

Carbopalladation across carbon–carbon multiple bonds is a powerful strategy for inter- and intramolecular C–C bond formation.[1] Generally, these processes are well-studied for catalytic cascade transformations involving aryl palladium or vinylpalladium species.[2] In contrast, carbopalladations of Pd enolates, which represent another important class of reactive Pd intermediates widely used in organic synthesis,[3] are exceedingly rare. To date, only a few examples of stoichiometric Pd carbopalladation reactions of carbon–carbon double bonds with Pd enolates have been reported [Eq. (1)].[4] To our knowledge, there are no reports on a catalytic version of this reaction. Herein, we report the Pd-catalyzed cascade transformation featuring the catalytic generation of the Pd enolate by a C–H functionalization strategy, coupled with carbopalladation across the C–C triple bond [Eq. (2)].
 |
(1) |
 |
(2) |
Recently, we reported a hydroarylation reaction of o-alkynyl biaryls A to produce fluorenes B.[5] It was proposed that this reaction proceeds by exclusive 5-exo-dig carbopalladation of a triple bond (to give IV) in aryl palladium intermediate I, which is formed from A by aromatic ortho C–H bond activation in the adjacent arene ring (Scheme 1). We hypothesized that the coordination of palladium to a triple bond and a ketone moiety of 1 would eventually lead to the palladium enolate intermediate II, which upon intramolecular carbopalladation would lead to vinylpalladium species III. Subsequent protiodepalladation of the latter should lead to indanone 2 and regenerate the palladium catalyst. To this end, we examined cyclization of 1-(2-(phenylethynyl)phenyl)ethanone (1a) in the presence of several palladium catalysts. [(Ph3P)2PdCl2] and [(MeCN)2PdCl2] were tested first. However, no desired product 2a was formed under these reaction conditions (Table 1, entries 1 and 2). Next, employment of the Pd(OAc)2 catalyst, widely used in C–H activation reactions, in THF resulted in complete decomposition of 1a. Switching to less polar toluene as solvent prevented the decomposition of the starting material, but it did not support the cyclization reaction even at elevated temperatures (Table 1, entry 4). Further optimizations revealed that the employment of the Pd(OAc)2 catalyst in combination with Ph3P ligand enabled the exclusive 5-exo-dig carbocyclization of 1a, producing 2a in 20 % yield, as a single regio-[6] and stereoisomer (Table 1, entry 5), albeit with unexpected E-geometry of the double bond! Employment of BINAP resulted in a slight improvement of the reaction yield (Table 1, entry 6). Use of 1,1′-bis(diphenylphosphino)ferrocene (dppf) led to further improvement of the reaction yield (Table 1, entry 7). Switching to electron-rich 1,1′-bis(diisopropylphosphino)ferrocene (d-i-prpf) resulted in the dramatic improvement of the reaction outcome (Table 1, entry 8).[5] Finally, employment of 6 mol% d-i-prpf allowed us to obtain 2a in 95 % yield (Table 1, entry 9).
Scheme 1.

Proposed route for the catalytic formation of Pd enolate.
Table 1.
Optimization of the reaction conditions.[a]
 | |||||
|---|---|---|---|---|---|
| Entry | [Pd] source | Ligand (mol %) | Solvent[b] | T [°C] | 2a [%][c] |
| 1 | [(Ph3P)2PdCl2] | toluene | 120 | 0 | |
| 2 | [(MeCN)2PdCl2] | toluene | 120 | 0 | |
| 3 | Pd(OAc)2 | THF | 50 | 0[d] | |
| 4 | Pd(OAc)2 | toluene | 120 | 0 | |
| 5 | Pd(OAc)2 | Ph3P (10) | toluene | 120 | 20 |
| 6 | Pd(OAc)2 | binap (5) | toluene | 120 | 39 |
| 7 | Pd(OAc)2 | dppf (5) | toluene | 120 | 50 |
| 8 | Pd(OAc)2 | d-i-prpf (5) | toluene | 120 | 89 |
| 9 | Pd(OAc)2 | d-i-prpf (6) | toluene | 120 | 95 |
Reaction conditions: alkynyl ketone 1 (0.5 mmol), palladium catalyst (0.05 mmol), 12 h.
All reactions were performed at an alkyne concentration of 0.5 M.
Yields were determined by GC-MS analysis versus internal standard.
Decomposition of the starting material was observed. binap = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, dppf = 1,1′-bis(diphenylphosphino)ferrocene, d-i-prpf = 1,1′-bis(diisopropylphosphino)ferrocene.
Next, the scope of this Pd-catalyzed cyclization was examined (Table 2). We found that this reaction was efficient for a wide range of alkynyl ketones 1, providing the alkylidene indanones 2a–l as single E isomers in good to excellent yields (Table 2). Various functional groups, such as F (Table 2, entries 4, 9), OMe (Table 2, entries 3, 7, 14), Cl (Table 2, entry 6), and CN (Table 2, entry 7) were perfectly tolerated under the reaction conditions. Cyclization of pyridine derivatives of o-alkynyl ketones occurred uneventfully as well, producing 2k (Table 2, entry 11) and 2l (Table 2, entry 12) in 86 % and 52% yield, respectively. Notably, the Pd-catalyzed cyclization of o-alkynyl aryl ketones possessing aryl substituents α to the carbonyl group (R1 = Ar) resulted in formation of isomeric indenone structures 2m (Table 2, entry 13) and 2n (Table 2, entry 14) in good yields. The cyclization of a substrate possessing a fluorine substituent ortho to the carbonyl function was effective as well, providing indanone 2o in 75% yield (Table 2, entry 15).
Table 2.
Pd-catalyzed 5-exo-dig carbocyclization of 1.
 | ||
|---|---|---|
| Entry | Product | Yield [%][a] |
| 1 |
 2a |
92 |
| 2 |
 2b |
98 |
| 3 |
 2c |
93 |
| 4 |
 2d |
98 |
| 5 |
 2e |
93 |
| 6 |
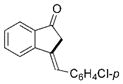 2f |
69 |
| 7 |
 2g |
76 |
| 8 |
 2h |
71 |
| 9 |
 2i |
98 |
| 10 |
 2j |
86 |
| 11 |
 2k |
86 |
| 12 |
 2l |
52 |
| 13 |
 2m |
73 |
| 14 |
 2n |
81 |
| 15 |
 2o |
75 |
Yield of isolated product. See the Supporting Information for details.
Naturally, we were intrigued by the unexpected E-geometry of the double bond in the product 2. Different potential reaction pathways could account for the formation of E-alkylidene indanone 2 from 1 in this Pd-catalyzed cyclization reaction (Scheme 2). The first scenario (path A) features a well-known π-philic activation of the triple bond in 1 with PdII catalyst (3)[2a,7] and subsequent intramolecular 5-exo-dig carbocyclization of the enol tautomer 4 to produce trans-addition intermediate 5. Protiodepalladation of the latter furnishes E-alkylidene indanone 2 directly. Alternatively, the reaction may occur by the initial coordination of the palladium catalyst to the carbonyl group in alkynyl ketone 1 to form 6, which upon deprotonation would produce a Pd enolate species 7 (path B). The subsequent coordination of Pd in 7 to the triple bond would form the alkyne-coordinated Pd enolate 8. This species, upon intramolecular carbopalladation of the triple bond, would lead to a vinylpalladium species 9. The subsequent E–Z isomerization of the double bond[8] would furnish isomeric vinylpalladium intermediate 5, which upon protiodepalladation would produce E isomer 2.
Scheme 2.

Plausible reaction pathways for carbocyclization of 1.
To verify the possibility of the electrophilic mechanism for this transformation (Scheme 2, path A), the carbocyclization of alkynyl ketone 1a was tested in the presence of several π-philic metal salts, including AuCl, AuCl3, PtCl2, electrophilic [Ph3PAuOTf], and Cu(OTf)2,[9] known to mediate cyclizations by π-philic activation of the alkyne moiety. However, no formation of the desired product 2a under these reaction conditions was observed.[10] This observation, together with the shown earlier low efficiency of Pd(OAc)2[9f] alone to catalyzed this transformation, did not support the possibility of the electrophilic path A for this transformation.
To further shed light on the possible reaction mechanism of the Pd-catalyzed cyclization of o-alkynyl ketones, density functional theory (DFT) calculations of the possible reaction pathways A and B were performed (Scheme 3).[11] Accordingly, pathway A was ruled out owing to the high free energy (49.3 kcal mol−1) of the corresponding transition state from 4 to 5.[12] In contrast, pathway B, having the lower energy profile and consisting of two major steps with nearly equivalent energy barriers (TS-1 and TS-2), was found to be the most probable pathway for this transformation.[12] Thus, reaction starts with the formation of a complex 6 upon slightly endothermic coordination of compound 1 with the palladium catalyst. The subsequent intramolecular deprotonation of the methyl group of the ketone by the acetate ligand of the PdII catalyst, via transition state TS-1 (ΔG⧧ = 31.2 kcalmol−1), produces 10 (28.6 kcalmol−1), which, upon loss of the AcOH molecule, easily converts into palladium π complex 7 (11.0 kcal mol−1). Notably, this process is quite different from the concerted metalation–deprotonation (CMD) step in the PdII-catalyzed coupling reactions, in which proton transfer to the base and Pd–C bond formation occurs at the same time.[14] Next, the reaction intermediate 7 transforms into the key alkyne-coordinated Pd enolate 8. The subsequent rate-limiting carbopalladation of the triple bond (TS-2, ΔG⧧ = 31.6 kcal mol−1) produces vinyl palladium species 9, which upon E–Z isomerization (TS-3, ΔG⧧ = 9.6 kcalmol−1) transforms into the isomer 5 (ΔG = −5.4 kcalmol−1). Protiodepalladation of the latter leads to the E product 2a (ΔG = −28.6 kcalmol−1; Scheme 3).
Scheme 3.

Gibbs free energies for the Pd-catalyzed carboyclization of 1 a (L = PMe3).[13]
In conclusion, we have developed the Pd-catalyzed regio- and stereoselective carbocyclization of o-alkynyl ketones, featuring the carbopalladation of the triple bond with Pd enolate species. This methodology allows for the synthesis of a variety of alkylidene indanones as single E stereoisomers in good to excellent yields. DFT calculations of the reaction mechanism revealed that the formation of the Pd enolate intermediate occurs by deprotonation assisted by the PdII acetate ligand. It was also demonstrated that the intramolecular carbopalladation of the alkyne with Pd enolate produces E-vinylpalladium species, which upon Z–E isomerization of the double bond transforms into thermodynamically more favorable Z isomer. Protiodepalladation from the latter produces indanones with unexpected E geometry of the methylene double bond.
Experimental Section
An oven-dried 3 mL Wheaton V-vial containing a stirring bar, was charged with 2′-alkynyl ketone 1 (0.5 mmol), Pd(OAc)2 (5.6 mg, 0.025 mmol), and d-i-prpf (13 mg, 0.03 mmol) under N2 atmosphere. Dry toluene (1 mL) was added, and the reaction vessel was capped with a pressure screw cap. The reaction mixture was heated at 100–120°C for 2–8 h until judged complete by GC/MS analysis. The resulting mixture was cooled down to room temperature and filtered through a layer of silica gel with the aid of EtOAc. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel (eluent: hexanes/EtOAc 20:1 to 10:1), thus affording the corresponding indenone product 2.
Supplementary Material
Footnotes
The support of the NIH (GM-64444) and the NSF (CHE-0710749) of US, and CCRI of Canada is gratefully acknowledged.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201006751.
Contributor Information
Natalia Chernyak, Department of Chemistry, University of Illinois at Chicago, 845 W. Taylor St., Rm 4500 SES, Chicago, IL 60607 (USA).
Dr. Serge I. Gorelsky, Centre for Catalysis Research and Innovation (CCRI), Department of Chemistry, University of Ottawa, 10 Marie Curie, Ottawa, ON K1N 6N5 (Canada).
Prof. Vladimir Gevorgyan, Department of Chemistry, University of Illinois at Chicago, 845 W. Taylor St., Rm 4500 SES, Chicago, IL 60607 (USA).
References
- 1.For reviews on carbopalladation of multiple bonds, see: Negishi Ei, Coperet C, Ma S, Liou S-Y, Liu F. Chem Rev. 1996;96:365. doi: 10.1021/cr950020x.Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048.Zimmer R, Dinesh CU, Nandanan E, Khan FA. Chem Rev. 2000;100:3067. doi: 10.1021/cr9902796.
- 2.For reviews on Pd-catalyzed cascade transformations, see: Balme G, Bouyssi D, Lomberget T, Monteiro N. Synthesis. 2003:2115.Abu Sohel SM, Liu RS. Chem Soc Rev. 2009;38:2269. doi: 10.1039/b807499m.
- 3.For selected references on transformations involving Pd enolates, see: Biscoe MR, Buchwald SL. Org Lett. 2009;11:1773. doi: 10.1021/ol900295u.Mart%n R, Buchwald SL. Org Lett. 2008;10:4561. doi: 10.1021/ol8017775.Hamashima Y, Sasamoto N, Umebayashi N, Sodeoka M. Chem Asian J. 2008;3:1443. doi: 10.1002/asia.200800120.Liao X, Weng Z, Hartwig JF. J Am Chem Soc. 2008;130:195. doi: 10.1021/ja074453g.Sasamoto N, Dubs C, Hamashima Y, Sodeoka M. J Am Chem Soc. 2006;128:14010. doi: 10.1021/ja065646r.Hamashima Y, Suzuki T, Takano H, Shimura Y, Sodeoka M. J Am Chem Soc. 2005;127:10164. doi: 10.1021/ja0513077.Hamashima Y, Hotta D, Umebayashi N, Tsuchiya Y, Suzuki T, Sodeoka M. Adv Synth Catal. 2005;347:1576.Hamashima Y, Somei H, Shimura Y, Tamura T, Sodeoka M. Org Lett. 2004;6:1861. doi: 10.1021/ol0493711.Hennessy EJ, Buchwald SL. J Am Chem Soc. 2003;125:12084. doi: 10.1021/ja037546g.Culkin DA, Hartwig JF. Acc Chem Res. 2003;36:234. doi: 10.1021/ar0201106.Hamashima Y, Yagi K, Takano H, Tamás L, Sodeoka M. J Am Chem Soc. 2002;124:14530. doi: 10.1021/ja028464f.Hamada T, Chieffi A, Åhman J, Buchwald SL. J Am Chem Soc. 2002;124:1261. doi: 10.1021/ja011122+.Fox JM, Huang X, Chieffi A, Buchwald SL. J Am Chem Soc. 2000;122:1360.
- 4.For selected examples of stoichiometric intramolecular carbopalladations of alkenes, see: Ito Y, Aoyama H, Hirao T, Mochizuki A, Saegusa T. J Am Chem Soc. 1979;101:494.Ito Y, Aoyama H, Saegusa T. J Am Chem Soc. 1980;102:4519.Kende AS, Roth B, Sanfilippo PJ. J Am Chem Soc. 1982;104:1784.Kende AS, Roth B, Sanfilippo PJ, Black-lock TJ. J Am Chem Soc. 1982;104:5808.Larock RC, Ho Lee N. Tetrahedron Lett. 1991;32:5911.
- 5.a) Chernyak N, Gevorgyan V. J Am Chem Soc. 2008;130:5636. doi: 10.1021/ja8006534. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chernyak N, Gevorgyan V. Adv Synth Catal. 2009;351:1101. doi: 10.1002/adsc.200800765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected examples of palladium catalyzed oxocyclization of ketoalkynes, see: Kobayashi K, Hashimoto K, Fukamachi S, Konishi H. Synthesis. 2008:1094.Lemhadri M, Doucet H, Santelli M. Tetrahedron. 2005;61:9839.
- 7.For selected examples of π-philic activation of alkynes by PdII species, see: Tunge JA, Foresee LN. Organometallics. 2005;24:6440.Nevado C, Charruault L, Michelet V, Nieto-Oberhuber C, Muñoz M, Méndez M, Rager MN, Genêt JP, Echavarren A. Eur J Org Chem. 2003:706.
- 8.For examples of E–Z isomerization of alkenes in Pd-catalyzed reactions, see: Dyker G, Kellner A. Tetrahedron Lett. 1994;35:7633.Cacchi S, Fabrizi G, Marinelli F, Moro L, Pace P. Tetrahedron. 1996;52:10225.Amatore C, Bensalem S, Ghalem S, Jutand A. J Organomet Chem. 2004;689:4642.
- 9.For selected examples of cyclization of alkynyl carbonyl compounds in the presence of π-philic metals, see: Binder JT, Crone B, Haug TT, Menz H, Kirsch SF. Org Lett. 2008;10:1025. doi: 10.1021/ol800092p.Gao Q, Zheng BF, Li JH, Yang D. Org Lett. 2005;7:2185. doi: 10.1021/ol050532q.Kennedy-Smith JJ, Staben ST, Toste FD. J Am Chem Soc. 2004;126:4526. doi: 10.1021/ja049487s.Asao N, Nogami T, Lee S, Yamamoto Y. J Am Chem Soc. 2003;125:10921. doi: 10.1021/ja036927r.Patil NT, Wu H, Yamamoto Y. J Org Chem. 2005;70:4531. doi: 10.1021/jo050191u.Asao N, Nogami T, Takahashi K, Yamamoto Y. J Am Chem Soc. 2002;124:764. doi: 10.1021/ja017366b.Asao N, Takahashi K, Lee S, Kasahara T, Yamamoto Y. J Am Chem Soc. 2002;124:12650. doi: 10.1021/ja028128z.
- 10.Frisch MJ, et al. DFT calculations were performed at the B3LYP/DZVP level of theory using Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 11.See the Supporting Information for details.
- 12.Structures and free energies for alternative reaction pathways are shown in Figures S1 and S2 of the Supporting Information.
- 13.For selected references on concerted metalation–deprotonation pathway in transition-metal-catalyzed C–H functionalizations, see: Rousseaux S, Gorelsky SI, Chung BKW, Fagnou K. J Am Chem Soc. 2010;132:10692. doi: 10.1021/ja103081n.Liégault B, Petrov I, Gorelsky SI, Fagnou K. J Org Chem. 2010;75:1047. doi: 10.1021/jo902515z.Liégault B, Lapointe D, Caron L, Vlassova A, Fagnou K. J Org Chem. 2009;74:1826. doi: 10.1021/jo8026565.Liégault B, Lapointe D, Caron L, Vlassova A, Fagnou K. J Org Chem. 2009;74:1826. doi: 10.1021/jo8026565.Özdemir I, Demir S, Çetinkaya B, Gourlaouen C, Maseras F, Bruneau C, Dixneuf PH. J Am Chem Soc. 2008;130:1156. doi: 10.1021/ja710276x.Gorelsky SI, Lapointe D, Fagnou K. J Am Chem Soc. 2008;130:10848. doi: 10.1021/ja802533u.Ackermann L, Vicente R, Althammer A. Org Lett. 2008;10:2299. doi: 10.1021/ol800773x.García-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J Am Chem Soc. 2006;128:1066. doi: 10.1021/ja056165v.Davies DL, Donald SMA, Al-Duaij O, Macgregor SA, Pölleth M. J Am Chem Soc. 2006;128:4210. doi: 10.1021/ja060173+.Davies DL, Donald SMA, Macgregor SA. J Am Chem Soc. 2005;127:13754. doi: 10.1021/ja052047w.Biswas B, Sugimoto M, Sakaki S. Organometallics. 2000;19:3895.Lavin M, Holt EM, Crabtree RH. Organometallics. 1989;8:99.
- 14.PdL2 species were used in calculations because only bidentate ligands were effective in this transformation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.