (green) or O4 (green) were used to label SCPs and immature Schwann cells, respectively, from E4 to E7 in the ventral root (VR) labeled by a trkB antibody (red). DRG denotes dorsal root ganglia. DAPI stains cell nuclei (blue). Scale bar, 50 μm. B, Treatment with the NRG1 antagonist at days E5 and E6 prevented the expression of O4 at E7 (bottom, green) in both the ventral root (VR) and the dorsal root (DR) highlighted by the dashed lines. The normal developmental loss of AP2α (top, green) expression in the ventral root (VR) was not affected by the antagonist treatment. DAPI staining shows cell nuclei (blue). Scale bar, 50 μm. C, BrdU incorporation (green) in Schwann cells at E7 in the ventral root (VR, dash lines) counterstained with trkB antibody (red) is slightly increased after 2 d treatment with the NRG1 antagonist compared with a saline control. DAPI staining shows cell nuclei (blue). Scale bar, 20 μm. D, Quantification of the percentage of BrdU-positive Schwann cells in motor axons at E7 shows a small increase in proliferation rate with antagonist treatment (n = 5, 4 for each condition, respectively; *p < 0.002).
Soluble NRG1 was formerly called glial growth factor because of its strong mitogenic effect on Schwann cells in vitro (Dong et al., 1995; Morrissey et al., 1995). However, its effects on proliferation in vivo are less clear, with some data suggesting anti-proliferative effects that are stage dependent (Winseck et al., 2002). We therefore next measured Schwann cell proliferation in the presence and absence of the NRG1 antagonist using BrdU labeling. The density of BrdU-positive Schwann cells along motor axons in E7 animals treated with the antagonist from E5 to E6 in fact is slightly increased, suggesting that NRG1 has a much stronger differentiation rather than proliferation effect on SCPs in vivo (Fig. 4C,D). This small increase in proliferation rate could be simply attributable to the presence of more SCPs, even in the presence of increased apoptosis. Thus, when NRG1-induced differentiation is disrupted, proliferation is higher from these more mitogenically active SCPs.
Axonal trkB signaling regulates Schwann cell survival through localized soluble NRG1 release
The results above suggest that the number of Schwann cells that survive and differentiate is directly regulated by the amount of soluble NRG1 released during this critical period of development, producing a precise matching of Schwann cells needed for each axonal segment. One way for this matching process to occur is through Schwann-cell-derived factors that regulate NRG1 release from axons. We have shown previously that Schwann-cell-derived neurotrophic factors, including BDNF and GDNF, promote the rapid release of soluble NRG1 from both motor and sensory axons (Esper and Loeb, 2004). To explore the possibility that BDNF/trkB signaling indirectly modulates Schwann cell survival in vivo by promoting the release of soluble NRG1 from axons, we treated embryos with exogenous BDNF. As shown in Figure 5A, this treatment significantly promoted SCP survival at E5. To determine whether this BDNF effect was on axons versus Schwann cells, we determined the location of trkB and p75 receptors (low-affinity receptor) in the developing nerve by confocal microscopy (Figs. 5C,D). We found that BDNF receptors trkB and p75 are localized specifically on axons, not Schwann cells. When we downregulated type I/II NRG1 by electroporation, BDNF no longer demonstrated any survival effects (Fig. 5B), suggesting that BDNF regulates Schwann cell survival indirectly through promoting the axonal release of soluble NRG1.
Figure 5.
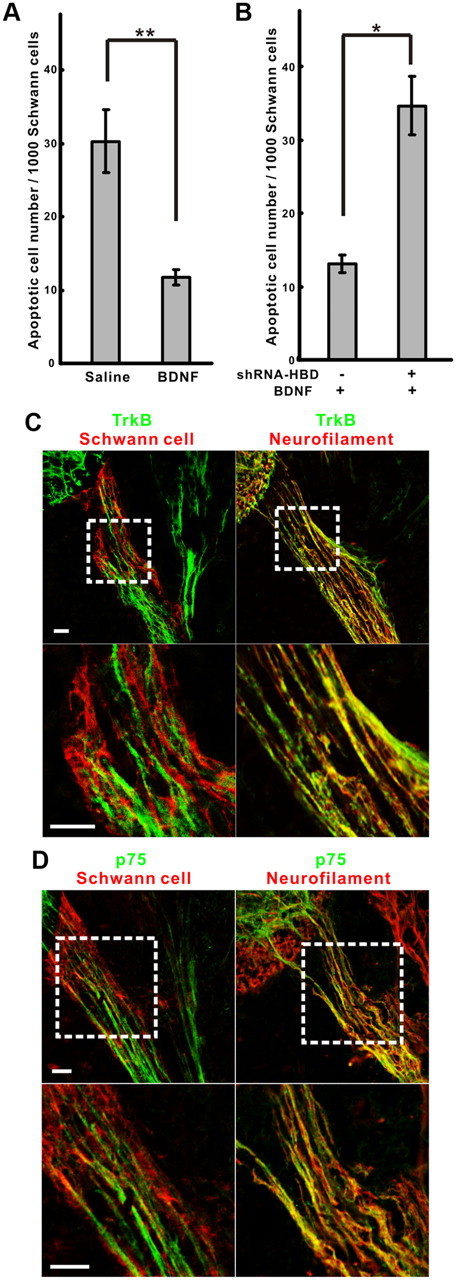
BDNF promotes Schwann cell survival through axonal receptors and soluble NRG1. A, BDNF in ovo treatment significantly decreased Schwann cell death compared with a saline control at E5 (n = 5, 8 for saline and BDNF, respectively; **p < 0.0005). B, Knocking down soluble NRG1 with shRNA–HBD electroporation on one side of the spinal cord abolished the survival effect of BDNF seen on the opposite side (n = 5, *p < 0.005). C, D, Confocal microscopy of the ventral root at E5 with trkB (green) in C or p75 (green) in D, together with Schwann cells (P0 protein, red, left) or neurofilament (red, right) demonstrate the axonal localization of both BDNF receptors. The bottom panels in each show high-power images of corresponding areas highlighted by the dashed lines. Scale bars, 20 μm.
To investigate further as to whether endogenous trkB signaling is required, we examined the effect of disrupting axonal trkB signaling on Schwann cell survival by shRNA in ovo electroporation in the chick as well in trkB knock-out mice. In the electroporation experiments, the opposite side of the spinal cord and GFP electroporation alone were used as controls (Fig. 6A). The shRNA against chick trkB produced a significant reduction of trkB immunoreactivity in GFP-positive cells in the ventral horn (Fig. 6A,B) and was associated with an increase in Schwann cell death at E7. This effect could be rescued with exogenous soluble NRG1 (Fig. 6C), suggesting that endogenous axonal trkB signaling supports Schwann cell survival through axon-derived soluble NRG1.
Figure 6.

Axonal trkB signaling promotes Schwann cell survival through soluble NRG1. A, Electroporation (EP) of an shRNA against trkB together with GFP reduced trkB staining (red) in the ventral horn (circle), relative to both the contralateral side of the spinal cord (CON) and GFP electroporation alone. DAPI staining shows cell nuclei (blue). Scale bar, 50 μm. B, Quantification of the ratio of relative trkB fluorescence between the electroporated versus control side of the spinal cords shows that the shRNA–trkB knocked down trkB expression by 60% per cell in the ventral horn (n = 5, 3 for each group, respectively; *p < 0.005). C, Although Schwann cell death at E7 significantly increased after knocking down trkB, adding back soluble NRG1 to these embryos rescued Schwann cells on both electroporated and control sides (n = 3, 7, 6 for each group, respectively; *p < 0.02, for CON side of shRNA–trkB vs shRNA–trkB plus NRG1; **p < 0.0005, for EP side of shRNA–trkB vs shRNA–trkB plus NRG1 and vs CON side of shRNA–trkB).
Because shRNA knockdowns are never 100% complete, we also examined the effect of the complete absence of trkB at E13.5 in trkB knock-out mice. Knock-out mice show a significant increase of Schwann cell apoptosis at lumbar level motor axons compared with wild-type littermates (Fig. 7A,B). E13.5 in the mouse corresponds to the same stage in the chick at which the SCP-immature Schwann cell transition occurs (E13–E15) (Jessen and Mirsky, 2005). Before this transition, at E12.5, we found no significant difference in Schwann cell death (data not shown), suggesting that Schwann cell survival during this important transition period is selectively regulated by trkB signaling. Together, these results support the presence of a positive feedback loop mediated by local signaling of Schwann-cell-derived BDNF on axonal trkB that promotes NRG1 release from axons that, in turn, leads to the survival of properly positioned Schwann cells.
Figure 7.

Schwann cell death is increased at E13.5 in TrkB knock-out mice. A, TUNEL staining of motor axons (green) at E13.5 shows more TUNEL-positive Schwann cells (red, arrows) in knock-out (KO) compared with wild-type (WT) littermates. DAPI stains cell nuclei (blue). Scale bar, 20 μm. B, Significantly increased numbers of apoptotic Schwann cells are seen in the knock-out embryos (n = 5 for each group; *p < 0.005).
Modulation of BDNF signaling has no effect on NRG1 expression in motor neurons
The above results suggest that BDNF–trkB signaling regulates Schwann cell survival by promoting the localized release of soluble NRG1 forms but cannot rule out additional effects of neurotrophic factor signaling on NRG1 synthesis (Loeb and Fischbach, 1997). To determine whether BDNF–trkB signaling in vivo also affects NRG1 mRNA and protein levels in motor neurons, we compared NRG1 expression [both HBD (type I/II) and CRD (type III) forms] after either BDNF treatment or electroporation with shRNA–trkB. Neither BDNF treatment nor trkB knockdown significantly changed NRG1 expression in the spinal cord at both the mRNA and protein levels (Fig. 8A–F), suggesting that BDNF–trkB signaling is working locally at the axoglial interface to modulate the release of NRG1 in vivo.
Figure 8.

NRG1 isoform expression in spinal cord is not significantly affected by BDNF–trkB signaling in vivo. A, qPCR showed no significant changes in spinal cord mRNA levels for both type I HBD–NRG1 and type III CRD–NRG1 at HH stage 27 after BDNF treatment (n = 5 for each group). B, C, NRG1 protein was also unchanged after BDNF treatment by Western blot. Quantification of the bands ranging from 50 to 150 kDa was done by normalization to actin after reprobing (n = 3, for each group). D, After electroporation with shRNA–trkB, the EP and CON side of lumbar level spinal cord were isolated based on GFP expression. mRNA levels for both HBD–NRG1 and CRD–NRG1 were unchanged by qPCR (n = 5). E, F, NRG1 protein expression was also unchanged by Western blot and quantified as described above (n = 3) with normalization to both actin and neurofilament (NF). Reprobing with a GFP antibody shows the presence of GFP expression only in the electroporated side. EP, Electroporation; CON, control.
Discussion
An axoglial feedback loop between soluble NRG1 and BDNF/TrkB signaling
Although there are extensive in vitro and in vivo studies that implicate both neuronally derived NRG1 and neurotrophic factor signaling in Schwann cell development and myelination (Chan et al., 2001; Cosgaya et al., 2002; Ng et al., 2007), here we have linked these two areas together, providing in vivo evidence for a positive feedback loop between axonal NRG1 and Schwann-cell-derived neurotrophins, such as BDNF. Figure 9 builds a developmental, stage-dependent model of Schwann cell development that incorporates our findings together with other known roles of NRG1 from the literature. In this study, we have shown that endogenous BDNF signaling through axonal trkB receptors regulates SCP survival and differentiation indirectly through modulating the amount of soluble NRG1 released at the axon–glial interface. This is consistent with our previous observations showing that BDNF and other neurotrophic factors rapidly promote soluble NRG1 release from axons in vitro through protein kinase Cδ-induced phosphorylation on the cytoplasmic tail of proNRG1 (Esper and Loeb, 2004; Esper and Loeb, 2009). This reciprocal regulatory pathway is stage dependent and occurs after the period of maximal naturally occurring SCP death at E5.5. A key advantage of this localized communication system is that it does not require communication back to the motor neuron cell body to optimize axoglial interactions. Consistently, we found that the modulation of BDNF–trkB signaling did not change NRG1 mRNA or protein expression in the spinal cord. SCPs that survive from programmed cell death, differentiate into immature Schwann cells, and receive sustained NRG1 signaling through the deposition of developmentally regulated HSPGs on their cell surface (Fig. 9). At later developmental stages, those axons that produce sufficient levels of the type III, membrane-bound NRG1 isoforms are then used required to promote axonal myelination (Taveggia et al., 2005).
Figure 9.

A stage-dependent model for axoglial NRG1 signaling. From E5 to E7 in chick embryos, SCP survival is dependent on a concentration gradient of soluble NRG1 that can be increased at local axon segments through axonal trkB receptor activation (the red spheres represent the heparin-binding domain, and the blue spheres are the EGF-like domain of type I/II NRG1). Those SCPs that do not receive sufficient levels of NRG1 undergo apoptosis. Those that survive require additional NRG1 signaling to differentiate into immature Schwann cells. The immature Schwann cells turn on the expression of cell-surface HSPGs (dashed lines) that act as a “sink” for heparin-binding forms of NRG1, which in turn provides sustained NRG1 signaling at E7. Later, the ensheathment fate (myelination) of axons at E14 requires type III CRD–NRG1.
The need for multiple splice variants of NRG1 in developing axons
The NRG1 gene is perhaps one of the largest and most complex growth factor signaling genes by virtue of the multiplicity of its alternatively spliced forms (Falls, 2003). A given neuron can express both membrane-bound (type III) as well as secreted forms (types I/II). Here, we demonstrate that soluble forms of NRG1 are needed at specific developmental stages, depending on the spatial relationship of axons and glia. All known secreted forms of NRG have an HBD, which significantly augments NRG1 activity and tissue-specific localization through targeting HSPGs (Loeb et al., 1999; Li and Loeb, 2001; Pankonin et al., 2005; Pankonin et al., 2009) and are expressed in spinal cord motor neurons and dorsal root ganglia sensory neurons very early in embryonic development (Meyer et al., 1997; Loeb et al., 1999). For the developing peripheral nerve, soluble forms of NRG1 are initially adherent to axons. SCPs that are close enough to those axons receive sufficient soluble NRG1 and survive to become immature Schwann cells. At E6, NRG1 signaling to developing Schwann cells is stabilized through an accumulation of matrix-bound NRG1. A similar role for soluble NRG1 has been proposed for neuromuscular junction development, in which presynaptically released soluble NRG1 can promote postsynaptic AChR expression (Sandrock et al., 1997). Once synapses have matured and passed the competitive survival stage of synapse elimination, NRG1 becomes highly concentrated in the synaptic basal lamina through the concentration of agrin and other HSPGs in the basal lamina factors (Li and Loeb, 2001; Loeb et al., 2002; Li et al., 2004a). This process also appears to be controlled by a feedback loop between presynaptic NRG1 and postsynaptic neurotrophic factors.
Part of the complexity in understanding the functions of various alternatively spliced forms of NRG1 during development comes from having many diverse roles at many different stages. The chick system has a unique advantage that enables the modulation of specific forms of NRG1 in specific regions at specific stages of development. Knock-out studies in mice, although sometimes more difficult to interpret, have also lead to significant insights. Knocking out all NRG1 isoforms and their receptors leads to a dramatic loss of neural crest cell-derived Schwann cell precursors and the deficiency of sympathetic gangliogenesis (Meyer and Birchmeier, 1995; Meyer et al., 1997; Riethmacher et al., 1997; Morris et al., 1999; Woldeyesus et al., 1999). In contrast, mice specifically deficient in the type III NRG1 isoform show a milder phenotype with the presence of a reduced number of SCPs that still differentiate to Schwann cells and line up along growing axons (Wolpowitz et al., 2000). This finding supports our observations here that type I/II NRG1 forms are needed for SCP survival and differentiation. Other studies have also indirectly implicated the importance of soluble NRG1 in the initial development of the sympathetic nervous system and the induction of muscle spindle differentiation (Britsch et al., 1998; Hippenmeyer et al., 2002).
NRG1 signaling as a differentiation factor but not a mitogen for early Schwann cell development
Our findings suggest a dual role for NRG1 for both survival and differentiation of SCPs. Perhaps one of the most dramatic findings of our targeted NRG1 antagonist was the complete inhibition of O4 expression as a marker of immature Schwann cell differentiation. This effect on differentiation is consistent with previous in vitro experiments showing NRG1 signaling accelerates the SCP–Schwann cell transition (Brennan et al., 2000). The downregulation of AP2α, a marker of SCPs, was not affected with the NRG1 antagonist, suggesting that SCP differentiation to immature Schwann cells is regulated by multiple steps and that NRG1 does not appear to be required for its downregulation.
Given the known mitogentic effects of NRG1 on cultured Schwann cells (Dong et al., 1995), one surprising result was from BrdU experiments showing that, during the transition of SCPs to immature Schwann cells, NRG1 is anti-proliferative. Treatment with the NRG1 antagonist in fact produced an increase, rather than a decrease, in Schwann cell proliferation in the developing nerve. The increased proliferation may have been an indirect effect of the strong anti-differentiation effect of the antagonist, leading to higher numbers of proliferating SCPs. The cancer literature similarly shows both proliferation and differentiation effects of NRG1 on breast epithelial cells that vary as a function of their level of malignant transformation. For example, as breast epithelial cells become more malignant, NRG1 signaling changes from an anti-proliferative to a proliferative effect (Li et al., 2004b).
Effect of soluble NRG1 on myelination, nerve injury, and the potential therapeutic use of a targeted NRG1 antagonist
Because type III NRG1 remains membrane bound, even after proteolytic cleavage from its precursor (Wang et al., 2001), it is ideally positioned to determine the ensheathment fate of axons and regulate myelin sheath thickness in peripheral nerves (Michailov et al., 2004; Taveggia et al., 2005). Interestingly, although high concentrations of soluble NRG1 inhibit Schwann cell myelination, low concentrations that switch from Erk to phosphatidylinositol 3-kinase–Akt activation actually promote myelination (Zanazzi et al., 2001; Syed et al., 2010). Furthermore, it has recently been suggested that, once the peripheral nerve forms, NRG1 is dispensable under normal conditions but critical for nerve repair after injury (Fricker et al., 2011). Together, these studies suggest that factors that modify the localization and concentration of NRG1 can dramatically alter the biological functions of the ligand. Control of NRG1 signaling through both alternative splicing of membrane-bound and secreted forms, the expression patterns of adherent HSPGS, and local gradients of neurotrophic factors from surrounding cells can thus fine tune NRG1 signaling to achieve its many goals in both development and in disease.
Understanding these variables could have important therapeutic implications in human diseases at the axoglial interface such as peripheral neuropathy and demyelinating disorders (Loeb, 2007). Along these lines, although in some disease situations it may be advantageous to increase NRG1 signaling, in others, NRG1 signaling may be detrimental and needs to be blocked. In fact, the NRG1 antagonist we used in this study has been shown to block NRG1-induced microglial activation and the development of chronic pain after peripheral nerve injury (Calvo et al., 2010, 2011).
Footnotes
This work was supported by NIH Grant RO1 NS059947 and the Hiller Amyotrophic Lateral Sclerosis Center. We thank Drs. F. Lefcort (Montana State University) and C. Krull (University of Michigan) for the pCAX and pMES vectors. We also acknowledge the Developmental Studies Hybridoma Bank under the auspices of the NICHD and maintained by the University of Iowa for the monoclonal antibodies used in this study.
References
- Bhattacharyya A, Frank E, Ratner N, Brackenbury R. P0 is an early marker of the Schwann cell lineage in chickens. Neuron. 1991;7:831–844. doi: 10.1016/0896-6273(91)90285-8. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, Oppenheim RW, Prevette D, Moore BW, Brackenbury R, Ratner N. S100 is present in developing chicken neurons and Schwann cells and promotes motor neuron survival in vivo. J Neurobiol. 1992;23:451–466. doi: 10.1002/neu.480230410. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Nave KA. Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia. 2008;56:1491–1497. doi: 10.1002/glia.20753. [DOI] [PubMed] [Google Scholar]
- Brennan A, Dean CH, Zhang AL, Cass DT, Mirsky R, Jessen KR. Endothelins control the timing of Schwann cell generation in vitro and in vivo. Dev Biol. 2000;227:545–557. doi: 10.1006/dbio.2000.9887. [DOI] [PubMed] [Google Scholar]
- Britsch S, Li L, Kirchhoff S, Theuring F, Brinkmann V, Birchmeier C, Riethmacher D. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 1998;12:1825–1836. doi: 10.1101/gad.12.12.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo M, Zhu N, Tsantoulas C, Ma Z, Grist J, Loeb JA, Bennett DL. Neuregulin–ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J Neurosci. 2010;30:5437–5450. doi: 10.1523/JNEUROSCI.5169-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo M, Zhu N, Grist J, Ma Z, Loeb JA, Bennett DL. Following nerve injury neuregulin-1 drives microglial proliferation and neuropathic pain via the MEK/ERK pathway. Glia. 2011;59:554–568. doi: 10.1002/glia.21124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JR, Cosgaya JM, Wu YJ, Shooter EM. Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc Natl Acad Sci U S A. 2001;98:14661–14668. doi: 10.1073/pnas.251543398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciutat D, Calderó J, Oppenheim RW, Esquerda JE. Schwann cell apoptosis during normal development and after axonal degeneration induced by neurotoxins in the chick embryo. J Neurosci. 1996;16:3979–3990. doi: 10.1523/JNEUROSCI.16-12-03979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science. 2002;298:1245–1248. doi: 10.1126/science.1076595. [DOI] [PubMed] [Google Scholar]
- Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR. Neu differentiation factor is a neuron-glia signal and regulates survival, proliferation, and maturation of rat Schwann cell precursors. Neuron. 1995;15:585–596. doi: 10.1016/0896-6273(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Dong Z, Sinanan A, Parkinson D, Parmantier E, Mirsky R, Jessen KR. Schwann cell development in embryonic mouse nerves. J Neurosci Res. 1999;56:334–348. doi: 10.1002/(SICI)1097-4547(19990515)56:4<334::AID-JNR2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Eberhart J, Swartz ME, Koblar SA, Pasquale EB, Krull CE. EphA4 constitutes a population-specific guidance cue for motor neurons. Dev Biol. 2002;247:89–101. doi: 10.1006/dbio.2002.0695. [DOI] [PubMed] [Google Scholar]
- Esper RM, Loeb JA. Rapid axoglial signaling mediated by neuregulin and neurotrophic factors. J Neurosci. 2004;24:6218–6227. doi: 10.1523/JNEUROSCI.1692-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper RM, Loeb JA. Neurotrophins induce neuregulin release through protein kinase Cdelta activation. J Biol Chem. 2009;284:26251–26260. doi: 10.1074/jbc.M109.002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Rosen KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429–458. doi: 10.1146/annurev.neuro.20.1.429. [DOI] [PubMed] [Google Scholar]
- Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, Fageiry SK, Jenkins M, Garratt AN, Birchmeier C, Bennett DL. Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci. 2011;31:3225–3233. doi: 10.1523/JNEUROSCI.2568-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garratt AN, Britsch S, Birchmeier C. Neuregulin, a factor with many functions in the life of a Schwann cell. Bioessays. 2000;22:987–996. doi: 10.1002/1521-1878(200011)22:11<987::AID-BIES5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- George L, Chaverra M, Todd V, Lansford R, Lefcort F. Nociceptive sensory neurons derive from contralaterally migrating, fate-restricted neural crest cells. Nat Neurosci. 2007;10:1287–1293. doi: 10.1038/nn1962. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morph. 1951;88:49–92. [PubMed] [Google Scholar]
- Hippenmeyer S, Shneider NA, Birchmeier C, Burden SJ, Jessell TM, Arber S. A role for neuregulin1 signaling in muscle spindle differentiation. Neuron. 2002;36:1035–1049. doi: 10.1016/s0896-6273(02)01101-7. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci. 1999;22:402–410. doi: 10.1016/s0166-2236(98)01391-5. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Kopp DM, Trachtenberg JT, Thompson WJ. Glial growth factor rescues Schwann cells of mechanoreceptors from denervation-induced apoptosis. J Neurosci. 1997;17:6697–6706. doi: 10.1523/JNEUROSCI.17-17-06697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull CE. A primer on using in ovo electroporation to analyze gene function. Dev Dyn. 2004;229:433–439. doi: 10.1002/dvdy.10473. [DOI] [PubMed] [Google Scholar]
- Li Q, Loeb JA. Neuregulin-heparan-sulfate proteoglycan interactions produce sustained erbB receptor activation required for the induction of acetylcholine receptors in muscle. J Biol Chem. 2001;276:38068–38075. doi: 10.1074/jbc.M104485200. [DOI] [PubMed] [Google Scholar]
- Li Q, Esper RM, Loeb JA. Synergistic effects of neuregulin and agrin on muscle acetylcholine receptor expression. Mol Cell Neurosci. 2004a;26:558–569. doi: 10.1016/j.mcn.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Li Q, Ahmed S, Loeb JA. Development of an autocrine neuregulin signaling loop with malignant transformation of human breast epithelial cells. Cancer Res. 2004b;64:7078–7085. doi: 10.1158/0008-5472.CAN-04-1152. [DOI] [PubMed] [Google Scholar]
- Liu JP. The function of growth/differentiation factor 11 (Gdf11) in rostrocaudal patterning of the developing spinal cord. Development. 2006;133:2865–2874. doi: 10.1242/dev.02478. [DOI] [PubMed] [Google Scholar]
- Loeb JA. Neuroprotection and repair by neurotrophic and gliotrophic factors in multiple sclerosis. Neurology. 2007;68:S38–S42. doi: 10.1212/01.wnl.0000275231.97764.43. discussion S43–S54. [DOI] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J Cell Biol. 1995;130:127–135. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. Neurotrophic factors increase neuregulin expression in embryonic ventral spinal cord neurons. J Neurosci. 1997;17:1416–1424. doi: 10.1523/JNEUROSCI.17-04-01416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Khurana TS, Robbins JT, Yee AG, Fischbach GD. Expression patterns of transmembrane and released forms of neuregulin during spinal cord and neuromuscular synapse development. Development. 1999;126:781–791. doi: 10.1242/dev.126.4.781. [DOI] [PubMed] [Google Scholar]
- Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL. Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. J Neurosci. 2002;22:2206–2214. doi: 10.1523/JNEUROSCI.22-06-02206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Li Q, An H, Pankonin MS, Wang J, Loeb JA. Targeting human epidermal growth factor receptor signaling with the neuregulin's heparin-binding domain. J Biol Chem. 2009;284:32108–32115. doi: 10.1074/jbc.M109.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- Meyer D, Yamaai T, Garratt A, Riethmacher-Sonnenberg E, Kane D, Theill LE, Birchmeier C. Isoform-specific expression and function of neuregulin. Development. 1997;124:3575–3586. doi: 10.1242/dev.124.18.3575. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- Morrissey TK, Levi AD, Nuijens A, Sliwkowski MX, Bunge RP. Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbB2. Proc Natl Acad Sci U S A. 1995;92:1431–1435. doi: 10.1073/pnas.92.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Ng BK, Chen L, Mandemakers W, Cosgaya JM, Chan JR. Anterograde transport and secretion of brain-derived neurotrophic factor along sensory axons promote Schwann cell myelination. J Neurosci. 2007;27:7597–7603. doi: 10.1523/JNEUROSCI.0563-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankonin MS, Gallagher JT, Loeb JA. Specific structural features of heparan sulfate proteoglycans potentiate neuregulin-1 signaling. J Biol Chem. 2005;280:383–388. doi: 10.1074/jbc.M402645200. [DOI] [PubMed] [Google Scholar]
- Pankonin MS, Sohi J, Kamholz J, Loeb JA. Differential distribution of neuregulin in human brain and spinal fluid. Brain Res. 2009;1258:1–11. doi: 10.1016/j.brainres.2008.12.047. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- Sandrock AW, Jr, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- Syed N, Reddy K, Yang DP, Taveggia C, Salzer JL, Maurel P, Kim HA. Soluble neuregulin-1 has bifunctional, concentration-dependent effects on Schwann cell myelination. J Neurosci. 2010;30:6122–6131. doi: 10.1523/JNEUROSCI.1681-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Miller SJ, Falls DL. The N-terminal region of neuregulin isoforms determines the accumulation of cell surface and released neuregulin ectodomain. J Biol Chem. 2001;276:2841–2851. doi: 10.1074/jbc.M005700200. [DOI] [PubMed] [Google Scholar]
- Winseck AK, Oppenheim RW. An in vivo analysis of Schwann cell programmed cell death in embryonic mice: the role of axons, glial growth factor, and the pro-apoptotic gene Bax. Eur J Neurosci. 2006;24:2105–2117. doi: 10.1111/j.1460-9568.2006.05107.x. [DOI] [PubMed] [Google Scholar]
- Winseck AK, Caldero J, Ciutat D, Prevette D, Scott SA, Wang G, Esquerda JE, Oppenheim RW. In vivo analysis of Schwann cell programmed cell death in the embryonic chick: regulation by axons and glial growth factor. J Neurosci. 2002;22:4509–4521. doi: 10.1523/JNEUROSCI.22-11-04509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldeyesus MT, Britsch S, Riethmacher D, Xu L, Sonnenberg-Riethmacher E, Abou-Rebyeh F, Harvey R, Caroni P, Birchmeier C. Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev. 1999;13:2538–2548. doi: 10.1101/gad.13.19.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- Zanazzi G, Einheber S, Westreich R, Hannocks MJ, Bedell-Hogan D, Marchionni MA, Salzer JL. Glial growth factor/neuregulin inhibits Schwann cell myelination and induces demyelination. J Cell Biol. 2001;152:1289–1299. doi: 10.1083/jcb.152.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]