

Functionalized aryl and heteroaryl alkynes are highly valuable classes of compounds widely used in contemporary organic synthesis and materials science. Such compounds are commonly formed by a Sonogashira cross-coupling reaction between a hetero(aryl) halide and a terminal alkyne. However, there has been growing interest in the development of a complementary strategy, an “inverse Sonogashira coupling” involving the direct alkynylation of unreactive C–H bonds with readily available alkynyl halides. A historical outline of the development of this transformation promoted or catalyzed by various main-group and transition metals is depicted in Scheme 1.
Scheme 1.

Development of the direct alkynylation of (hetero)arenes.
The first practical example of this type of alkynylation of an aromatic heterocycle, the sydnone derivative 1, was disclosed by Kalinin et al. in 1992.[1] This formal direct alkynylation involved the use of a stoichiometric amount of CuI to generate the organocopper intermediate 2, which underwent palladium(0)-catalyzed cross-coupling with alkynyl bromides 3 to give alkynyl sydnones 4 [Eq. (1)].
Later, Trofimov and co-workers, who introduced the term “inverse Sonogashira coupling”, reported that a variety of
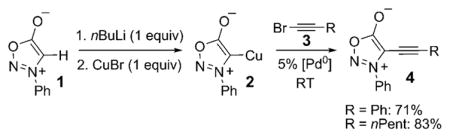 |
(1) |
pyrroles and indoles 5 underwent alkynylation promoted by greater than stoichiometric amounts of Al2O3 to give C2-alkynylated pyrroles and C3-alkynylated indoles in good yields [Eq. (2)].[2] This reaction is specific to electron-deficient alkynyl ketones and esters 6, as it features the trans addition of nucleophilic heterocycles 5 to Michael acceptors 6, followed by a subsequent dehydrobromination to form 8. Besides Al2O3, other main-group metal oxide active surfaces, such as BaO and ZnO,[2b] and K2CO3 efficiently promoted this transformation.
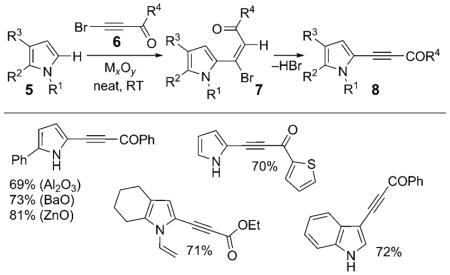 |
(2) |
In 2002, Yamaguchi and co-workers reported the first example of a catalytic direct alkynylation of aromatic compounds: phenols 9 (X = O) were coupled with the chloroalkyne 10 in the presence of a catalytic amount of the main-group-metal salt GaCl3 and the bases nBuLi and 2,6-di(tert-butyl)-4-methylpyridine (DtBMP) [Eq. (3); Bn = benzyl].[3a] A variety of alkynyl phenols 12 (X = O), including halosubstituted derivatives, were accessed in this way with exclusive ortho selectivity. The authors proposed that this reaction occurs via the vinyl–gallium intermediate 11 generated upon the carbogallation of 10 with gallium phenoxide; a subsequent β elimination yielded 12. Later, the same group adopted this chemistry for a direct alkynylation of N-benzylanilines 9 (X = NBn).[3b]
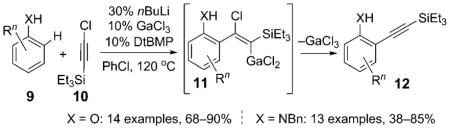 |
(3) |
This field did not experience major growth, however, until 2007, when the first example of a transition-metal-catalyzed direct alkynylation of electron-rich N-fused heterocycles was reported by our research group (Scheme 2).[4] We showed that in the presence of a palladium catalyst, indolizine, pyrroloquinoline, pyrroloisoquinoline, and pyrrolooxazole cores 13 were highly efficiently and regioselectively alkynylated with bromoalkynes 3 containing a broad range of substituents. The crucial conceptual advance was the recognition that the reactivity of the alkynyl–palladium intermediate 15, generated through the oxidative addition of Pd0 into the C–Br bond of 3, resembled that of the aryl–palladium species 15′, which is known to participate in the arylation of indolizines through an electrophilic mechanism[5] (of the type 13→16→17; Scheme 2).
Scheme 2.

Palladium-catalyzed alkynylation of N-fused heterocycles. TMS = trimethylsilyl.
Subsequently, Gu and Wang applied this chemistry to the direct palladium-catalyzed regioselective C3 alkynylation of indoles 18 with various aryl- and alkenyl-substituted alkynyl bromides 3 [Eq. (4)].[6] An electrophilic mechanism was also suggested in this case by the authors for the alkynylation reaction.
Further benefits of the use of transition metals were revealed by Chatani and co-workers in an alkynylation of anilides that is complementary to the transformation described by Yamaguchi and co-workers[3b] (Scheme 3).[7] Thus, a variety of anilides 20 underwent the palladium(II)-catalyzed
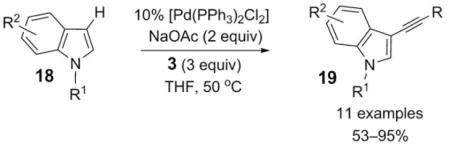 |
(4) |
directed ortho alkynylation to furnish aryl alkynes 22 in moderate to high yields. The authors proposed that the reaction proceeded by the ortho palladation of 20 with an electrophilic palladium catalyst to give palladacycle 23; the palladation was enhanced by the requisite addition of a silver salt. Next, two possibilities were envisioned. The first, similar to the proposal of Yamaguchi and co-workers,[3] involved carbopalladation (→25), followed by trans β elimination. An alternative path featured the PdII/PdIV cycle: the oxidative addition of 21 to 23 was followed by reductive elimination from 24. Importantly, since no Pd0 species was involved in the catalytic cycle, halogen substituents (Cl, Br) could be present. Thus, subsequent elaboration of the products by standard cross-coupling reactions is possible.
Scheme 3.

Direct alkynylation of anilides. Tf = trifluoromethanesulfonyl, TIPS = triisopropylsilyl.
Recently, nickel(0)- and copper(I)-catalyzed variations of the inverse Sonogashira reaction of azoles 26 with different alkynyl bromides 3 were reported by Miura and co-workers[8] and Besselièvre and Piguel[9] [Eq. (5); cod =1,5-cyclooctadiene, dppbz =1,2-bis(diphenylphosphanyl)benzene, dpephos = bis(2-(diphenylphosphanyl)phenyl) ether]. These reactions proceeded in moderate to high yields with an array of azole cores [see Eq. (5)]. Mechanistically, the direct alkynylation developed by Miura and co-workers proceeds through a catalytic version of the formal cross-coupling reaction described by Kalinin et al. [see Eq. (1)]. The alkynyl–nickel intermediate formed by the oxidative addition of the Ni0 catalyst to 3 undergoes a transmetalation/reductive elimination sequence with a heteroaryl copper or lithium species I,[1a] which is generated in situ through the metalation of 26. Independently, Besselièvre and Piguel[9] postulated the same heteroaryl–copper intermediate I, the subsequent transformation of which was proposed to involve a CuI/CuIII cycle resembling the PdII/PdIV cycle proposed by Chatani and coworkers.[7]
Gold is a recent addition by Waser and co-workers to the arsenal of transition-metal catalysts employed in the inverse Sonogashira reaction.[10] Unprecedented functional-group tolerance and mild reaction conditions were demonstrated
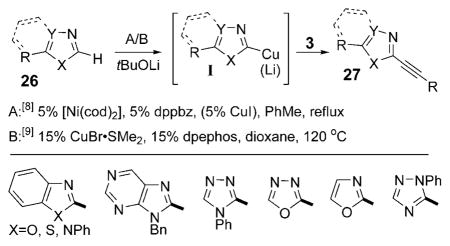 |
(5) |
in the gold(I)-catalyzed alkynylation of indole (C3) and pyrrole (C2) cores 28 with the recyclable hypervalent alkynyl iodine reagent 29 [Eq. (6)]. The observed regioselectivity of alkynylation could be overruled by blocking the C3 (C2) position of the indole (pyrrole), or by the introduction of a bulky triisopropylsilyl (TIPS) group at the pyrrole N atom. Several mechanistic hypotheses featuring trans addition/elimination and AuI/AuIII catalytic cycles were suggested by the authors for this reaction.
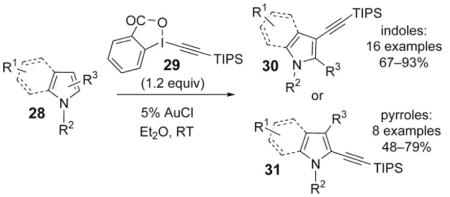 |
(6) |
In summary, recent findings in the field of direct alkynylation reactions open up new exciting opportunities for the functionalization of C–H bonds. Although the development of more general and efficient catalytic systems and the expansion of the scope of this reaction are still highly wanted, the current advances augur the continuing growing interest in and broad application of this method in synthesis.
Footnotes
The support of the National Institutes of Health (GM-64444) is gratefully acknowledged.
References
- 1.Kalinin VK, Pashchenko DN, She FM. Mendeleev Commun. 1992;2:60. for the palladium-catalyzed direct alkynylation of 3-phenylsydnone (1) in 34% yield, see: Rodriguez A, Fennesy RV, Moran WJ. Tetrahedron Lett. 2009;50:3942.
- 2.Trofimov BA, Stepanova ZV, Sobenina LN, Mikhaleva AI, Ushakov IA. Tetrahedron Lett. 2004;45:6513.Trofimov BA, Sobenina LN, Stepanova ZV, Vakul’skaya TI, Kazheva ON, Aleksandrov GG, Dyachenko OA, Mikhaleva AI. Tetrahedron. 2008;64:5541. and references therein.
- 3.a) Kobayashi K, Arisawa M, Yamaguchi M. J Am Chem Soc. 2002;124:8528. doi: 10.1021/ja026108r. [DOI] [PubMed] [Google Scholar]; b) Amemiya R, Fujii A, Yamaguchi M. Tetrahedron Lett. 2004;45:4333. [Google Scholar]
- 4.Seregin IV, Ryabova V, Gevorgyan V. J Am Chem Soc. 2007;129:7742. doi: 10.1021/ja072718l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For reviews, see: Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173. doi: 10.1039/b606984n.Campeau LC, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35.
- 6.Gu Y, Wang X. Tetrahedron Lett. 2009;50:763. [Google Scholar]
- 7.Tobisu M, Ano Y, Chatani N. Org Lett. 2009;11:3250. doi: 10.1021/ol901049r. [DOI] [PubMed] [Google Scholar]
- 8.Matsuyama N, Hirano K, Satoh T, Miura M. Org Lett. 2009;11:4156. doi: 10.1021/ol901684h. [DOI] [PubMed] [Google Scholar]
- 9.Besselièvre F, Piguel S. Angew Chem. 2009;121:9717. doi: 10.1002/anie.200904776. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9553. [Google Scholar]
- 10.Brand JP, Charpentier J, Waser J. Angew Chem. 2009;121:9510. doi: 10.1002/anie.200905419. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9346. [Google Scholar]