

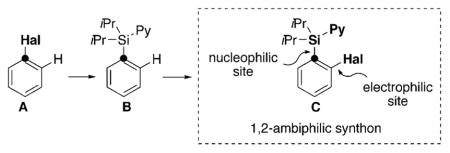
Ambiphilic aromatic synthons—compounds possessing both electrophilic and nucleophilic centers in the same molecule—are important building blocks that are widely used for a modular construction of complex molecules in organic synthesis, medicinal chemistry, and materials science.[1] Traditionally, they are accessed through multistep syntheses. One of the most efficient strategies toward 1,2-ambiphilic structures involves directed ortho-metalation (DOM) approach.[2] Our research group has recently developed the palladium-catalyzed directed ortho-acyloxylation of pyridyldiisopropylsilyl (PyDipSi) arenes B[3] [Eq. (1)] based on a C–H activation process.[4] Most importantly, we have shown that the PyDipSi directing group[5] could efficiently participate in a variety of reactions as a nucleophilic entity. Because the acyloxy group is known to serve as an electrophilic coupling partner,[6] the o-acyloxylated PyDipSi-arenes can be formally considered as 1,2-ambiphiles. Taking into account the immense synthetic potential of aryl halides as electrophilic reagents, we aimed at the development of a general strategy for the synthesis of ortho-halogenated aryl silanes C, which are much more powerful 1,2-ambiphiles. Herein, we report the palladium-catalyzed ortho-halogenation reaction of easily accessible PyDipSi-arenes B into 1,2-ambiphiles C and their further transformations to a variety of valuable building blocks.
 |
(1) |
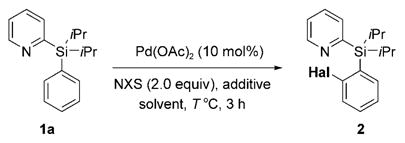
First, we tested PyDipSi-arene 1a under a variety of halogenation reaction conditions in the presence of 10 mol% of Pd(OAc)2 (Table 1).[4a,7] Initially, the palladium-catalyzed bromination with 2 equivalents of NBS (N-bromosuccinimide) in PrCN at 80°C afforded 50% of the desired product 2 (Table 1, entry 1; Hal =Br). Further increase of temperature to 100°C led to a slight improvement of the reaction outcome (Table 1, entry 2). Addition of 50 equivalents of acetic acid[7a,b] resulted in significant decrease of the reaction yield (Table 1, entry 3). The employment of a stoichiometric amount of Cu(OAc)2 additive gave only traces of brominated product (Table 1, entry 4). Remarkably, addition of 1.5 equivalents of PhI(OAc)2 dramatically improved the reaction, and provided the bromination product in 80% yield (Table 1, entry 5). Performing the reaction at the elevated temperature (100°C), however, gave a lower yield of 2 (Table 1, entry 6). Gratifyingly, switching solvent to 1,2-dichloroethane allowed for a better reaction yield (85%) at lower temperature (60°C; Table 1, entry 7). Employment of NIS (N-iodosuccinimide) as a halogen source under these reaction conditions produced iodinated aryl silane 2 in 95% yield (Table 1, entry 8; Hal =I). On the other hand, employment of NCS (N-chlorosuccinimide) gave the chlorinated product in a moderate yield only (Table 1, entry 9; Hal =Cl).
Table 1.
Optimization of ortho-halogenation reaction. X =halide.
 | |||||
|---|---|---|---|---|---|
| Entry | Additive (equiv) | Hal | Solvent | T [8C] | Yield [%][a] |
| 1 | none | Br | PrCN | 80 | 50 |
| 2 | none | Br | PrCN | 100 | 65 |
| 3 | AcOH (50) | Br | PrCN | 80 | 15 |
| 4 | Cu(OAc)2 (1) | Br | PrCN | 100 | trace |
| 5 | PhI(OAc)2 (1.5) | Br | PrCN | 80 | 80 |
| 6 | PhI(OAc)2 (1.5) | Br | PrCN | 100 | 65 |
| 7 | PhI(OAc)2 (1.5) | Br | C2H4Cl2 | 60 | 85 |
| 8 | PhI(OAc)2 (1.5) | I | C2H4Cl2 | 65 | 95 |
| 9 | PhI(OAc)2 (1.5) | Cl | C2H4Cl2 | 65 | 42 |
Yield determined by NMR spectroscopy.
Next, the generality of the palladium-catalyzed ortho-halogenation of PyDipSi-arenes 1 was examined. The iodination reaction with NIS in the presence of 1.5 equivalents of PhI(OAc)2 was studied first. We found this transformation to be efficient for a wide range of substrates, which allowed for the synthesis of monoiodinated aryl silanes 2a–w in good to excellent yields (Scheme 1). It was found that a variety of groups, including OMe (2b, 2k), F (2d, 2n), Cl (2e), Br (2 f, 2l), ester (2g), and amide (2h) were perfectly tolerated under the halogenation reaction conditions. Iodination of para-substituted aryl silanes possessing both electron-donating (2b) and electron-withdrawing (2d–h) substituents proceeded with equal efficiency. meta-Substituted substrates displayed excellent site selectivity in the iodination reaction, and provided monoiodinated compounds as single regioisomers (2i–l). In addition, ortho-iodination of m-, p-disubstituted aryl silanes (2m,n), and 2-naphthyl derivative (2o) occurred uneventfully and furnished the desired products as sole regioisomers in high yields. Next, the bromination reaction of 1 allowed for efficient synthesis of o-bromo aryl silanes 2p–r. Notably, chlorination of electron-rich aryl silane, possessing an OMe group para to the functionalization site, was found to be more efficient than that of electron-neutral 1a (Table 1, entry 9), thus producing chloro-derivative 2s in 69% yield. Finally, PyDipSi derivatives of various heterocycles, such as benzofuran (2t), carbazole (2u), indole (2v), and benzoxazole (2w), were monoiodinated in good yields. We find these results remarkable, as 6-halo derivatives of most of these heterocycles are not readily available and require multistep preparation. These derivatives now can be accessed from the 5-haloprecursors of the corresponding PyDipSi-heterocycles, which are either commercially available or can be easily synthesized in one step.
Scheme 1.

Palladium-catalyzed ortho-halogenation of aryl silanes. [a] Yield of isolated product. [b] See Supporting Information for experimental details. [c] Reaction was performed in PrCN at 100°C. [d] Reaction was performed without PhI(OAc)2 and with 1 equivalent of NIS. Boc = tert-butoxycarbonyl.
Naturally, after the development of efficient palladium-catalyzed halogenation of aryl silanes, we investigated possible transformations of the PyDipSi directing group (Scheme 2).[8] First, the reaction of 2c with AgF/H2O (2:3) in THF resulted in efficient removal of the directing group, thus affording m-iodobiphenyl (6) in 97% yield.[9] Interestingly, the overall three-step transformation of p-bromobi-phenyl into m-iodobiphenyl constitutes an example of a formal Finkelstein/“1,2-halogen dance” reaction. Next, the iododesilylation reaction of chlorobromoaryl silane 2e with NIS in the presence of AgF in THF allowed for efficient preparation of 1-cloro-3-bromo-4-iodobenzene (3), which is a synthetically useful and versatile building block for modular functionalization of the benzene ring. Furthermore, iodoaryl silane 2i was efficiently converted into o-iodoaryl boronate 4,[10] which is another powerful 1,2-ambiphile, in 87% yield by a one-pot sequence involving borodesilylation with BCl3, and subsequent protection with pinacol.[11,12] Furthermore, borodesilylation of 2i and subsequent oxidation with H2O2/NaOH afforded o-iodophenol 5 in 80% yield.
Scheme 2.
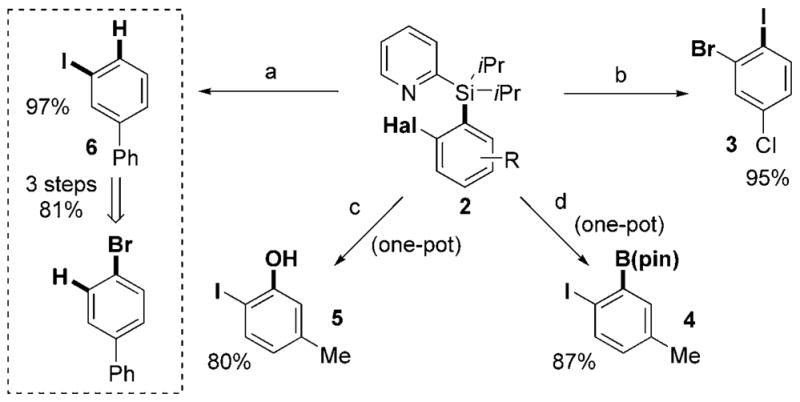
Transformations of the PyDipSi group in haloarene derivatives. Reagents and conditions: a) AgF (4 equiv), H2O (6 equiv), THF, RT, 12 h; b) AgF (4 equiv), NIS (4 equiv), THF, RT, 12 h; c) 1. BCl3 (4.4 equiv), DCM, 0 °C, RT, 6 h; 2. 30 wt% H2O2/3 wt % NaOH (excess), H2O, RT, 12 h; d) 1. BCl3 (4.4 equiv), DCM, 0 °C, RT, 6 h; 2. pinacol (excess), Et3N/DCM (1:1), RT, 12 h. THF = tetrahydrofuran.
Further utility of o-halogenated PyDipSi-arene derivatives was demonstrated by a convergent synthesis of unsymmetrically substituted benzo[b]silole 10 and dibenzosilole 15 (Scheme 3). First, treatment of 2i with HF at room temperature led to selective substitution of the pyridine group with fluoride,[13] thus providing fluorosilane 7 in excellent yield. Next, o-iodoaryl fluorosilane 7 was alkynylated with potassium phenylethynyltrifluoroborate under Suzuki reaction conditions[14] and produced 8 in 66% yield. Alternatively, alkynylated aryl silane 8 can be accessed from 2i through a sequence involving Sonogashira reaction[15] with phenylacetylene and subsequent substitution of the pyridine group with fluoride. A subsequent reduction of silylfluoride 8 with LiAlH4 furnished silylhydride 9. 5-Endo-dig cyclization of the latter in the presence of KH in DME[16] provided 10 in 72% yield. En route to dibenzosilole derivative 15, o-iodoaryl silane 2i was subjected to Suzuki coupling[17] with 4-methoxyphenylboronic acid and gave biphenylsilane 12 in 89% yield. Next, substitution of the pyridine group in 12 with fluoride produced silylfluoride 13 quantitatively. Smooth reduction of 13 into hydride 14 and its subsequent electrophilic cyclization reaction with trityl tetrakis(pentafluorophenyl)borate[18] resulted in formation of dibenzosilole 15 in 71% yield (Scheme 3).
Scheme 3.
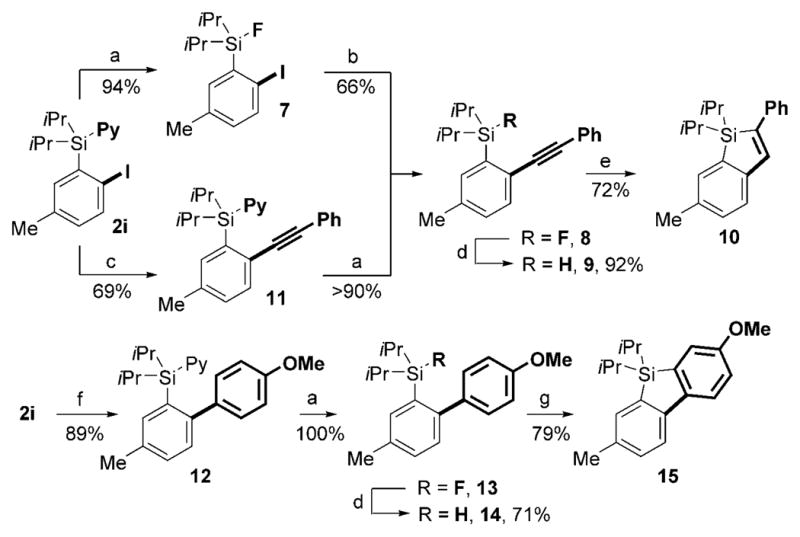
Synthesis of benzannulated siloles 10 and 15. Reagents and conditions: a) HF, THF, RT, 1 h; b) PhCCH, [{Pd(CH3CN)2}Cl2] (3 mol%), tBu3P (6 mol%), CuI (2 mol%), iPr2NH, 1,2-dioxane, 60 °C, 12 h; c) PhCCBF3K, [{Pd(dppf)}Cl2]·DCM (10 mol%), Cs2CO3, THF, reflux, 48 h; d) LiAlH4 (2.5 equiv), THF, reflux, 12 h; e) KH (1.4 equiv), DME, 5 h; f) 4-MeO-C6H4B(OH)2 (1.2 equiv), [Pd2(dba)3] (5 mol%), tBu3P (10 mol%), K3PO4, 1,2-dioxane, 70°C, 12 h; g) Ph3CB(C6F5)4, 1,6-lutidine, CH2Cl2, RT, 1 h. dba = trans,trans-dibenzylideneacetone, DME = 1,2-dimethoxyethane, dppf =1,1′-bis(diphenylphosphanyl)ferrocene, Py =pyridine.
Definitely, o-benzyne is one of the most synthetically attractive 1,2-ambiphiles.[19] Because o-silylphenyliodonium triflates are known to efficiently generate benzynes in the presence of TBAF,[20,21] we decided to convert the iodide functionality in PyDipSi-arenes 2 into a better leaving iodonium group. Accordingly, substrate 2e, after exchange of the pyridine group to fluoride, was smoothly converted into the corresponding iodonium tetrafluoroborate 16 (Scheme 4).[22] Treatment of the latter with TBAF in CH2Cl2 allowed for the efficient generation of benzyne 17, trapping of which with furan provided 1,4-epoxydihydronaphthalene 18 in 89% yield. To the best of our knowledge, the above sequence, taken together with the o-iodination of PyDipSi-arenes, represents the first example of benzyne synthesis featuring C–H activation strategy.
Scheme 4.

Conversion of PyDipSi-iodoarenes into benzyne. Reagents and conditions: a) 1. 48 wt % HF (excess), THF, RT, 1 h; 2. m-CPBA (1.2 equiv), DCM, then BF3·Et2O (2.5 equiv), RT, 1 h; 3. PhB(OH)2 (1.1 equiv), 0°C, RT, 30 min ; b) TBAF (1.2 equiv), furan (5 equiv), DCM, RT, 1 h. m-CPBA =meta-chloroperbenzoic acid, TBAF =tetra-n-butylammonium fluoride.
In conclusion, we have developed a general and efficient strategy for the synthesis of 1,2-ambiphilic aromatic and heteroannulated aromatic synthons. This method features installation of the removable/modifiable PyDipSi directing group on haloarenes and subsequent palladium-catalyzed directed ortho-halogenation reaction to give the o-halogenated PyDipSi-arene derivatives. Synthetic usefulness of these 1,2-ambiphilic building blocks was demonstrated in a variety of transformations, involving participation of both nucleophilic aryl silane and electrophilic aryl iodide moieties. These transformations include protio-, halo-, borodesilylations, and conversion of the PyDipSi group into the OH functionality, as well as Suzuki and Sonogashira cross-coupling reactions of the aryl iodide unit. Finally, the unique reactivity of these 1,2-ambiphiles was illustrated in convergent syntheses of benzannulated silole derivatives, as well as in the efficient generation of o-benzyne.
Supplementary Material
Footnotes
The support of the NIH (GM-64444) and of the NSF (CHE-0710749) is gratefully acknowledged.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201004426.
References
- 1.For selected examples on importance of ambiphilic synthons, see: Yudin AK, Hili R. Chem Eur J. 2007;13:6538. doi: 10.1002/chem.200700710.Tejedor D, Méndez-Abt G, González-Platas J, Ramírez MA, García-Tellado F. Chem Commun. 2009:2368. doi: 10.1039/b819613c.Samarakoon TB, Hur MY, Kurtz RD, Hanson PR. Org Lett. 2010;12:2182. doi: 10.1021/ol100495w.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716. doi: 10.1021/ja0716204.and references therein.
- 2.For a review on directed ortho-metalation, see: Snieckus V. Chem Rev. 1990;90:879.for recent examples of employment of DOM group in cross-coupling reactions, see: Quasdorf KW, Riener M, Petrova KV, Garg NK. J Am Chem Soc. 2009;131:17748. doi: 10.1021/ja906477r.Antoft-Finch A, Blackburn T, Snieckus V. J Am Chem Soc. 2009;131:17750. doi: 10.1021/ja907700e.
- 3.Chernyak N, Dudnik AS, Huang C, Gevorgyan V. J Am Chem Soc. 2010;132:8270. doi: 10.1021/ja1033167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For selected recent reviews on transition-metal-catalyzed C–H activation of arenes, see: Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem. 2009;121:5196. doi: 10.1002/anie.200806273.Angew Chem Int Ed. 2009;48:5094.Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n.Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173. doi: 10.1039/b606984n.Ackermann L, Vicente R, Kapdi AR. Angew Chem. 2009;121:9976. doi: 10.1002/anie.200902996.Angew Chem Int Ed. 2009;48:9792.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058.
- 5.For employment of pyridyldimethylsilyl directing group in Heck arylations, see: Itami K, Mitsudo K, Kamei T, Koike T, Nokami T, Yoshida JI. J Am Chem Soc. 2000;122:12013.Itami K, Nokami T, Yoshida JI. J Am Chem Soc. 2001;123:5600. doi: 10.1021/ja015655u.for employment of dimethylhydrosilyl directing group in iridium-catalyzed C–H borylations, see: Robbins DW, Boebel TA, Hartwig JF. J Am Chem Soc. 2010;132:4068. doi: 10.1021/ja1006405.Boebel TA, Hartwig JF. J Am Chem Soc. 2008;130:7534. doi: 10.1021/ja8015878.
- 6.For review on cross-coupling reactions of pivalates, see: Gooßen LJ, Gooßen K, Stanciu C. Angew Chem. 2009;121:3621.Angew Chem Int Ed. 2009;48:3569.
- 7.For representative examples of palladium-catalyzed directed C–H halogenation of arenes, see: Kalyani D, Dick AR, Anani WQ, Sanford MS. Tetrahedron. 2006;62:11483. doi: 10.1021/ol060747f.Kalyani D, Dick AR, Anani WQ, Sanford MS. Org Lett. 2006;8:2523. doi: 10.1021/ol060747f.Mei TS, Giri R, Maugel N, Yu JQ. Angew Chem. 2008;120:5293. doi: 10.1002/anie.200705613.Angew Chem Int Ed. 2008;47:5215.Powers DC, Ritter T. Nat Chem. 2009;1:302. doi: 10.1038/nchem.246.Mei TS, Wang DH, Yu JQ. Org Lett. 2010;12:3140. doi: 10.1021/ol1010483.Kakiuchi F, Kochi T, Mutsutani H, Kobayashi N, Urano S, Sato M, Nishiyama S, Tanabe T. J Am Chem Soc. 2009;131:11310. doi: 10.1021/ja9049228.Andrienko OS, Goncharov VS, Raida VS. Russ J Org Chem. 1996;32:79.
- 8.For an excellent example of ruthenium-catalyzed o-silylation of arenes using a removable boron-tethered directing group, see: Ihara H, Suginome M. J Am Chem Soc. 2009;131:7502. doi: 10.1021/ja902314v.
- 9.a) Yanagisawa A, Kageyama H, Nakatsuka Y, Asakawa K, Matsumoto Y, Yamamoto H. Angew Chem. 1999;111:3916. doi: 10.1002/(sici)1521-3773(19991216)38:24<3701::aid-anie3701>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1999;38:3701. [Google Scholar]; b) Itami K, Mineno M, Kamei T, Yoshida JI. Org Lett. 2002;4:3635. doi: 10.1021/ol026573t. [DOI] [PubMed] [Google Scholar]
- 10.Recently, it was shown that boronic acids could be ortho-iodinated under Friedel–Crafts reaction conditions. However, the reported method is limited to electron-rich substrates only, see: Al-Zoubi RM, Hall DG. Org Lett. 2010;12:2480. doi: 10.1021/ol100537x.
- 11.a) Itami K, Kamei T, Yoshida JI. J Am Chem Soc. 2003;125:14670. doi: 10.1021/ja037566i. [DOI] [PubMed] [Google Scholar]; b) Kamei T, Itami K, Yoshida JI. Adv Synth Catal. 2004;346:1824. [Google Scholar]; c) Zhao Z, Snieckus V. Org Lett. 2005;7:2523. doi: 10.1021/ol0506563. [DOI] [PubMed] [Google Scholar]; d) Rottländer M, Palmer N, Knochel P. Synlett. 1996:573. [Google Scholar]
- 12.For a review on C–B bond formation through a C–H activation, see: Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p.
- 13.Brown RS, Slebocka-Tilk H, Buschek JM, Ulan JG. J Am Chem Soc. 1984;106:5979. [Google Scholar]
- 14.Molander GA, Katona BW, Machrouhi F. J Org Chem. 2002;67:8416. doi: 10.1021/jo0262356. [DOI] [PubMed] [Google Scholar]
- 15.Hundertmark T, Littke AF, Buchwald SL, Fu GC. Org Lett. 2000;2:1729. doi: 10.1021/ol0058947. [DOI] [PubMed] [Google Scholar]
- 16.Ilies L, Tsuji H, Nakamura E. Org Lett. 2009;11:3966. doi: 10.1021/ol9015282. [DOI] [PubMed] [Google Scholar]
- 17.Littke AF, Dai C, Fu GC. J Am Chem Soc. 2000;122:4020. [Google Scholar]
- 18.Furukawa S, Kobayashi J, Kawashima T. J Am Chem Soc. 2009;131:14192. doi: 10.1021/ja906566r. [DOI] [PubMed] [Google Scholar]
- 19.For reviews, see: Wenk HH, Winkler M, Sander W. Angew Chem. 2003;115:518. doi: 10.1002/anie.200390151.Angew Chem Int Ed. 2003;42:502.Pellissier H, Santelli M. Tetrahedron. 2003;59:701.
- 20.Our initial attempts on direct conversion of the o-iodo derivatives 2 into benzynes were unsuccessful.
- 21.For selected examples on employment of o-(trimethylsilyl)phenyliodonium triflates as precursors for the preparation of benzyne, see: Kitamura S, Yamane M. J Chem Soc Chem Commun. 1995:983.Kitamura T, Yamane M, Inoue K, Todaka M, Fukatsu N, Meng Z, Fujiwara Y. J Am Chem Soc. 1999;121:11674.
- 22.Bielawski M, Aili D, Olofsson B. J Org Chem. 2008;73:4602. doi: 10.1021/jo8004974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.