

Abstract
As the mainstay in the treatment of various cancers for several decades, chemotherapy is successful but still faces challenges including non-selectivity and high toxicity. Improving the selectivity is therefore a critical step to improve the therapeutic efficacy of chemotherapy. Prodrug is one of the most promising approaches to increase the selectivity and efficacy of a chemotherapy drug. The classical prodrug approach is to improve the pharmaceutical properties (solubility, stability, permeability, irritation, distribution, etc.) via a simple chemical modification. This review will focus on various targeted prodrug designs that have been developed to increase the selectivity of chemotherapy drugs. Various tumor-targeting ligands, transporter-associated ligands, and polymers can be incorporated in a prodrug to enhance the tumor uptake. Prodrugs can also be activated by enzymes that are specifically expressed at a higher level in tumors, leading to a selective anti-tumor effect. This can be achieved by conjugating the enzyme to a tumor-specific antibody, or delivering a vector expressing the enzyme into tumor cells.
Keywords: prodrug, cancer therapy, targeted delivery, polymer, transporter, ADEPT, GDEPT
I. Introduction
Chemotherapy is successful but still faces challenges due to lack of selectivity and associated toxicity. Chemotherapy drugs act through anti-proliferative mechanism or by arresting cell cycle at a specific phase, rather than producing a toxic effect to particular types of cancer cells. [1] Therefore, these drugs, due to the poor selectivity, affect all rapidly proliferating and dividing cells such as red blood cells, hair follicles, gut epithelia, bone marrow, and lymphatic system, making chemotherapy drugs unsuitable for a long-term treatment. Furthermore, chemotherapy drugs are not efficient in treating slowly growing solid tumors, whereas most human solid tumor cells do not proliferate rapidly.[2] As a result, high-dose chemotherapy is generally required to effectively inhibit the tumor proliferation, especially the resistant solid tumors. However, non-selectivity of the chemotherapy drugs could result in lethal damage to the adjacent normal proliferating cells, leading to discontinuation of the therapy before all malignant cells are killed. Hence, improving the selectivity is the critical step to improve the therapeutic efficacy of a chemotherapy drug.
Among different strategies designed to improve the selectivity of chemotherapy drugs, targeted prodrugs represent a promising approach for highly selective chemotherapy. Prodrugs are defined as chemically modified, biologically inert, small-molecule drugs that are transformed in vivo to release the pharmacologically active drug.[3] Prodrug approach provides a remarkable tool to improve the pharmaceutical properties of the active pharmacologic agents via a simple chemical modification. Traditional prodrug design aims to: i) improve solubility in water or lipid membrane, chemical stability, oral or local absorption, and brain permeability; ii) reduce unacceptable taste, irritation or pain, pre-systemic metabolism, and toxicity.[4, 5] Current review will focus on unmet needs of traditional pro-drug design to improve the selectivity and targeting features.
Prodrugs can be designed to target specific antigens, peptide transporters, or enzymes that are over-expressed on tumor cells in comparison to other normal cells. This can be achieved by conjugating a tumor-specific ligand or a polymer to the chemotherapy drug via a cleavable linker [6]. The general design of a prodrug is depicted in Figure 1. A chemotherapy prodrug may contain as many as four components: i) the parent drug or its derivative that exhibits the pharmacologic effect; ii) a metabolically labile chemical linker which links the functional group (hydroxyl, carboxylic, amine, carbonyl, and phosphate groups, etc.) of the parent drug to the rest part of the prodrug designated as the “promoiety”; iii) a polymer spacer, or an enzymatically cleavable spacer that can release the parent drug in the presence of a tumor-specific enzyme; iv) a targeting moiety for specific delivery to tumor cells.
Figure 1.

General design of a prodrug.
II. Chemical linker in prodrug
To construct a prodrug, there must be a functional group on the parent drug that can be used to form a chemical bond with the promoiety. Generally, the linker should be self-immolatable or cleavable so that parent drug can be released spontaneously or under a certain triggerable condition such as the presence of an enzyme or a change in pH. The promoiety affiliated to the parent drug provides the ability to improve the drug-like properties or overcome the barriers in delivering the drug to its target cells.[3] Commonly used linkers in prodrug design are listed in Table 1.
Table 1.
Common linkers used in prodrug conjugation
| Linker | Chemical structure | |
|---|---|---|
| Ester | Carboxyl ester | 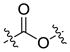 |
| Carbamate ester | 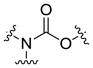 |
|
| Carbonate ester | 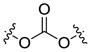 |
|
| Phosphate ester | 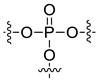 |
|
| Amide | Peptide bond | 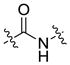 |
| Other linkers | Oxime and imine | 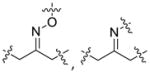 |
| Disulfide bond | ||
| Thioether bond | ||
Ester is the most common linkage in prodrug design. It is easy to synthesize and its function groups such as hydroxyl and carboxyl acid group that are widely available in most parent drugs as well as promoiety molecules. Moreover, esterases are ubiquitously distributed in the body. Once administrated, the ester bond can be readily hydrolyzed by esterases in the blood, liver and other organs, leading to the release of the parent drug [7]. Depending on different structures of the prodrug or environment conditions, half-life of the ester bond varies from several minutes to several hours [8, 9]. For example, both EBZ-2208 and IT-101 are composed of the similar parent drug and the same ester linkage. EBZ-2208 has a half-life of only 12.3 minutes in human plasma[10], while the half-life of IT-101 is around 1.7 hours in human plasma[11, 12]. EBZ-2208 is a prodrug of the camptothecin derivative SN38 with a PEG of 40 kDa through a glycine spacer.[10] It is presumed that the linear structure of PEG and its hydrophilic property make the ester bond of the EBZ-2208 easily accessible to the activity site of esterase. On the contrary, IT-101 has a micelle-like structure that may protect the ester bond from esterases. Different types of the ester bond also exhibit different stabilities in the body. For instance, carbamate ester is more stable in comparison to carboxyl ester, phosphate ester, and carbonate ester [3].
Amide bond is another commonly used linkage in prodrug. It is the derivative of an amine and a carboxyl groups. Amide has a relatively higher enzymatic stability than ester bond. Most of the amide bonds are stable for several hours or even several days in the plasma in the absence of specific enzymes. However, majority of the amide bonds in prodrugs are designed to be cleavable by a specific enzyme to increase the target-ability or reduce the toxicity. Peptide linkers such as GFLG and SSKYQ are probably the best examples of this type of amide bond. The tetrapeptide linker GFLG is specifically cleaved by lysosomal enzyme in tumors. SSKYQ is a substrate peptide of the prostate specific antigen (PSA), an enzyme that is only active in prostate tumors. This type of linker shows a reliable stability in the blood circulation and only release the drug in target cells. GFLG has been successfully adopted in PK1, PK2, PNU166945, DX-8951 and other prodrugs that are under clinical trials [13]. Several SSKYQ involved prodrugs such as L-377202 also enter Phase I/II clinical trials [14].
In addition to ester and amide bonds, several other types of linkers including oximes/imines[15], disulfide bond [16] or uncleavable thioether bond [17], have also been used in prodrug. Generally, the linker between parent drug and promoiety should be stable enough to remain intact in the blood circulation until it reaches the target cells. Optimization of the linkage is highly suggested during the prodrug development to achieve the maximal stability and efficacy. For instance, the hydrazone linker is an acid labile linker that is relatively stable at neutral pH, but unstable at pH 5. Following internalization of the prodrug, the hydrazone linker is cleaved in the acidic endosomes and lysosomes to release the active drug.[18] Similarly, peptide linker is stable in the serum, but can be readily cleaved in intracellular compartments by specific tumor-associated protease.[19] Disulphide linkers are more tumor-specific as they are cleaved by intracellular thiol such as glutathione that is over-expressed in tumor cells compared to normal cells [20]. However, it should be noteed that although the cleavable linkages are preferable for most prodrugs to release parent drugs in the body shortly after the administration, uncleavable linkers may provide a better therapeutic efficacy if the targeting moiety itself has a pharmacological activity.[17]
III. Targeting-ligand conjugated prodrug
Tumor cells tend to overexpress specific antigens, receptors, or transporters that can be targeted using various moieties including antibodies, antibody fragments, peptides, aptamers, and small-molecule ligands. Selectivity of a chemotherapy drug for its intended target can be achieved by conjugating a tumor-specific ligand to the cytotoxic drug.
(a) Antibody conjugated prodrug
Among the numerous targeting moieties, monoclonal antibody is the first type of ligand used for targeted prodrug due to its high affinity. The antibody-drug conjugate (ADC) is generally inactive without further transformation in the body and internalized through a receptor mediated endocytosis, followed by release of the active drug in the cells [21]. It is important to note that the ratio of drug to antibody should be optimized to achieve maximal and specific efficacy [22]. Tumor specific antibodies provide the means to target therapeutic agents to tumor cells without harming adjacent normal cells. However, several problems including immunogenicity, selectivity, and permeability into solid tumors need to be addressed before the successful clinical application. The immunogenicity associated with murine mAb has been overcome by using humanized mAb, whereas the selectivity can be improved by identifying new tumor antigens and related antibodies. Lack of efficient cell permeability is the major hurdle for the antibody approach because therapeutic agents must cross various biological barriers and the high interstitial pressure to reach their target cells [23]. This can be sikved by using antibody fragments such as scFv and Fab that can penetrate into tumor cells with higher efficiency compared to the intact antibody [24]. A wide variety of tumor-specific antigens have been identified. See the review article by Teicher for a detailed discussion on the cell surface antigens that have been targeted primarily by ADC. These tumor-specific antigens include CD19, CD22, TAG-72, MUC16, PSMA, EGFR, αv integrin and various others that are over-expressed on a wide range of tumor cells. Some of these antigens are more specific to a specific type of cancer than the others.[25] For example, MUC16 is more profoundly expressed in ovarian (or other reproductive organ) cancer cells rather than the tumors of other origins,[26] while PSMA is preferably expressed in prostate cancer tissues [27]. On the contrary, certain antigens are associated with a specific type of cells regardless of the tumor origin. For instance, TAG-72 is highly expressed in human adenocarcinomas including colorectal, pancreatic, gastric, ovarian, endometrial, breast, and non-small cell lung cancers [25]. Numerous chemotherapy agents including antifolates [22], vinca alkaloids [28], anthracyclines [18], taxane [29], monomethyl auristatin E [18], calichemicin [30, 31], and mytansine derivative DM1 [32] have been conjugated to tumor-targeting antibodies. Some of the antibody-drug conjugates were listed in Table 2.
Table 2.
Antibody-conjugated prodrug
| Prodrug | Antigen | Antibody | Tumor target | Phase status |
|---|---|---|---|---|
| Mylotarg (Gemtuzumab-ozogamicin) | CD33 | Gemtuzumab (anti-CD33) | Acute myeloid leukemia (AML) | II/III/IV* |
| Trastuzumab-DM1 (T- DM1) | HER-2 | Anti-HER2 mAb | HER-2 positive Breast cancer | II/III* |
| IMGN901 (huN901-DM1) | CD33 | Gemtuzumab (anti-CD33 mAb) | Acute myeloid leukemia (AML) | II & III |
| MLN2704 | PSMA | Humanized anti- PSMA mAb | PSMA positive Breast cancer | I & II |
| huC242-DM1 | CD56 | Anti-CD56 Ab | CD56 positive small cell carcinoma | II |
| huC242-DM4 | PSMA | Anti-PSMA mAb | PSMA positive prostate cancer | I & II* |
| cAC10-vcMMAE | CanAg | Anti-CanAg mAb (huC242) | CanAg positive solid tumors | I |
| cAC10-vcMMAE | CanAg | Anti-CanAg mAb Chimeric mAb | Gastric Cancer | I & II |
| cAC10-vcMMAE | CD30 | cAC10 | Hodgkin disease | Xenograft model |
Information was gathered from 1(http://www.immunogen.com); 2(http://clinicaltrials.gov); 3(http://www.immunogen.com).
While there are around 20 antibody-drug conjugates in clinical trials [25], Gemtuzumab (Mylotarg, Wyeth) is the only FDA approved antibody-drug conjugate for the treatment of acute myeloid leukemia (AML). As depicted in Fig. 2A, Mylotarg consists of calicheamicin, a cytotoxic drug, linked to the humanized anti-CD33 monoclonal antibody (clone P67.6) via a hydrolysable bifunctional linker.[33] The CD33 antigen is expressed in more than 90% of the AML patients, whereas normal human tissues and pluripotent hematopoietic stem cells do not express the CD33 antigen. Therefore, Mylotarg specifically binds to the CD33 antigen on tumor cells with subsequent internalization of the prodrug.[34] Pharmacokinetic study of different doses indicated that the CD33 antigen binding sites were saturated when 7.5 mg/m2 of Mylotarg was administrated. The pharmacokinetic parameters of adult patients receiving 9 mg/m2 Mylotarg are Cmax: 5.86 ± 1.35 (mg/ml), t1/2: 72.4 ± 42.0 (h), AUC: 123 ± 105(mg·h/ml), CL: 0.15 ± 0.13 (L/h/m2), and Vss: 0.30 ± 0.31 (L/kg). Similar pharmacokinetic profile was observed in both children and adult patients [35]. Twenty-six percent of the 277 patients in a phase II clinical trial demonstrated an overall response after using Mylotarg.[33] This prodrug was approved in 2000 under the FDA’s accelerated approval program, and a postmarketing study was begun in 2004. Unfortunately, Mylotarg was withdrawn from the market in 2010 due to the ineffectiveness and severe side effects found in the post-approval clinical trial.[36]
Figure 2.

Structure of the prodrugs Mylotart, Trastuzumab-DM1, and CMC-544.
Nevertheless, there are several other antibody-conjugated prodrugs that are undergoing clinical trials at different phases. Among them, Trastuzumab-amide-MCC-DM1 (Trastuzumab-DM1) and Inotuzumab ozogamicin (CMC-544) are approaching the final stage of clinical trial.[37, 38]
Human epidermal growth factor receptor (HER2) is highly expressed in 20~25% of human breast cancers. Overexpression of HER2 is associated with poor prognosis and aggressive tumor phenotypes, making it an attractive therapeutic target. The humanized monoclonal HER2 antibody, trastuzumab, was approved by FDA in 1998 for the treatment of metastatic HER2 positive breast cancer.[17, 39] Although trastuzumab signifies the most remarkable advancement in the application of monoclonal antibody for cancer therapy, a significant proportion of the patients do not respond to trastuzumab.[17] Subsequently, adjuvant chemotherapy drugs are often required for the trastuzumab therapy. Direct conjugation of a chemotherapy drug to the trastuzumab is therefore becoming an attractive alternative to the antibody monotherapy. Trastuzumab-DM1 (Fig. 2) is such a novel antibody-drug conjugate that uses trastuzumab as the targeting moiety to specifically deliver DM1 (derivative of maytansine 1), a highly potent antimitotic agent, to HER2 positive breast cancer cells. Lewis Phillips and his colleagues evaluated the trastuzumab-DM1 with different linkers, and found that the nonreducible thioether linkage exhibited the highest activity in comparison to cleavable disulfide linkages.[17] Phase I clinical trial revealed that Trastuzumab-DM1 had a maximum tolerated dose (MTD) of 3.6 mg/kg. At MTD, the half-life(t1/2) was 3.5 days, and the clearance was 12.9 ml/d/kg which is much slower than that at low doses (<1.2 mg/kg).[40] In a recent phase II clinical trial, 32.7% of the 110 patients who received the trastuzumab-DM1 demonstrated a significant improvement with acceptable side effects.[37]
CMC-544 is another promising antibody-conjugated drug composed of a humanized anti-CD22 monoclonal antibody linked to N-acetyl gamma calicheamicin dimethylhydrazide (CalichDMH), a DNA-damaging antibiotic agent, via an acid-hydrolysable linkage. CMC-544 uses the same parent drug and linkage as Mylotarg, but with a different targeting moiety CD22. CD22 is expressed on the surface of both mature and malignant B lymphocytes, but not on other non-B lineages. Upon binding to CD22 on malignant B-cells, CMC-544 is rapidly internalized to deliver CalichDMH into the cells.[38] In the preliminary study, the maximum tolerated dose (MTD) of CMC-544 was reported to be 1.8 mg/m2, and the half-life was about 24 hours at MTD. After repeated administrations (2~3 cycles), the half-life was increased to 4 days and the clearance was decreased 8~14 fold compared to the 1st dose.[41] CMC-544 is currently being evaluated in a phase III study for the treatment of B-lymphoid malignancies.
(b) Peptide-drug conjugate
Compared to antibody, peptide is a more appropriate targeting moiety due to its small molecular weight, excellent cell permeability, flexibility in chemical conjugation, and ease of production.[39] Peptide ligands can be designed against a tumor-specific antigen or a peptide transporter that is over-expressed in cancer cells. Such peptide ligands can be directly conjugated to chemotherapy drugs to achieve a targeted delivery to cancer cells. Table 3 summarizes three commonly used peptide ligands.
Table 3.
Peptide-drug conjugate
Minko and her colleagues have reported a successful application of the LHRH (Lutenizing Hormone Releasing Hormone) peptide conjugated prodrug for cancer therapy.[42, 43] LHRH receptors are over-expressed in several cancer cells including breast, prostate, ovarian and endometrial cancers. Although this receptor is also expressed in healthy prostate, breast and ovarian tissues, the expression is much lower than that in cancer cells.[43] The LHRH peptide was conjugated to camptothecin, a cytotoxic drug, via a PEG as the spacer. The results demonstrated a substantially enhanced efficacy of the peptide-conjugated prodrug along with minimized side effects on healthy organs. In addition, the LHRH peptide conjugated-prodrug has a targeting potential for solid tumor tissue as well as a single tumor cell. Hence, this prodrug can efficiently target the spreading or metastatic tumor cells with a low toxicity to normal tissues.[42]
Similarly, Neamati and co-workers conjugated a cyclic peptide E[c(RGDyK)]2 (RGD) to paclitaxel, an antimicrotubule agent commonly used in the treatment of metastatic breast cancer. The RGD peptide specifically binds to αv integrin receptors that are over-expressed in metastatic cancer cells and activated endothelia cells, but not in other normal tissues and resting endothelial cells. In vivo studies showed a specific tumor uptake at 4 hours post administration. [44]
Yoneda et al. have explored the use of glucose-regulated protein 78 (GRP78) as a tumor-specific antigen for targeted delivery of paclitaxel and doxorubucin.[45] GRP78 is over-expressed on many tumor cells, including colon, skin, prostate, and breast cancers. By contrast, its expression on normal tissues is very low [46]. Pep42, a cyclic 13-mer oligopeptide (CTVALPGGYVRVC) specifically binds to GRP78, followed by internalization through the GRP78 receptor-mediated endocytosis and then trafficked to the lysosome that contains protease cathepsin B. Therefore, a cleavable linker, the Val-Cit motif, was used to link the Pep42 to anti-cancer drugs. The Val-Cit linker is fairly stable in the plasma, but can be cleaved by cathepsin B.[47] Cytotoxicity of the Pep42-prodrug was estimated in osteosarcoma cells, SJSA-1, and both Pep42-paclitaxel and Pep42-doxorubicin showed an enhanced toxicity in comparison to the free drug.[45]
(c) Aptamer-drug conjugate
Aptamer is a type of single-stranded RNA or DNA that binds to a specific target molecule with high specificity and affinity (Kd in the nM to pM range) owing to its three dimensional structure.[48] Aptamer represents another important targeting moiety because of its small molecular weight, more synthetic accessibility, ease of production, lack of immunogenicity, good stability, and flexibility in chemical conjugation.[49] Therefore, aptamers are commonly used as non-protein based alternatives to antibodies for targeted delivery of various molecules to cancer cells.[50] In addition to its high specificity and affinity, aptamer can be chemically synthesized based on its sequence, making it an ideal candidate for the therapeutic application where high quality control is needed.[51]
Aptamers are generated by a process called Systemic Evolution of Ligands by Exponential Enrichment (SELEX) through screening of a complex nucleic acid library. Prostate specific membrane antigen (PSMA) that is profoundly expressed in prostate cancer cells [52], and neovasculature of non-prostate solid tumors including breast and lung cancers [53, 54] have been targeted by conjugating the PSMA-specific aptamer (A10 RNA aptamer, Apt) to nanoparticles containing cisplatin.[55, 56] The aptamer conjugated cisplatin polymeric nanoparticles (Pt-NP-Apt) exhibited a higher toxicity in PSMA-positive prostate cancer cells (LNCaP) with an IC50 value of 0.03 μM, compared to the unmodified nanoparticles (Pt-NP) with an IC50 value of 0.13 μM. Moreover, the effectiveness of Pt-NP-Apt NP in LNCaP cells was approximately 10-fold higher than that of free cisplatin [55].
Recently Huang et al. conjugated doxorubicin, the most utilized anticancer drug, to a DNA aptamer sgc8c for the treatment of human T-cell acute lymphoblastic leukemia (T-cell ALL).[57] The sgc8c aptamer recognizes the protein tyrosine kinase 7 (PTK7), a transmembrane receptor highly expressed on T-cell ALL cells, such as CCRF-CEM (Human T cell lymphoblast-like cell line).[58] TDO5 aptamer, which binds specifically to the human Burkitt’s lymphoma cell line and Ramos cells, was used as a negative control to estimate the specificity of the sgs8c-Dox conjugate. It was observed that cells treated with TDO5 showed a much less cellular uptake in comparison to the evenly distributed red fluorescence in the cells treated with sgc8c-dox, indicating a high affinity and specificity of the sgc8c-dox to the CCRF-CEM cells.[57]
(d) Folic acid-drug conjugate
As a member of the vitamin B family, folic acid is one of the most commonly used targeting moiety to specifically deliver various imaging agents, therapeutic agents, and nano-scaled systems to tumor cells. Folic acid binds to its receptor, folate receptor (FR), with a very high affinity (KD 0.1 ~ 1 nM). Folate receptor is over-expressed on the surface of many malignant cells including breast, ovarian, lung, kidney, and endometrial cancers. The expression of folate receptor on other normal tissues is low and only restricted to some epithelial cells. In addition, folic acid has a low immunogenicity and relatively simple chemistry compared to other targeting moieties such as antibody, peptide and aptamer.[20, 59–61]
A variety of chemotherapy drugs have been conjugated with folic acid to target folate receptors on tumor cells. Upon binding to folate receptors, folic acid conjugated prodrugs enter cells via receptor-mediated endocytosis. Philip and his co-workers have conjugated folic acid to campothecin, a poor water-soluble and highly toxic chemotherapy agent, via a hydrophilic peptide containing a disulfide bond. Folic acid increases the specificity of the prodrug, while the cleavable spacer increases the solubility of campothecin and provides an efficient release of campothecin within tumor cells via the disulfide reduction. The folic acid conjugated campothecin exhibited high affinity for a human cervical cancer cell line (KB cells) that overexpresses folate receptors, leading to a significant inhibition of cell proliferation (IC50 10 nM). Moreover, pretreatment of the KB cells with excess folic acids abolished the activity of the prodrug, suggesting that the uptake of the prodrug is folate receptor mediated.[59]
Recently doxorubicin has also been conjugated to folic acid to enhance the specificity to tumor cells. Doxorubicin (Dox) is an anthracyclinic drug for a wide variety of different cancers. However, short half-life, poor solubility and extremely high toxicity limit its therapeutic efficacy. Therefore, in addition to the folic acid (FOL) modification, D-α-tocopheryl polyethylene glycol succinate (TPGS) was also conjugated to doxorubicin to increase the solubility and drug permeability across cell membrane. The TPGS-Dox-FOL prodrug exhibited increased half-life, high anti-tumor efficacy (45-fold more effective than the unmodified Dox), and less accumulation in the heart, which is the major organ affected by doxorubicin’s side effects.[61]
IV. Membrane transporter-associated prodrug
Membrane transporters are integral membrane proteins that mediate the movement of sugar, nucleosides, amino acids and peptides across cell membrane.[4] Traditionally, membrane transporters have been used to improve the bioavailability of polar drugs via a prodrug approach. Recently, the role of membrane transporters in targeted cancer therapy has been exploited, and some of the examples are listed in Table 4.
Table 4.
Membrane Transporters-conjugated prodrug
| Organ | Disease Condition | Membrane Transporter | Prodrug | Reference |
|---|---|---|---|---|
| HeLa-PEPT1 tumor cells | PEPT1 | Bestatin | [66] | |
| Colon Liver | Colon cancer metastatis Hepatic metastasis |
PEPT1 | Floxuridine | [64] |
| Breast, Lung, Ovary, Pancreas, Bladder | Solid tumor | PEPT1 and PEPT2 | Gemcitabin | [67] |
| Ovary | Ovarian cancer cell | SMVT | Biotinylated Camptotheci | [70] |
| ID8 murine cell line | SMVT | Biotinylated HPMA | [72] | |
| HeLa Cell line | SMVT | Biotinylated PAMAM | [71] |
Membrane transporters include glucose transporter, peptide transporter, and amino-acid transporter. Among them, peptide transporter is the most attractive and widely used transporter for the prodrug design. Peptide transporters are proton dependent in nature and responsible for the uptake of di- and tripeptides. Peptide transporters of mammalian origin are divided into two categories: (i) peptide transporters PEPT1 and PEPT2; and (ii) peptide/histidine transporters PHT1 and PHT2.[62] PEPT1 and PEPT2 transport all possible di- and tri-peptides in a sequence-independent but stereoselective manner.[62] PEPT1 transporter is over-expressed in several cancer cells including the malignant ductal pancreatic cancer cell lines AsPc-1[63] and Capan-2,[64] and human fibrosarcoma cell line HT-1080, [65] but not in normal cells.
Nakanishi et al. have reported a specific accumulation of Bestatin, a peptide-mimetic anticancer drug and a substrate of PEPT1, in PEPT1 expressing tumor cells HeLa-PEPT1. Bestatin also induced a significant suppression of tumor growth in another cancer cell line that naturally over-expresses PEPT1. All these results verified that the peptide-mimetic drug is absorbed specifically through the peptide transporter PEPT1.[66] Amidon and his colleagues conjugated a PEPT1 substrate to floxuridine, an anticancer drug for metastatic colon cancer and hepatic metastases, via an ester bond. This prodrug exhibited a higher uptake in PEPT1 over-expressing tumor cells. Accordingly, a selective growth inhibition was observed in tumor cells over-expressing PEPT1, but not in PEPT1-negative tumor cells.[64]
Gemcitabine (2′, 2′-difluoro-2′-deoxycytidine) is a nucleoside analog compound that is used clinically as an efficient anti-neoplastic agent. It is effective in treating solid tumor and is clinically available to treat breast, lung, ovarian, pancreatic and bladder cancers. Amino acid ester conjugates of Gemcitabin were evaluated for binding with transporters using a radiolabeled substrate uptake assay. The result showed that these compounds serve as substrate for either one or both of the peptide transporters PEPT1 and PEPT2.[67]
Another important membrane transporter that has been studied for targeted prodrug is the sodium-dependent multivitamin transporter (SMVT). SMVT is expressed along the small and large intestines and is responsible for the uptake of essential nutrients such as biotin, lipoate, pantothenate [68], and peptides.[69] Minko’s group have shown that the PEG conjugated anticancer drug Camptothecin (CPT) containing biotin increased the cytotoxicity of CPT in human ovarian cancer cells via the SMVT mediated uptake.[70] The human ovarian cancer cell line, A2780 (sensitive) and its multidrug resistant strain A2780/AD, express high levels of SMVT that can be exploited to enhance the cytotoxicity of the anticancer prodrug.[70] Similarly, the biotinylated polyimidoamine (PAMAM) dendrimer-FITC conjugate, and biotinylated hydroxypropylmethacrylate (HPMA) polymer exhibited much higher uptake into HeLa cell and ID8 murine cancer cells, respectively through interaction between biotin and the SMVT.[71, 72]
V. Polymeric prodrug
(a) PEG-drug conjugate
Polyethylene glycol (PEG) is a water-soluble, amphiphilic polymer that has been approved by FDA for oral, parenteral and topical administrations. It is extremely safe and can be eliminated from the kidney (<50 kDa) or liver (>50 kDa) [73]. PEGylation is primarily used to improve the physicochemical properties of macromolecules including antibodies, enzymes and growth factors. Recent advances demonstrated that PEG can also be conjugated to small molecule drugs to increase the targetability. Based on the molecular weight of PEG, pegylation of small molecule drugs can be divided into two platforms, the high-molecular-weight PEG prodrug and the low-molecular-weight PEG prodrug.
The high-molecular-weight PEG conjugated prodrug always involves PEG with a range of molecular weight from 30 kDa to 60 kDa or even higher. In this strategy, PEG is simply used to increase the size of the prodrug, leading to an increased circulation half-life and modified biodistribution profile. The increased exposure of prodrug to tumor cells is primarily caused by the enhanced permeation and retention (EPR) effect, a passive tumor targeting mechanism. The large PEG conjugated prodrug probably is the most successful platform for PEGylated small molecule drugs. Several PEGylated prodrugs are currently undergoing clinical trials. NKTR-102, for instance, is a PEGylated irinotecan for the treatment of colorectal cancer and other cancers. Irinotecan is a potent anticancer agent that is metabolized to its active metabolite SN-38 in the body. A large PEG is conjugated to irinotecan via a cleavable linker that will release irinotecan gradually in vivo. In two mouse xenograft models, concentration of the active metabolite SN-38 in tumors was compared between NKTR-102 and free irinotecan. NKTR-102 increases the half-life of SN-38 90-fold (from 4h to 15 days) in colorectal cancer and 10 fold in lung cancer. The exposures of SN-38 in colorectal and lung tumor tissues were increased by 390 fold and 58 fold, respectively, after pegylation [74]. Preclinical study also demonstrated that NKTR-102 has a superior anti-tumor effect in a colorectal cancer model [75]. In the phase I clinical trial, the pharmacokinetics and anti-tumor activity of NKTR-102 were evaluated in patients who have failed the prior standard treatment [76]. NKTR-102 showed a prolonged half-life in the plasma for 50 days, compared to 47 hours of the free irinotecan formulation. Thirty-eight percent of the patients showed a significant or partial response. Moreover, the response was observed in various tumor origins including breast, ovarian, and cervical cancers. In addition to NKTR-102, high-molecular-weight PEG have also been conjugated to other drugs such as docetaxel (NKTR-105), SN-38 (EZN-2208), and camptothecin (Pegamotecan) [10]. Clinical data of these prodrugs confirm the value of this platform in extending the half-life and improving the targetability of the parent drug.
The architecture of PEG is an important factor in determining the drug loading capacity. The architecture of PEG can be liner, branched or even dendritic. Branched structure not only increases the steric hindrance and extends the half-life of the prodrug, but also provides more conjugation sites for parent drug. Take 40 kDa PEG as an example, drug loading of the linear methoxy PEG is only ~1.25 wt%, while the linear diol PEG of 40 kDa increases the value to ~1.7 wt% (e.g. Pegamotecan)[77]. If the 4-arm PEG is used, drug loading can reach a value of 4.3~5.0 wt% (e.g. EZN-2208) [78].
The low-molecular-weight PEG prodrug contains PEG with a molecular weight less than 5000 Da. This platform is primarily designed to increase the water solubility and oral bioavailability, as well as modify the biodistribution of small molecule drugs by decreasing the penetration of a specific barriers such as Blood-Brain Barrier (BBB). One typical example of this type prodrug is NKTR-118, a PEG-naloxol conjugates. Naloxol is a derivative of the opioid antagonist drug naloxone. NKTR-118 was developed to treat opioid bowel dysfunction, and opioid induced constipation which is caused by opioid overdose. Phase I clinical trial results demonstrated a 10-fold increase in the oral bioavailability of NKTR-118 compared to that of naloxone. In addition, NKTR-108 demonstrates a significant lower BBB penetration than naloxone, which will help to maintain the pain relief effect of opioid [79, 80].
(b) Other polymer-drug conjugate
Although PEG has been successfully exploited as a polymer for prodrug, it has a limited number of functional groups for drug conjugation, which substantially reduces its drug loading capacity. Instead, some other polymers including N-(2-hydroxypropyl)methacrylamide (HPMA), polyglutamic acid (PGA) have more functional groups that can increase the drug loading to as high as 37 wt% [81].
FCE28068 (PK1) is a HPMA-doxorubicin prodrug containing the Gly-Phe-Leu-Gly (GFLG) peptide linker (8 wt%). FCE28068 exhibited substantially reduced anthracycline-associated side effects even when the doxorubicin equivalent dose reaches 1680 mg/m2. There was also no observed cardiotoxicity, which is the most common toxicity associated with doxorubicin. Pharmacokinetic study showed that FCE28068 had a distribution half-life of 1.8 h and an elimination half-life of 93h [82]. Although the antitumor activity was observed in patients with colorectal cancer, non-small-cell lung cancer (NSCLC) or breast cancer during a phase I clinical trial, only the patients with NSCLC or breast cancers showed a partial response in the phase II clinical trial [83].
PGA is another promising polymer backbone for polymer-drug conjugate. Unlike PEG and HPMA, PGA polymer chain is biodegradable. Therefore, longer PGA polymer chain (>60 kDa) can be used in prodrug even though its molecular weight is beyond of the renal elimination limits. Moreover, PGA conjugated prodrug always has a very high drug loading capacity. PGA conjugated to paclitaxel (CT-2103, Xyotax) has a drug loading of 37 wt%[81], while the loading capacity of another PGA-camptothecin (CT-2106) also reaches 33~35 wt% [84]. Both of these PGA prodrugs have entered into Phase I/II trials [81, 85, 86].
(c) Polymeric prodrug nanoparticles
Polymeric prodrug can self-assemble to form a nano-scaled structure such as polymeric micelle that is formed by a bi-block copolymer consisting of a hydrophilic component and a hydrophobic core formed by the drug/polymer conjugate. For example, hydrophobic drug can be conjugated to the aspartic acid segment of a block copolymer to form the hydrophobic core of the micelle, while the hydrophilic part will surround the hydrophobic core as a hydrophilic shell. Most of these polymeric micelles use PEG as the hydrophilic shell, while the hydrophoblic core varies depending on the design. Size of the micelle generally ranges from 20 nm to 100 nm that is sufficient to increase the accumulation of drugs in tumor tissues by the EPR effect. Polymeric micelle is proven to be stable enough to circulate in the blood for a long time, while the isolated polymer chain is expected to be eliminated by renal filtration. There are several polymeric prodrug micelles under clinical evaluation including paclitaxel-incorporating micelle (NK105), cisplatin-incorporating micelle (NC-6004) and SN-38-incorporating micelle (NK012) that have been extensively reviewed by Matsumur.[87]
Another type of polymeric prodrug micelle is formed by a linear, cyclodextrin-containing polymer. For example, IT-101 contains a single polymer chain of ~70k Da and self-assembles into micelles in the range of 30~40 nm. Instead of hydrophobic interaction, the micellar core is formed by the interaction between cyclodextrin and camptothecin. Therefore, addition of organic solvent such as DMSO does not disassemble the micelle structure. However, the micelle collapses after the addition of soluble adamantine (AD) presumably by displacing camptothecin from the inclusion complex.[12] In the phase I clinical trial, the half-life of IT-101 was improved to around 40 hours. Twelve patients were treated with i.v. infusion every other week in the dose range of 6~18 mg/m2/wk. Three of them, with pancreatic cancer, renal cancer and NSCLC, completed a 6-cycle treatment and showed a progressive-free survival time of 18, 14, and 11 months, respectively [88]. Phase II clinical trial in ovarian cancer was already initiated in 2008.
Prodrug can also be encapsulated in nanoparticles, micelles and lipid complexes. For instance, it is difficult to formulate paclitaxel into lipid-based nanoparticles due to its limited lipid solubility. Subsequently, cholesterol was conjugated to paclitaxel to form the prodrug Tax-chol with an increased lipophilicity. Tax-chol was successfully encapsulated into lipid nanoparticles composed of distearoyl phosphatidylcholine (DSPC)/triolein/Chol oleate/PEGChol/f-PEG-Chol (40:40:18:2.0:0.5, mole/mole) with a diameter of 130 nm. Encapsulation efficiency was greater than 90%, and the lipid complex exhibited an excellent colloidal stability. Moreover, conjugation of the nanoparticles with folic acid dramatically increased the cellular uptake and cytotoxicity in folate receptor positive cells [89].
(d) Pharmacokinetics of polymeric prodrug
Polymeric prodrug generally exhibits an improved pharmacokinetical profile than small molecule drug. Polymeric prodrug either has a large molecular weight itself (10~100 kDa), or assembles into nano-scaled structure. Therefore, polymeric prodrug demonstrates a longer half-life as well as the passive targeting ability by the EPR effect. For example, the pegylated prodrug NKTR-102 showed a half-life of 15 days in colorectal cancer, leading to prolonged tumor exposure of the parent drug SN-38.[90] On the other hand, the clearance of polymeric prodrug is an important issue during the prodrug design. As a large molecule, the polymeric prodrug is hard to be cleared from the tissue if it is not biodegradable. Therefore, most of the PEG molecules used in prodrug approach have a molecular weight less than 60 kDa, which is the limit for renal filtration.
VI. Enzyme cleavable prodrug
Enzyme cleavable prodrug is one of the most advanced strategies to design a targeted prodrug. This approach further increases the targetability of a prodrug by releasing the parent drug in tumor cells. Generally, the enzyme should be specifically located in tumors so that only the prodrug in the target tumor cells can be cleaved to release the active agent, leading to minimal side effects. However, only a limited number of enzymes have been proven to successfully activate prodrugs till now. Most of the enzymes are either widely distributed or weak in the catalytic ability to cleave prodrugs in a short time.
Prostate specific antigen (PSA) and prostate membrane specific antigen (PSMA) might be the two most popular enzymes that have been widely exploited for prodrug. PSA is a serine protease that specifically cleaves seminal fluid protein semenogelin I and II [91]. Over-expression of PSA is an established prognostic marker for prostate cancer. Moreover, the enzymatic activity of PSA only exists in prostate cancer cells, but not in the systemic circulation because of the presence of PSA inhibitors (α1-antichymotrypsin and α2-macroglobulin) in the blood. Prodrugs that are specifically cleaved by PSA result in accumulation in tumor tissues rather than other normal cells. Several peptide sequences have been identified as PSA substrates, and the most widely used sequences are HSSKLQ and SSKYQ. PSA recognizes these sequences and cleave the peptide at the carboxyl group of tyrosine. Kumar et. al., reported that SSKYQ is a much more efficient substrate that has a 10-fold higher Kcat/Km value than that of HSSKLQ [92]. Several drugs including 5-fluorodeoxyuridine (5FudR)[93], paclitaxel [94], cyclopamine[92], doxorubicin[95] and thapsigargin[96] have been conjugated to these peptides. Another type of the PSA substrate peptide consists of non-native amino acids. This substitution is believed to stabilize the conjugates and avoid undesired proteolytic hydrolysis by chymotrypsin-like proteases in the plasma. Simple substitution at one or two amino acids can maintain the cleavage efficiency without compromising its specificity.[97]
Glutaryl-Hyp-Ala-Ser-Chg-Gln-Ser-Leu-Doxorubicin (L-377202) is an enzyme cleavable prodrug [14]. N-terminal of the peptide was capped with a Glutaryl group that inhibits the proteolytic hydrolysis caused by exopeptidase, and improves the solubility. Incorporating the non-native amino acids (Hyp and Chg) into the sequence minimized the undesirable hypotensive effect which was caused by the release of histamine [97]. This hybrid sequence also increased the conjugation stability and PSA specificity. In the preclinical study, L-377202 showed a comparable activity to doxorubicin in PSA-positive prostate tumor cells, but not in normal epithelial cells or PSA-negative prostate tumor cells. Moreover, L-377202 demonstrated a 15-fold more potent anti-tumor effect than doxorubicin in a nude mouse model. The improved therapeutic index was correlated with the selective localization of the prodrug in the PSA-positive prostate tumor cells. Compared to conventional doxorubicin, L-377202 exhibited 5~10 fold higher concentration of drug in tumor tissues, while a 10-fold less accumulation in the heart.[14]
PSMA is a type II membrane glycoprotein (100 kd) with an extensive extracellular domain of 706 amino acids, a transmembrane region of 24 amino acids, and a short intracellular domain of 18 amino acids.[98] It is a well known tumor antigen and the expression of PSMA in prostate cancer cells, especially the most advanced androgen-resistant prostate cancer cells, are about a thousand-fold greater than the expression in normal tissues.[98] The over-expression of PSMA is actually a predictive indicator for recurrence in primary prostate cancer.[99] Taken together, PSMA is an ideal target for the development of a targeted prostate cancer prodrug [100]. Moreover, as an exopeptidase, PSMA catalyzes the hydrolysis of N-Acetylaspartylglutamate (NAAG) to Glutamate and N-Acetylaspartate, making it a good candidate for the enzyme-cleavable prodrug design. However, only one PSMA substrate peptide has been identified, which limits its application in prodrug [101].
VII. Enzyme activated prodrug
Some anti-cancer drugs might be effective and curative only if given at a much higher dose than currently used in the clinic. Prodrugs activated by enzymes that are specifically expressed in tumors can efficiently deliver chemotherapy drugs at least 50-fold the maximum recommended dose and subsequently kill the tumor cells [1]. In this approach the site-specific drug delivery is achieved by activation of the prodrug by enzymes that are either unique to the target tumor cells, or present at a much higher concentration in tumor cells compared to other tissues.[6] However, human tumors that express a significantly high level of activating enzymes are rare [102].
Two strategies have been developed to overcome this limitation. The first one is known as the antibody-directed enzyme prodrug therapy (ADEPT). The activating enzyme, conjugated to a tumor-specific antibody, is delivered into tumor cells, followed by administration of the prodrug that is inert until it is activated by the enzyme. The second strategy is to deliver the gene encoding the activating enzyme into tumor cells, followed by the prodrug administration. This approach is known as gene-directed enzyme prodrug therapy (GDEPT) or virus-directed enzyme prodrug therapy (VDEPT). Both ADEPT and GDEPT specifically activate the prodrug in tumor cells, thus avoids the systemic toxicity associated with most chemotherapy drugs.
a) Antibody-directed enzyme prodrug therapy (ADEPT)
In this approach, the activating enzyme, not the drug, is specifically delivered into tumor cells using a tumor-specific antibody. The enzyme is linked to an antibody either chemically or using the recombinant DNA technology to produce a fusion protein consisting of the enzyme and antibody. Targeted delivery of the enzyme-antibody conjugates to tumor cells creates a micro-environment in which the enzyme converts the prodrug to an active agent. The major advantage of this approach is that a single molecule of enzyme can generate hundreds of active molecules per second from the prodrug. Secondly, not all cells in the tumor tissue are required to take up the enzyme-antibody conjugates because the drug molecules released by the enzyme would diffuse to adjacent cells that do not take up the enzyme [1]. Different combinations of enzymes, antibodies, and prodrugs have been successfully adopted to target a variety of human cancers that are otherwise quite resistant to the conventional chemotherapy.[103]
Disadvantages of this approach include: i) immunogenicity of the enzyme-antibody; ii) possible activation of prodrugs by the unbound enzyme-antibody circulating in the blood; and iii) the conjugation heterogeneity.[104, 105] The immunogenicity associated with murine antibodies and bacterial enzymes can be solved by replacing them with humanized antibodies.[106] For example, the murine anti-TAG-72 mAb (CC49) used in a phase I clinical trial for ovarian cancer induced human anti-mouse-antibody response [107]. However, another pilot clinical trial demonstrated that the humanized anti-TAG-72 antibody did not induce any immunogenic response, and the antibody is suitable for tumor targeting in human patients with high specificity and affinity [108]. Moreover, the same group demonstrated that this anti-TAG-72 humanized antibody (HuCC49ΔCH2) conjugated β-galactosidase selectively activated the geldanamycin prodrug (17-AG-C2_Gal). The released active drug 17-AG-C2 entered the cancer cells, bound to Hsp90 and induced AKT degradation up to 70%, leading to an enhanced anticancer activity by more than 25 folds compared to the prodrug [109]. Table 5 summarizes various antigenes and antibodies that have been used in ADEPT.
Table 5.
Antigenes and antibodies used in ADEPT prodrugs
| Prodrug | Antigen | Antibody | Tumor Target | Phase status |
|---|---|---|---|---|
| MFECP1 + ZD2767P | Murine anti-CEA scFv | CEA positive tumors | I/II | |
| A5CP + ZD2767P | Murine anti-CEA F(ab)2 | Advanced CRC | I [110] | |
| Vinca-cephalosporin | Carcinogenic embryogenic antigen (CEA) | CEM2314 | Colon carcinoma | Cell line [104] |
| Phenylenediamine mustard-cephalosporin | L6 | Lung adenocarcinoma | Cell line and animal model [104] | |
| Doxorubicin phosphate | L6 | Lung adenocarcinoma | Cell line [104] | |
| Phenolmustard phosphate | L6 | Lung adenocarcinoma | Cell line [104] | |
| Etoposide phosphate | CEA | BW431 | Colon carcinoma | Cell line [104] |
| Mitomycin C phosphate | L6 | Lung adenocarcinoma | Cell line [104] |
b) Gene-directed enzyme prodrug therapy (GDEPT)
GDEPT, also known as VDEPT or gene prodrug activation therapy (GPAT), is an effective prodrug approach to target tumors by delivering a gene encoding an activating enzyme into tumor cells, followed by a systemic administration of an inactive prodrug which is converted to its active parent drug by the expressed enzyme in the tumor cells. GDEPT is similar to ADEPT in that both use a non-endogenous enzyme to activate a prodrug. The difference lies in the location where the prodrug is activated. The prodrug is activated extracellularly in ADEPT, but intracellularly in GDEPT [1, 6]. Therefore, the enzyme/prodrug combination that is suitable for ADEPT may not be suitable for GEDPT.
Bacterial or viral enzymes are preferred for the GDEPT approach due to following reasons: i) intracellular expression of the enzyme only induces a negligible immune reaction; ii) most of these enzymes have no corresponding activities in humans; iii) bacterial proteins tend to have fewer requirements for the post-translational modification, whereas a more complex eukaryotic protein may not fold properly in different species.[1, 111]
Cytotoxic effects of the active drugs on non-transduced neighboring cells is called the bystander effect. This occurs when the active drugs leave the transduced cell and kill the neighboring cells that do not express the activating enzyme. There are several explanations for the mechanism: i) the active drug remains entrapped in the vesicles of dead tumor cells, which are then phagocytosed by nontransduced cells [112]; ii) the active parent drug can diffuse through the gap junction between adjacent cells [113]; and iii) the active parent drug is small and soluble (for example, 5-fluorouracil) enough to diffuse across cell membrane to affect adjacent cells.[114]
A phase I clinical trial for the treatment of breast cancer demonstrated that GDEPT could be safely applied in human patients without local or systemic complications. The gene expression vector contains the proximal erbB-2 promoter to specifically express the cytosine deaminase enzyme in erbB-2 positive tumor cells, followed by systemic infusion of the prodrug 5-Fluorcytosine that is converted into its toxic metabolite 5-fluorouracil. [115] Also, they verified that expression of this enzyme was highly selective in the tumor cells. Tumor regression was observed in patients who received this GEDPT. Furthermore, they did not observe any side effects or complications associated with this treatment, such as insertional mutagenesis, anti-DNA antibody formation, local infection or tumor nodule ulceration.[115]
Similar results were also observed by another group. Ring et al. constructed a chimeric promoter consisting of the erbB-2 promoter and the MUC1 enhancer, resulting in an increased sensitivity of MUC1-positive tumor cells to ganciclovir. This promoter/enhancer was used to drive the expression of the suicide gene thymidine kinase. In pancreatic and breast cancer cells that were transfected with the retrovirus containing this expression cassette, only the MUC1 positive cells were killed by the prodrug ganciclovir. [116, 117]
Using GDEPT one can also increase tumor selectivity by incorporating a tumor-specific regulatory element in the gene expression vector.[118, 119] For example, GDEPT virus vector has been successful used to target prostate cancer cells by using the PSA-specific promoter or the PSMA-specific promoter/enhancer [120]. Another enzyme that has been used to target certain cancers is cytochrome p450, also known as CYP. The anticancer drugs such as IFA [121], AQ4N [122] and anthracycline MMDX [123] have been used with the vector encoding the p450 enzyme. These prodrugs are metabolized by the p450 enzyme to the active agents. Intratumoral expression of the p450 enzyme dramatically increased the antitumor activity of MMXD in a SCID mouse xenograft model [123]. Table 6 summarizes the enzyme activated prodrugs undergoing clinical trials.
Table 6.
Enzyme activated prodrugs in clinical trials
| Enzyme | Prodrug | Cytotoxic drug | Application | Phase status |
|---|---|---|---|---|
| Cytosine deaminase | Gancyclovir, 5-FC | Ganciclovir, triphosphate, 5-FU | GDEPT | I [1, 115, 124, 125] |
| Thymidine Kinase | Ganciclovir | GDEPT, | I, II [126–128] | |
| Cytochrome-P450 | Cyclophosphamide, Ifosfamide, | Phosphamide mustard, Acrolein | GDEPT | I, II [129, 130] |
| Nitroreductase | 5-(Aziridine-1-yl)-2,4-dinitrobenzamide (CB1954) 4-Nitrobenzyloxycarbonyl derivative |
5-(Aziridine-1-yl)-4-hydroxylamino-2-nitrobenzamide | GDEPT | I [131–133] |
| Carboxypeptidase G2 (CPG2/CMDA) | Benzoic acid mustard glutamate, e.g. CMDA ZD2767P | Benzoic acid mustards CJS11 | ADEPT, GDEPT | I, II [134, 135] |
| B-Lactamase | Vinca-cephalosporin Phenylenediamine mustard-cephalosporin Nitrogen-mustard-cephalosporin |
4-Desacetylvinblastine-3-carboxhydrazide, Phenylenediamine mustard, Nitrogen mustards | ADEPT | |
| Alkaline phosphatase | Phenolmustard phosphate, Doxorubicin phosphate, Mitomycin phosphate, Etoposide phosphate | Phenolmustard, Doxorubicin, Mitomycin alcohol, Etoposide | ADEPT |
VIII. CONCLUSION
Prodrug provides a powerful and versatile tool to improve the pharmaceutical properties of a drug via various chemical modifications or conjugations with a promoiety. Despite the remarkable progress achieved in the classic prodrug design, the targetd prodrug is still in its infancy, and more efforts are needed to establish its role in cancer therapy. The first-ever FDA-approved antibody-drug conjugate (Mylotarg) was withdrawn from the market in 2010 due to its ineffectiveness and severe side effects found in the post-approval clinical trial. Nevertheless, numerous targeted prodrugs are undergoing clinical trials after the proof-of-concept has been convinced in preclinical studies. Immunogenicity, cell permeability, and cell specificity are the three major obstacles in the development of an efficient targeted prodrug.
Immunogenicity can be addressed by using humanized antibody or other alternative ligands such as peptides and aptamers. Similarly, using antibody fragments or small ligands could improve the permeability of a prodrug. The central hypothesis of a targeted prodrug is the specific expression of an antigen on tumor cells that can be recognized by natural or artificial ligands. Consequently, the most critical aspect of developing an efficient targeted prodrug is the selection of a tumor-specific antigen and its ligands that can be conjugated to the parent drug or activating enzyme. Unfortunately, there are only a limited number of tumor-specific antigens that have been identified and many of them are endogenously expressed in other normal tissues, leading to a nonspecific toxicity of the prodrug. Hence, it is essential for scientists to validate the specificity of the antigen in animal studies rather than in vitro studies. Moreover, identifying targeting ligands with is another challenge. Due to the various limitations of intact antibodies, antibody fragments or small artificial ligands such as peptides and aptamers are preferred for the targeted prodrug approach. However, screening of peptide or aptamer ligands with a high affinity is time consuming and very difficult. In the future, focusing on identification of tumor-specific ligands with high affinity, high specificity, high stability, low immunogenicity, and high permeability will be important for the success of a targeted prodrug.
Figure 3.

Schematic diagram of the ADEPT. The antibody conjugated enzyme specifically binds to antigens on tumor cells. Next, prodrug is administered and then activated by the conjugated enzyme to the active parent drug which induces cytotoxic effects on tumor cells.
Figure 4.

Schematic diagram of the GDEPT. A gene expression vector that encodes an activating enzyme is delivered into tumor cells. The activating enzyme is then expressed intracellularly to activate a prodrug into its active parent drug. The active drug can migrate to adjacent nontransfected tumor cells, which is called the bystander effect.
Acknowledgments
This work was supported by a grant (1R21CA143683-01) from the National Cancer Institute at NIH, and a grant (1R21AA017960-01A1) from the National Institute of Alcohol Abuse and Alcoholism at NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Knox RJ, Connors TA. Prodrugs in Cancer Chemotherapy. Pathol Oncol Res. 1997;3(4):309–324. doi: 10.1007/BF02904292. [DOI] [PubMed] [Google Scholar]
- 2.Denny WA. Prodrug strategies in cancer therapy. Eur J Med Chem. 2001;36(7–8):577–95. doi: 10.1016/s0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- 3.Rautio J, et al. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7(3):255–70. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 4.Singh Y, Palombo M, Sinko PJ. Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem. 2008;15(18):1802–26. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller CE. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem Biodivers. 2009;6(11):2071–83. doi: 10.1002/cbdv.200900114. [DOI] [PubMed] [Google Scholar]
- 6.Han HK, Amidon GL. Targeted prodrug design to optimize drug delivery. AAPS Pharm Sci. 2000;2(1):E6. doi: 10.1208/ps020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liederer BM, Borchardt RT. Enzymes involved in the bioconversion of ester-based prodrugs. J Pharm Sci. 2006;95(6):1177–95. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]
- 8.Yang JZ, Chen W, Borchardt RT. In vitro stability and in vivo pharmacokinetic studies of a model opioid peptide, H-Tyr-D-Ala-Gly-Phe-D-Leu-OH (DADLE), and its cyclic prodrugs. J Pharmacol Exp Ther. 2002;303(2):840–8. doi: 10.1124/jpet.102.037135. [DOI] [PubMed] [Google Scholar]
- 9.Meyer-Losic F, et al. DTS-108, a novel peptidic prodrug of SN38: in vivo efficacy and toxicokinetic studies. Clin Cancer Res. 2008;14(7):2145–53. doi: 10.1158/1078-0432.CCR-07-4580. [DOI] [PubMed] [Google Scholar]
- 10.Pasut G, Veronese FM. PEG conjugates in clinical development or use as anticancer agents: an overview. Adv Drug Deliv Rev. 2009;61(13):1177–88. doi: 10.1016/j.addr.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Cheng J, et al. Synthesis of linear, beta-cyclodextrin-based polymers and their camptothecin conjugates. Bioconjug Chem. 2003;14(5):1007–17. doi: 10.1021/bc0340924. [DOI] [PubMed] [Google Scholar]
- 12.Davis ME. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptothecin. Adv Drug Deliv Rev. 2009;61(13):1189–92. doi: 10.1016/j.addr.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Wallace S. Polymer-drug conjugates: recent development in clinical oncology. Adv Drug Deliv Rev. 2008;60(8):886–98. doi: 10.1016/j.addr.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeFeo-Jones D, et al. A peptide-doxorubicin ‘prodrug’ activated by prostate-specific antigen selectively kills prostate tumor cells positive for prostate-specific antigen in vivo. Nat Med. 2000;6(11):1248–52. doi: 10.1038/81351. [DOI] [PubMed] [Google Scholar]
- 15.Kumaresan PR, et al. Evaluation of ketone-oxime method for developing therapeutic on-demand cleavable immunoconjugates. Bioconjug Chem. 2008;19(6):1313–8. doi: 10.1021/bc800024v. [DOI] [PubMed] [Google Scholar]
- 16.Cheng K, et al. Enhanced hepatic uptake and bioactivity of type alpha1(I) collagen gene promoter-specific triplex-forming oligonucleotides after conjugation with cholesterol. J Pharmacol Exp Ther. 2006;317(2):797–805. doi: 10.1124/jpet.105.100347. [DOI] [PubMed] [Google Scholar]
- 17.Lewis Phillips GD, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–90. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 18.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13(3):235–44. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 19.Doronina SO, et al. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjug Chem. 2008;19(10):1960–3. doi: 10.1021/bc800289a. [DOI] [PubMed] [Google Scholar]
- 20.Jaracz S, et al. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13(17):5043–54. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 21.Garnett MC. Targeted drug conjugates: principles and progress. Adv Drug Deliv Rev. 2001;53(2):171–216. doi: 10.1016/s0169-409x(01)00227-7. [DOI] [PubMed] [Google Scholar]
- 22.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5(2):147–59. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 23.Stohrer M, et al. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000;60(15):4251–5. [PubMed] [Google Scholar]
- 24.Adams GP, et al. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998;58(3):485–90. [PubMed] [Google Scholar]
- 25.Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009;9(8):982–1004. doi: 10.2174/156800909790192365. [DOI] [PubMed] [Google Scholar]
- 26.Gires O. Tumor-Associated Antigens: Identification, Characterization, and Clinical Applications. 2009 [Google Scholar]
- 27.Wang W, et al. PSMA expression in Schwannoma: a potential clinical mimicker of metastatic prostate carcinoma. Urol Oncol. 2009;27(5):525–8. doi: 10.1016/j.urolonc.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Spearman ME, Goodwin RM, Kau D. Disposition of the monoclonal antibody-vinca alkaloid conjugate, KS1/4-DAVLB (LY256787), in Fischer 344 rats and rhesus monkeys. Drug Metab Dispos. 1987;15(5):640–7. [PubMed] [Google Scholar]
- 29.Quiles S, et al. Synthesis and preliminary biological evaluation of high-drug-load paclitaxel-antibody conjugates for tumor-targeted chemotherapy. J Med Chem. 53(2):586–94. doi: 10.1021/jm900899g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamann PR, et al. An anti-MUC1 antibody-calicheamicin conjugate for treatment of solid tumors. Choice of linker and overcoming drug resistance. Bioconjug Chem. 2005;16(2):346–53. doi: 10.1021/bc049795f. [DOI] [PubMed] [Google Scholar]
- 31.Boghaert ER, et al. The oncofetal protein, 5T4, is a suitable target for antibody-guided anti-cancer chemotherapy with calicheamicin. Int J Oncol. 2008;32(1):221–34. doi: 10.3892/ijo.32.1.221. [DOI] [PubMed] [Google Scholar]
- 32.Krop IE, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 28(16):2698–704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 33.Sievers EL. Antibody-targeted chemotherapy of acute myeloid leukemia using gemtuzumab ozogamicin (Mylotarg) Blood Cells Mol Dis. 2003;31(1):7–10. doi: 10.1016/s1079-9796(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 34.van Der Velden VH, et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood. 2001;97(10):3197–204. doi: 10.1182/blood.v97.10.3197. [DOI] [PubMed] [Google Scholar]
- 35.Buckwalter M, et al. Pharmacokinetics of gemtuzumab ozogamicin as a single-agent treatment of pediatric patients with refractory or relapsed acute myeloid leukemia. J Clin Pharmacol. 2004;44(8):873–80. doi: 10.1177/0091270004267595. [DOI] [PubMed] [Google Scholar]
- 36.Mylotarg (gemtuzumab ozogamicin): Market Withdrawal. [cited; Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm216458.htm.
- 37.Krop I, et al. A phase 2 study of trastuzumab-dm 1 (t-dm 1), a novel her 2 antibody–drug conjugate, in her 2+ metastatic breast cancer (mbc) patients previously treated with conventional chemotherapy, lapatinib and trastuzumab. San Antonio Brest Cancer Symposium; 2009. [Google Scholar]
- 38.DiJoseph JF, et al. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin Cancer Res. 2006;12(1):242–9. doi: 10.1158/1078-0432.CCR-05-1905. [DOI] [PubMed] [Google Scholar]
- 39.Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Control Release. 146(3):264–75. doi: 10.1016/j.jconrel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krop IE, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28(16):2698–704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 41.Anjali Advani EG, Gisselbrecht Christian, Rohatiner Ama, Rosen Steven, Smith Mitchell, Boni Joseph, Lejeune Chantal, Patel Hemant. Preliminary Report of a Phase 1 Study of CMC-544, an Antibody-Targeted Chemotherapy Agent, in Patients with B-Cell Non-Hodgkin’s Lymphoma (NHL). American Society of Hematology Annual Meeting; 2005. [Google Scholar]
- 42.Khandare JJ, et al. Novel polymeric prodrug with multivalent components for cancer therapy. J Pharmacol Exp Ther. 2006;317(3):929–37. doi: 10.1124/jpet.105.098855. [DOI] [PubMed] [Google Scholar]
- 43.Dharap SS, et al. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci U S A. 2005;102(36):12962–7. doi: 10.1073/pnas.0504274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen X, et al. Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. J Med Chem. 2005;48(4):1098–106. doi: 10.1021/jm049165z. [DOI] [PubMed] [Google Scholar]
- 45.Yoneda Y, et al. A cell-penetrating peptidic GRP78 ligand for tumor cell-specific prodrug therapy. Bioorg Med Chem Lett. 2008;18(5):1632–6. doi: 10.1016/j.bmcl.2008.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin BK, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278(9):7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 47.Dubowchik GM, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg Med Chem Lett. 1998;8(23):3341–6. doi: 10.1016/s0960-894x(98)00609-x. [DOI] [PubMed] [Google Scholar]
- 48.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: an emerging class of therapeutics. Annu Rev Med. 2005;56:555–83. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, et al. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009;37(9):3094–109. doi: 10.1093/nar/gkp185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sefah K, et al. Development of DNA aptamers using Cell-SELEX. Nat Protoc. 5(6):1169–85. doi: 10.1038/nprot.2010.66. [DOI] [PubMed] [Google Scholar]
- 51.Chu TC, et al. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006;34(10):e73. doi: 10.1093/nar/gkl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farokhzad OC, et al. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64(21):7668–72. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 53.Morris MJ, et al. Phase I evaluation of J591 as a vascular targeting agent in progressive solid tumors. Clin Cancer Res. 2007;13(9):2707–13. doi: 10.1158/1078-0432.CCR-06-2935. [DOI] [PubMed] [Google Scholar]
- 54.Milowsky MI, et al. Vascular targeted therapy with anti-prostate-specific membrane antigen monoclonal antibody J591 in advanced solid tumors. J Clin Oncol. 2007;25(5):540–7. doi: 10.1200/JCO.2006.07.8097. [DOI] [PubMed] [Google Scholar]
- 55.Dhar S, et al. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105(45):17356–61. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolishetti N, et al. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 107(42):17939–44. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang YF, et al. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chem biochem. 2009;10(5):862–8. doi: 10.1002/cbic.200800805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shangguan D, et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci U S A. 2006;103(32):11838–43. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henne WA, et al. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg Med Chem Lett. 2006;16(20):5350–5. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]
- 60.Aronov O, et al. Folate-targeted PEG as a potential carrier for carboplatin analogs. Synthesis and in vitro studies. Bioconjug Chem. 2003;14(3):563–74. doi: 10.1021/bc025642l. [DOI] [PubMed] [Google Scholar]
- 61.Anbharasi V, Cao N, Feng SS. Doxorubicin conjugated to D-alpha-tocopheryl polyethylene glycol succinate and folic acid as a prodrug for targeted chemotherapy. J Biomed Mater Res A. 94(3):730–43. doi: 10.1002/jbm.a.32734. [DOI] [PubMed] [Google Scholar]
- 62.Rubio-Aliaga I, et al. Targeted disruption of the peptide transporter Pept2 gene in mice defines its physiological role in the kidney. Mol Cell Biol. 2003;23(9):3247–52. doi: 10.1128/MCB.23.9.3247-3252.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kodama T. Antibodies that inhibit transport activity of peptide transporters. US: 2010. [Google Scholar]
- 64.Landowski CP, et al. Targeted delivery to PEPT1-overexpressing cells: acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol Cancer Ther. 2005;4(4):659–67. doi: 10.1158/1535-7163.MCT-04-0290. [DOI] [PubMed] [Google Scholar]
- 65.Nakanishi T, et al. Carrier-mediated transport of oligopeptides in the human fibrosarcoma cell line HT1080. Cancer Res. 1997;57(18):4118–22. [PubMed] [Google Scholar]
- 66.Nakanishi T, et al. Cancer cell-targeted drug delivery utilizing oligopeptide transport activity. Int J Cancer. 2000;88(2):274–80. [PubMed] [Google Scholar]
- 67.Gallop MA. Gemcitabine Prodrugs, Pharmaceuticals Compositions and Uses Thereof. XenoPort, Inc; Santa Clare, CA (US): USA: 2010. pp. 1–52. [Google Scholar]
- 68.Stella VJ. Prodrugs: Challanges and Rewards. 2007;(Part 1) [Google Scholar]
- 69.Ramanathan S, et al. Targeting the sodium-dependent multivitamin transporter (SMVT) for improving the oral absorption properties of a retro-inverso Tat nonapeptide. Pharm Res. 2001;18(7):950–6. doi: 10.1023/a:1010932126662. [DOI] [PubMed] [Google Scholar]
- 70.Minko T, et al. Enhancing the anticancer efficacy of camptothecin using biotinylated poly(ethylene glycol) conjugates in sensitive and multidrug-resistant human ovarian carcinoma cells. Cancer Chemother Pharmacol. 2002;50(2):143–50. doi: 10.1007/s00280-002-0463-1. [DOI] [PubMed] [Google Scholar]
- 71.Russell-Jones G, et al. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J Inorg Biochem. 2004;98(10):1625–33. doi: 10.1016/j.jinorgbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 72.Yang W, et al. Targeting cancer cells with biotin-dendrimer conjugates. Eur J Med Chem. 2009;44(2):862–8. doi: 10.1016/j.ejmech.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 73.Yamaoka T, Tabata Y, Ikada Y. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci. 1994;83(4):601–6. doi: 10.1002/jps.2600830432. [DOI] [PubMed] [Google Scholar]
- 74.Eldon Michael A, C-MS, Viegas Tacey, Bentley Michael. NKTR-102, a novel PEGylated-irinotecan conjugate, results in sustained tumor growth inhibition in mouse models of human colorectal and lung tumors that is associated with increased and sustained tumor SN38 exposure. the 2007 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2007. p. C157. [Google Scholar]
- 75.Eldon Michael A, LA, Staschen Carl-Michael, Viegas Tacey, Singhal Anil, Heather Persson MR, Bentley Michael. Anti-tumor activity and pharmacokinetics of NKTR-102, a novel PEGylated-irinotecan conjugate, in irinotecan-resistant colorectal tumors implanted in mice. 14th European Cancer Conference; 2007. p. P-0722. [Google Scholar]
- 76.Von Hoff DD, Borad GSJMJ, Rosen LS, Utz4 J, Basche M, Alemany C, Dhar S, Acosta L, Barker T, Walling J, Hamm JT. First Phase I Trial of NKTR-102 (PEG-irinotecan) Reveals Early Evidence of Broad Anti-Tumor Activity in Three Different Schedules. 2008 EORTC-NCI-AACR Symposium; 2008. p. 595. [Google Scholar]
- 77.Ronit Satchi-Fainaro RD. Polymer therapeutics: polymers as drugs, conjugates and gene delivery systems. Advances in polymer science. 2006:36–37. [Google Scholar]
- 78.Sapra P, et al. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res. 2008;14(6):1888–96. doi: 10.1158/1078-0432.CCR-07-4456. [DOI] [PubMed] [Google Scholar]
- 79.Eldon Michael A, PDS, PhD, Neumann Theresa A, PhD, Wolff Ron, PhD, Cheng Lin, PhD, Viegas †Tacey X, PhD, Bentley †Michael D, PhD, Simone Fishburn C, PhD, Kugler Alan R., PhD NKTR-118 (Oral PEG-Naloxol), a PEGylated Derivative of Naloxone: Demonstration of Selective Peripheral Opioid Antagonism After Oral Administration in Preclinical Models. The American Academy of Pain Management 18th Annual Clinical Meeting; 2007. p. P-28. [Google Scholar]
- 80.Neumann Theresa A, PHvP, MD, Marcantonio Annette, MBA, Song Di, PhD, Morrison Paul J, Eldon Michael A, PhD, Kugler Alan R., PhD Clinical Investigation of NKTR-118 as a Selective Oral Peripheral Opioid Antagonist. the American Academy of Pain Management 18th Annual Clinical Meeting; 2007. p. P-27. [Google Scholar]
- 81.Sabbatini P, et al. Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. J Clin Oncol. 2004;22(22):4523–31. doi: 10.1200/JCO.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 82.Vasey PA, et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5(1):83–94. [PubMed] [Google Scholar]
- 83.Seymour LW, et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int J Oncol. 2009;34(6):1629–36. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- 84.Bhatt R, et al. Synthesis and in vivo antitumor activity of poly(l-glutamic acid) conjugates of 20S-camptothecin. J Med Chem. 2003;46(1):190–3. doi: 10.1021/jm020022r. [DOI] [PubMed] [Google Scholar]
- 85.Lin NU, et al. Phase II study of CT-2103 as first- or second-line chemotherapy in patients with metastatic breast cancer: unexpected incidence of hypersensitivity reactions. Invest New Drugs. 2007;25(4):369–75. doi: 10.1007/s10637-007-9034-y. [DOI] [PubMed] [Google Scholar]
- 86.Homsi J, et al. Phase I trial of poly-L-glutamate camptothecin (CT-2106) administered weekly in patients with advanced solid malignancies. Clin Cancer Res. 2007;13(19):5855–61. doi: 10.1158/1078-0432.CCR-06-2821. [DOI] [PubMed] [Google Scholar]
- 87.Matsumura Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv Drug Deliv Rev. 2008;60(8):899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 88.Oliver JCYY, Synold TW, Schluep T, Davis M. A dose-finding pharmacokinetic study of IT-101, the first de novo designed nanoparticle therapeutic, in refractory solid tumors. 2008 ASCO Annual Meeting Proceedings; 2008. [Google Scholar]
- 89.Stevens PJ, Sekido M, Lee RJ. A folate receptor-targeted lipid nanoparticle formulation for a lipophilic paclitaxel prodrug. Pharm Res. 2004;21(12):2153–7. doi: 10.1007/s11095-004-7667-5. [DOI] [PubMed] [Google Scholar]
- 90.Eldon Michael A, C-MS, Viegas Tacey, Bentley Michael. NKTR-102, a novel PEGylated-irinotecan conjugate, results in sustained tumor growth inhibition in mouse models of human colorectal and lung tumors that is associated with increased and sustained tumor SN38 exposure. the 2007 AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2007. p. C157. [Google Scholar]
- 91.Lilja H, Abrahamsson PA, Lundwall A. Semenogelin, the predominant protein in human semen. Primary structure and identification of closely related proteins in the male accessory sex glands and on the spermatozoa. J Biol Chem. 1989;264(3):1894–900. [PubMed] [Google Scholar]
- 92.Kumar SK, et al. Targeted inhibition of hedgehog signaling by cyclopamine prodrugs for advanced prostate cancer. Bioorg Med Chem. 2008;16(6):2764–8. doi: 10.1016/j.bmc.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mhaka A, et al. A 5-fluorodeoxyuridine prodrug as targeted therapy for prostate cancer. Bioorg Med Chem Lett. 2002;12(17):2459–61. doi: 10.1016/s0960-894x(02)00433-x. [DOI] [PubMed] [Google Scholar]
- 94.Kumar SK, et al. Modulating paclitaxel bioavailability for targeting prostate cancer. Bioorg Med Chem. 2007;15(14):4973–84. doi: 10.1016/j.bmc.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 95.Denmeade SR, et al. Enzymatic activation of a doxorubicin-peptide prodrug by prostate-specific antigen. Cancer Res. 1998;58(12):2537–40. [PubMed] [Google Scholar]
- 96.Denmeade SR, et al. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003;95(13):990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- 97.Garsky VM, et al. The synthesis of a prodrug of doxorubicin designed to provide reduced systemic toxicity and greater target efficacy. J Med Chem. 2001;44(24):4216–24. doi: 10.1021/jm0101996. [DOI] [PubMed] [Google Scholar]
- 98.Ghosh A, Heston WD. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem. 2004;91(3):528–39. doi: 10.1002/jcb.10661. [DOI] [PubMed] [Google Scholar]
- 99.Ross JS, et al. Correlation of primary tumor prostate-specific membrane antigen expression with disease recurrence in prostate cancer. Clin Cancer Res. 2003;9(17):6357–62. [PubMed] [Google Scholar]
- 100.Wright GL, Jr, et al. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48(2):326–34. doi: 10.1016/s0090-4295(96)00184-7. [DOI] [PubMed] [Google Scholar]
- 101.Mhaka A, et al. Use of methotrexate-based peptide substrates to characterize the substrate specificity of prostate-specific membrane antigen (PSMA) Cancer Biol Ther. 2004;3(6):551–8. doi: 10.4161/cbt.3.6.846. [DOI] [PubMed] [Google Scholar]
- 102.Bagshawe KD. Adv Pharmacol. Vol. 24. 1993. Antibody-directed enzyme prodrug therapy (ADEPT) pp. 99–121. [DOI] [PubMed] [Google Scholar]
- 103.Connor TA. Prodrugs in Cancer Chemotherapy. Stem Cells. 1995;13(5):501–511. doi: 10.1002/stem.5530130507. [DOI] [PubMed] [Google Scholar]
- 104.Melton RG, Sherwood RF. Antibody-enzyme conjugates for cancer therapy. J Natl Cancer Inst. 1996;88(3–4):153–65. doi: 10.1093/jnci/88.3-4.153. [DOI] [PubMed] [Google Scholar]
- 105.Melton RG. Antibody-directed enzyme prodrug therapy (ADEPT) Drugs Future. 1996;21:167. doi: 10.3892/ijo.9.3.567. [DOI] [PubMed] [Google Scholar]
- 106.Bagshawe KD. Antibody-Directed Enzyme Prodrug Therapy. 2007. [DOI] [PubMed] [Google Scholar]
- 107.Alvarez RD, et al. A Phase I study of combined modality (90)Yttrium-CC49 intraperitoneal radioimmunotherapy for ovarian cancer. Clin Cancer Res. 2002;8(9):2806–11. [PubMed] [Google Scholar]
- 108.Xiao J, et al. Pharmacokinetics and clinical evaluation of 125I-radiolabeled humanized CC49 monoclonal antibody (HuCC49deltaC(H)2) in recurrent and metastatic colorectal cancer patients. Cancer Biother Radiopharm. 2005;20(1):16–26. doi: 10.1089/cbr.2005.20.16. [DOI] [PubMed] [Google Scholar]
- 109.Fang L, et al. Enzyme specific activation of benzoquinone ansamycin prodrugs using HuCC49DeltaCH2-beta-galactosidase conjugates. J Med Chem. 2006;49(21):6290–7. doi: 10.1021/jm060647f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Francis RJ, et al. A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours. Br J Cancer. 2002;87(6):600–7. doi: 10.1038/sj.bjc.6600517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rigg AS, Lemoine NR. Genetic prodrug activation therapy for pancreatic cancer. Ann N Y Acad Sci. 1999;880:319–25. doi: 10.1111/j.1749-6632.1999.tb09535.x. [DOI] [PubMed] [Google Scholar]
- 112.Freeman SM, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53(21):5274–83. [PubMed] [Google Scholar]
- 113.Bi WL, et al. In vitro evidence that metabolic cooperation is responsible for the bystander effect observed with HSV tk retroviral gene therapy. Hum Gene Ther. 1993;4(6):725–31. doi: 10.1089/hum.1993.4.6-725. [DOI] [PubMed] [Google Scholar]
- 114.Huber BE, et al. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc Natl Acad Sci U S A. 1994;91(17):8302–6. doi: 10.1073/pnas.91.17.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pandha HS, et al. Genetic prodrug activation therapy for breast cancer: A phase I clinical trial of erbB-2-directed suicide gene expression. J Clin Oncol. 1999;17(7):2180–9. doi: 10.1200/JCO.1999.17.7.2180. [DOI] [PubMed] [Google Scholar]
- 116.Ring CJ, et al. Suicide gene expression induced in tumour cells transduced with recombinant adenoviral, retroviral and plasmid vectors containing the ERBB2 promoter. Gene Ther. 1996;3(12):1094–103. [PubMed] [Google Scholar]
- 117.Ring CJ, et al. Use of transcriptional regulatory elements of the MUC1 and ERBB2 genes to drive tumour-selective expression of a prodrug activating enzyme. Gene Ther. 1997;4(10):1045–52. doi: 10.1038/sj.gt.3300510. [DOI] [PubMed] [Google Scholar]
- 118.Hlavaty J, et al. Multiple modifications allow high-titer production of retroviral vectors carrying heterologous regulatory elements. J Virol. 2004;78(3):1384–92. doi: 10.1128/JVI.78.3.1384-1392.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Altaner C. Prodrug cancer gene therapy. Cancer Lett. 2008;270(2):191–201. doi: 10.1016/j.canlet.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 120.Wu L, et al. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Ther. 2001;8(18):1416–26. doi: 10.1038/sj.gt.3301549. [DOI] [PubMed] [Google Scholar]
- 121.Chen CS, Jounaidi Y, Waxman DJ. Enantioselective metabolism and cytotoxicity of R-ifosfamide and S-ifosfamide by tumor cell-expressed cytochromes P450. Drug Metab Dispos. 2005;33(9):1261–7. doi: 10.1124/dmd.105.004788. [DOI] [PubMed] [Google Scholar]
- 122.McCarthy HO, et al. Bioreductive GDEPT using cytochrome P450 3A4 in combination with AQ4N. Cancer Gene Ther. 2003;10(1):40–8. doi: 10.1038/sj.cgt.7700522. [DOI] [PubMed] [Google Scholar]
- 123.Lu H, Chen CS, Waxman DJ. Potentiation of methoxymorpholinyl doxorubicin antitumor activity by P450 3A4 gene transfer. Cancer Gene Ther. 2009;16(5):393–404. doi: 10.1038/cgt.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Freytag SO, et al. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol Ther. 2007;15(3):636–42. doi: 10.1038/sj.mt.6300068. [DOI] [PubMed] [Google Scholar]
- 125.Freytag SO, et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63(21):7497–506. [PubMed] [Google Scholar]
- 126.Klatzmann D, et al. A phase I/II study of herpes simplex virus type 1 thymidine kinase “suicide” gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Hum Gene Ther. 1998;9(17):2595–604. doi: 10.1089/hum.1998.9.17-2595. [DOI] [PubMed] [Google Scholar]
- 127.Fillat C, et al. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: fifteen years of application. Curr Gene Ther. 2003;3(1):13–26. doi: 10.2174/1566523033347426. [DOI] [PubMed] [Google Scholar]
- 128.Chevez-Barrios P, et al. Response of retinoblastoma with vitreous tumor seeding to adenovirus-mediated delivery of thymidine kinase followed by ganciclovir. J Clin Oncol. 2005;23(31):7927–35. doi: 10.1200/JCO.2004.00.1883. [DOI] [PubMed] [Google Scholar]
- 129.Kan O, Kingsman S, Naylor S. Cytochrome P450-based cancer gene therapy: current status. Expert Opin Biol Ther. 2002;2(8):857–68. doi: 10.1517/14712598.2.8.857. [DOI] [PubMed] [Google Scholar]
- 130.Braybrooke JP, et al. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin Cancer Res. 2005;11(4):1512–20. doi: 10.1158/1078-0432.CCR-04-0155. [DOI] [PubMed] [Google Scholar]
- 131.Searle PF, et al. Nitroreductase: a prodrug-activating enzyme for cancer gene therapy. Clin Exp Pharmacol Physiol. 2004;31(11):811–6. doi: 10.1111/j.1440-1681.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 132.Palmer DH, et al. Virus-directed enzyme prodrug therapy: intratumoral administration of a replication-deficient adenovirus encoding nitroreductase to patients with resectable liver cancer. J Clin Oncol. 2004;22(9):1546–52. doi: 10.1200/JCO.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 133.Chung-Faye G, et al. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: a phase I and pharmacokinetic study of its prodrug, CB1954. Clin Cancer Res. 2001;7(9):2662–8. [PubMed] [Google Scholar]
- 134.Martin J, et al. Antibody-directed enzyme prodrug therapy: pharmacokinetics and plasma levels of prodrug and drug in a phase I clinical trial. Cancer Chemother Pharmacol. 1997;40(3):189–201. doi: 10.1007/s002800050646. [DOI] [PubMed] [Google Scholar]
- 135.Napier MP, et al. Antibody-directed enzyme prodrug therapy: efficacy and mechanism of action in colorectal carcinoma. Clin Cancer Res. 2000;6(3):765–72. [PubMed] [Google Scholar]