

Abstract
Elongation factor G (EF-G) plays a crucial role in two stages of mRNA-(tRNA)2 translocation. First, EF-G•GTP enters the pre-translocational ribosome in its intersubunit-rotated state, with tRNAs in their hybrid (P/E and A/P) positions. Second, a conformational change in EF-G’s Domain IV induced by GTP hydrolysis disengages the mRNA-anticodon stem-loops of the tRNAs from the decoding center to advance relative to the small subunit when the ribosome undergoes a backward inter-subunit rotation. These events take place as EF-G undergoes a series of large conformational changes as visualized by cryo-EM and X-ray studies. The number and variety of these structures leave open many questions on how these different configurations form during the dynamic translocation process. To understand the molecular mechanism of translocation, we examined the molecular motions of EF-G in solution by means of molecular dynamics simulations. Our results show: (1) rotations of the super-domain formed by Domains III–V with respect to the super-domain formed by I–II, and rotations of Domain IV with respect to Domain III; (2) flexible conformations of both 503- and 575-loops; (3) large conformational variability in the bound form caused by the interaction between Domain V and the GTPase-associated center; (4) after GTP hydrolysis, the Switch I region seems to be instrumental for effecting the conformational change at the end of Domain IV implicated in the disengagement of the codon-anticodon helix from the decoding center.
Keywords: electron microscopy, flexible fitting, GTP hydrolysis, hybrid state, inter-subunit rotation, ribosome, translation, tRNA
INTRODUCTION
As a GTPase, elongation factor G (EF-G) in bacterial cells, corresponding to EF2 in eukaryotic and archaeal cells, plays a crucial role in polypeptide elongation. During this process, tRNAs and mRNA are translocated by one codon with respect to the ribosome, allowing the next codon to move into the A site and to be recognized by an incoming aminoacyl-tRNA. Translocation may be divided into two steps: translocation of the acceptor arms of tRNAs on the large subunit, followed by mRNA-(tRNA)2 translocation on the small subunit. The first step entails an inter-subunit rotation of the 30S subunit relative to the 50S subunit around an axis normal to their interface, while tRNAs move from their classical (A/A and P/P) into the hybrid (P/E and A/P) positions1–5 and mRNA remains tied to the small subunit. This intermediate state of the complex formed by the ribosome, tRNAs, and mRNA has been termed macrostate II (MS-II),6 to distinguish it from macrostate I (MS-I) characterized by an unrotated small subunit and classic tRNA positions. The second step of translocation entails a rotation of the small subunit head and a backward rotation of the 30S subunit, during which mRNA and the anticodon stem-loops of the tRNAs advance relative to the small subunit.7
The role of EF-G in this process, first thought to be instrumental for inducing inter-subunit rotation,1,8 has been clarified following experimental evidence for spontaneous, factor-free rotation in smFRET9 and cryo-EM studies.10,11 Accordingly, EF-G in the GTP-bound conformation enters the 70S ribosome when the latter is in its MS-II state and temporarily stabilizes this conformation. GTP hydrolysis is triggered on EF-G, and a conformational change in EF-G’s Domain IV disengages the mRNA-anticodon helix from the decoding center, allowing the second step of translocation to proceed.7 These events take place as EF-G undergoes a series of large conformational changes as visualized by cryo-EM8 (summarized in the Supplement to Frank et al.,13 see Fig. 1) and X-ray studies.12
Figure 1.

The conformations of EF-G in various bound states, as observed by both X-ray crystallography and cryo-EM: EF-G•GTP in unbound form (red); EF-G•GTP bound to the ribosome (yellow); EF-G•GDP bound to the ribosome (blue); and EF-G•GDP in unbound form (pink). As summarized by Frank et al.,13 the conformational changes involve a hinge-like movement of the C-terminal domains (III, IV, and V) with respect to the N-terminal domains (I, II, and G′). Reproduced with permission by the National Academy of Sciences, USA.
To date, structural data shedding light on the interaction of EF-G with the ribosome include the reconstruction of a complex of 70S•EF-G•GDPNP, where the non-hydrolyzable GTP analog GDPNP was used to stabilize the state for visualization.8,14 Another complex is composed of 70S•EF-G•GDP•fusidic acid, stabilized immediately after GTP hydrolysis, which was visualized by cryo-EM8 and X-ray crystallography.12 These complexes show differences (i) in the conformation of the 70S ribosome, namely the presence or absence of the intersubunit rotation, (ii) in the configuration of the P-site tRNA being in either the P/P or the P/E state, and (iii) in the relative inter-domain orientations of EF-G.15 The number and variety of these structures leave open many questions on how these different configurations form during the dynamic translocation process.
The dynamics during translocation was recently investigated by an smFRET study of a pre-translocational ribosome complex in the presence of EF-G•GDPNP.16 In this study, the authors found that the ribosome-EF-G complex moves dynamically between the classical non-rotated (MS-I) and rotated state (MS-II). This observation implies that EF-G in the GTP-bound form flexibly fits in two different conformations of the translocating ribosome. Evidently, its conformational flexibility allows EF-G to adapt to different structural contexts within the highly dynamic ribosome.
The conformations assumed by EF-G during the translocation process have been described by comparing the 70S-bound complexes with its unbound form, pointing to an extensive rotation of Domain IV and a rotation between the two super-domains comprising Domains I–II and III–V.8,13 In these studies, either the super-domains were treated as two rigid bodies13 or each domain was treated as a separate rigid body.8 Thus, motions within the domains were left for further investigation. In addition, the large variability observed raises the question if EF-G in isolation can realize spontaneously the whole conformational spectrum. To this end, we studied the molecular motions of EF-G in solution by means of molecular dynamics (MD) simulations. A large conformational range was sampled along 15-ns MD trajectories, showing (1) collective rotations of super-domain III–V with respect to super-domain I–II; (2) rotations of Domain IV with respect to Domain III; (3) flexible conformations of both 503- and 575-loops. It is found that the tip of Domain IV (the 503-loop) sweeps through an arc of more than 50 Å from the compacted ribosome-unbound conformation to the ribosome-bound forms. Evidently, the large conformational variability in the bound form is included through interaction between the GTPase-associated center and Domain V of EF-G, interaction between Domain IV and the tRNA, as well as helix 44 in the 30S subunit. Most importantly, the Switch I region (residues: 37–56) is involved in the super-domain rotation, passing the conformational changes caused by GTP hydrolysis on toward the end of Domain IV, which is located in the vicinity of the codon-anticodon pairing. This observation sheds light on the way GTP hydrolysis on EF-G is involved in translocation.
METHODS
MD simulations
The MD simulations were prepared and performed using the molecular simulation software package AMBER8 (University of California, San Francisco)17 and the Cornell force field.18 The X-ray atomic coordinates of EF-G from Thermotoga maritima were obtained from the Brookhaven Protein Data Bank (PDB code: 1FNM).19 The missing residues in the switch region I were grafted from an EF-G model.20 Net charges on EF-G were neutralized by adding sodium ions around the protein. In addition, an ionic condition of 100 Na+/100 Cl− was used for the simulation system. The system was placed in a water box with a water shell that was at least 10 Å away from the solute to the outer edge of the shell, using the TIP3P water model.21 The water box had a size of 112 × 69 × 76 Å 3 and contained 30,130 water molecules. The system was subjected to an energy minimization and justification of the size of the water box. Three independent simulation runs were performed lasting 15 ns each starting from independent initial velocity distributions (random seeds), see the detailed protocol by Li et al.22 The simulations were done in the NpT ensemble (p = 1 bar, T = 300 K). The equations of motion were integrated with a time step of 2 fs, with SHAKE23 used to restrain the bonds involving hydrogen atoms. A distance cut-off of 9 Å was applied to short-range non-bonded interactions, whereas long-range electrostatic interactions were treated using the particle-mesh Ewald method.24 The Ptraj program17 was used for trajectory analysis.
An atomic model of the Escherichia coli ribosome25 was obtained from a cryo-EM reconstruction of a 70S•EF-G•GDPNP complex (EMD-13638) using the molecular dynamics flexible fitting (MDFF) method,26 as previously described.27 An EF-G crystal structure (PDB code: 1FNM18) was flexibly fitted into a segmented density from EMD-1363 using MDFF in explicit solvent.28 In addition to the secondary structure restraints usually applied in MDFF simulations, harmonic restraints were employed to preserve hydrogen bonds involving EF-G backbone atoms present in secondary structure elements; the new hydrogen-bond restraints resulted in improved stability of EF-G beta sheets during the fitting process. After the EF-G model was refined using a segmented density map, it was incorporated into the ribosome model and the 70S:EF-G complex was further refined into the cryo-EM map using MDFF in explicit solvent.
RESULTS
Motion of the ribosome-unbound EF-G
The dynamics of EF-G in the ribosome-unbound form was examined using MD simulations. The simulations started with an all-atom model, which was built based on the X-ray structure of EF-G (PDB code: 1FNM), onto which the missing fragments in Domain I were grafted from a structure obtained from an EF-G model.19 In a solvated simulation system this atomic model was initially subjected to an energy minimization and a brief equilibration before the start of equilibration simulations. Then, three independent trajectories were obtained lasting for 15 ns each. For each of these trajectories, the beginning conformation was used as a reference for analyzing the conformational changes along the trajectory. The conformational changes in the overall structure were calculated including only the coordinate changes in the Cα atoms along each of the three trajectories, resulting in a maximum root mean-squared deviation (RMSD) of 5 Å [Fig. 2(A)]. The RMSD data imply that the X-ray conformation was neither stably kept nor anywhere closely resumed throughout the MD trajectories. The changes in the overall structure are found to be contributed by the structural components at both the domain level and the loop level. The overall conformational changes include frequent reorientations between the two super-domains [Fig. 2(B)], as well as a motion of Domain IV that is independent from the other domains. Local conformational changes include the Switch I region within Domain I [Fig. 2(C)] and the mobile 503-loop within Domain IV [Fig. 2(D)].
Figure 2.

The RMSD traces of three independent MD simulation trajectories. A: RMSD of entire EF-G. B: RMSD of super-domain II. C: RMSD of the switch region I. D: RMSD of the 503-loop.
At the loop level, the Switch I region within Domain I, containing about 20 residues (residues: 37–56), was only partially ordered in the X-ray structures in both the GTP-bound and the GDP-bound forms, indicating a high flexibility.29,30 This region was modeled in the context of EF-G bound to a translocating 70S ribosome, where it shows up as a large unstructured loop.20 Along the simulation trajectories, the unstructured loop of the Switch I region moves around the starting position, without following an identifiable pattern. Accounting for the Cα atoms, the maximum RMSD reaches 3.8 Å [Fig. 2(C)]. Such a large conformational fluctuation agrees with the biochemical findings that conformational changes in the Switch I region are involved in the exchange of GTP and GDP on EF-G. An interesting question to be asked is if the exchange between GTP and GDP would prompt a rally of conformational changes to other domains of EF-G. Indeed, we observed that several residues (numbers: 50–57) in the Switch I region are positioned within bonding distance to portions of Domains III, and thus their contact may affect the motion of Domain III. Throughout the simulation trajectories, this close contact is maintained, but there is no obvious structural correlation found between the fluctuations of the Switch I loops and the motion of Domain III.
The other highly mobile loop, the 503-loop, is composed of residues 503–506 in Domain IV, showing a large RMSD of their Cα atoms of about 1.7 Å [Fig. 2(D)]. This RMSD refers to only the very local conformational change within that loop, and does not include the collective motion along with the entire Domain IV. The 503-loop loop sweeps over more than 10 Å throughout the trajectories. The re-positioning of this loop is believed to be involved in the tRNA translocation; (see details in Discussion).
Changing orientation between the two super-domains (I–II and III–IV) of EF-G
As observed in previous cryo-EM studies, 70S-bound EF-G is in an extended conformation compared with its unbound X-ray form.8,13 These cryo-EM studies combine the five domains of the protein into two super-Domains: I–II and III–V. When each super-domain is treated as a rigid body, a rotation of super-domain III–V around its X-ray position by about 23° is found in the 70S-bound EF-G whereas super-domain I–II keeps its position unchanged.13 This observation indicated that binding to the ribosome is associated with some major conformational changes in EF-G, but left unclear if EF-G may form this conformation without interacting with the ribosome. In the current study, we re-examined the binding effect, but without treating the domains as rigid bodies. We found that the domain re-orientation, recognized in the previous study, remains the same after removing the rigidity restrains [Fig. 2(B)]. Domains I and II keep a virtually unchanged overall dimension although there are internal changes in the Switch I region. We observe a rotation of super-domain II along the simulation trajectories, defining a dihedral angle (ϕ = 615:Cα-433:Cα-116:Cα-128:Cα, see Fig. 3 for the measurement). The value of ϕ is 59° in the unbound form, as shown in the X-ray structure, and increases to near 99° in the 70S-bound form, as shown by the cryo-EM study.13 This large angle appears to be similar in both the 70S•EF-G•GDP•fusidic acid complex and the 70S•EF-G•GDPNP complex, indicating a substantial change caused by the binding of the factor to the ribosome.
Figure 3.

Illustration of rotation of super-domain III–V. A: Illustration of dihedral angle ϕ which is used to measure the rotation of super-domain III–V. B: Traces of the changes in angle ϕ along three MD simulation trajectories.
Along the simulation trajectories, the angle ϕ frequently varies in a range from 52° to 83° (Fig. 3), fluctuating around the position in the unbound structure. The substantial range of this rotation indicates a major motion of EF-G in the unbound form, which, however, does not quite reach the angle needed to form the position in the ribosome-bound form. A likely reason is the absence, in the simulation systems, of the interaction with the ribosome, which occurs in the experimentally observed 70S ribosome-bound structures. Another likely reason is the limited simulation time which might not allow the observation of the full functional range.
Motion of Domain IV
The major conformational difference between the ribosome-bound and -unbound EF-G, as shown by previous cryo-EM studies, is in the position of Domain IV (Fig. 1). The very end of Domain IV is 30 Å apart between the two conformations. This position is obviously affected by the rotation of super-domain II. In addition, Domain IV also rotates relative to the position of Domain III and V within super-domain II, as shown in Figure 4A. This internal super-domain motion was not observed in the previous fitting structure.8 It is of interest to observe how this extended position of Domain IV is formed. In this study, we designated the position of Domain IV using the distance d (distance between residue 343 in Domain III and 499 in Domain IV) to analyze the motion of Domain IV along three MD simulation trajectories. The results show (Fig. 4) that d varies in the unbound EF-G between 47 Å and 58 Å, fluctuating around the beginning position where it has a distance of 52 Å. Although the very end of Domain IV sweeps over a wide range of about 40 Å across the simulation trajectories (Fig. 4), the most extended position (58 Å) does not reach that in the bound EF-G. Furthermore, we see that the large fluctuation does not occur in the exact direction toward the bound form. Clearly, the direction of Domain IV’s motion is sensitive to the collective rotation of super-domain III–V. A larger rotational angle of super-domain III–V than the upper bound of ϕ, which we observed through the trajectories, would change the direction toward the ribosome-bound position.
Figure 4.

Illustration of dynamics of Domain IV. A: X-ray structure of 70S ribosome-bound EF-G (green)12 and the unbound form (pink).20 B: Snapshots of MD simulation (green and blue) show the range of Domain IV’s motion. C: Superimposition of ternary complex (gray) and EF-G (blue).
It is interesting to look at the large motion of Domain IV when comparing the entire configuration of EF-G with the tRNA•EF-Tu complex. The conformational flexibility in Domain IV of EF-G strikingly resembles the flexibility of its structural mimic in the 70S ribosome-bound aa-tRNA•EF-Tu complex. A superimposition of EF-G onto the aa-tRNA•EF-Tu•GDP ternary complex reveals that the position of Domain IV is equivalent to that of the anticodon stem loop in the aa-tRNA, which is positioned in the A/T-state significantly different from the ribosome-unbound tRNA (Fig. 4). The position of the anticodon stem is confined by the mobile D-stem loop while the T-loop interacts with the GTPase-associated center (composed of ribosomal protein L11 and its binding region in the 23S rRNA).31 This observation again suggests that the extended position of Domain IV in the bound form of EF-G might be the result of its interaction with the ribosome when both Domains IV and V are confined by the binding contexts at the two separately located sites.
Conformational changes in EF-G due to the inter-subunit rotation
Snapshots of EF-G bound to the 70S ribosome have been obtained by cryo-EM studies and X-ray crystallography.8,12 One of these is the cryo-EM reconstruction map of the 70S-EF-G•GDPNP complex, using the non-hydrolyzable analog GDPNP to preserve EF-G in the GTP state.8 In this complex, the 70S ribosome is in a rotated state is associated with a tRNA in the P/E hybrid state. The molecular conformation in this complex was obtained using the MDFF method.26 The MDFF model improves the structure-density matching for EF-G from 0.57 with the starting X-ray structure to 0.74. The conformational changes in EF-G are obvious, with an RMSD of 5.5 Å. In contrast, there are no obvious changes in the conformation of the ribosome from the initial model.
The other snapshot showing the 70S ribosome•EF-G•GDP•fusidic acid complex is available from both cryo-EM and X-ray crystallography.8,12 This state is formed immediately after GTP hydrolysis, as the result of which the intersubunit rotation is reversed, a tRNA occupies the P site, but EF-G stays in the complex due to the presence of fusidic acid. Thus, the two conformations, the MDFF model and the X-ray structure, provide two consecutive states of EF-G: before versus after GTP hydrolysis.
The major conformational differences in the 70S ribosome-bound EF-G•GDPNP and EF-G•GDP•fusidic acid states appear in the degrees of the rotation of super-domain II. By overlapping super-domain I in the two complexes, we find a rotation of super-domain II by about 14° (Fig. 5). In addition to this super-domain rotation, the other important difference between these two EF-G conformations is that the Switch I region is absent in the X-ray structure for the EFG•GDP state,9 implying the existence of large conformational flexibility in this region. As shown in the fitting model, this region appears at the interface between the two super-domains in the EF-G•GDPNP conformation (red piece in Fig. 5). This position makes it obvious that any conformational differences of Switch I region would perturb the relative orientation between the two super-domains.
Figure 5.

Illustration of EF-G’s conformation bound to the ribosome in the post-translocational state (green)12 which is superimposed by EF-G in the conformation bound with the ribosome in the rotated state (blue), as shown in the current MDFF model.
Another major difference in these two EF-G conformations is in the manner of binding between Domain V and the L11 lobe, including helices 43 and 44 of the 23S rRNA and protein L11. The base of A1067 is in a loose bonding distance to Ile631 in Domain V of EF-G-GDP in the ribosome-bound complex,12 but this Domain V-L11 lobe binding situation changes substantially in the 70S ribosome-EF-G•GDPNP complex when the ribosome is in the inter-subunit rotated state. Here the binding network is re-organized such that the A1095 base in the 23S rRNA and protein L11 move into a strong bonding network with Domain V of EF-G•GDPNP, in contrast to the situation in the other state where these elements are distant from EF-G. As shown in Figure 6, in the rotated state of the ribosome, residue 21 in the N-terminal domain of L11 directly contacts residue 639 in Domain V of EF-G.
Figure 6.
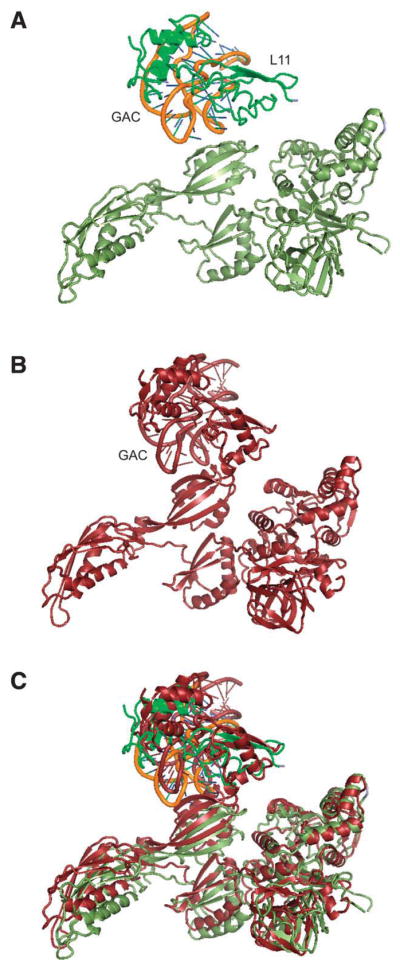
Illustration of EF-G interacting with the L11 lobe when the ribosome is in a non-rotated (upper) and the inter-subunit rotated (middle) states.
Beyond the difference in super-domain rotation observed in the two EF-G conformations, the end loops of Domain IV, the 503-loop and the 575-loop, show local conformational changes, with a separation in the 575-loop positions by 8 Å, and by 4 Å in the 503-loop. Apparently, the current MD simulations enable us to observe this dynamic motion, unlike the previous fitting approach treating the domains as individual rigid bodies.8 The re-positioning of the end loops of Domain IV is associated with the repositioning of the tRNA from the P site to the P/E configuration since the 503-loop interacts with the codon-anticodon binding region in the two states. In addition, the 575-loop of Domain IV also interacts with the ribosome during the tRNA translocation such that residues 578–579 in this loop contact the conserved A1493 residue of 16S rRNA in the decoding center, as shown in the X-ray structure. Evidently, these two loops need to be positioned flexibly so that they can move together with either the tRNA or h44 separately. Our MD simulations of free EF-G show that the distance between residues 503 and 575 in these two loops varies within a range of •4 Å around the initial position in the X-ray conformation. This range would allow the 503-loop to move with the P-site tRNA relatively independently from the other loop when the 575-loop moves, bound to h44. This type of motion is actually illustrated by a recent smFRET study,16 showing that, in the presence of EF-G•GDPNP, the P-site tRNA moves back and forth between the P-site and the hybrid P/E configuration before the translocation is completed.
DISCUSSION
In this study, we have used MD simulations to analyze the conformational variability of EF-G and use this information to understand the conformational changes that are associated with the binding of the factor to the 70S ribosome during translocation. Our results show that an unbound EF-G samples a wide space of conformations, with the most pronounced differences being in the orientation of Domain IV relative to the rest of the molecule. This observation is consistent with the cryo-EM data, as well as a more recent X-ray structure, showing the repositioning of Domain IV in the 70S-bound form. The rotation of super-domain II, evident from a comparison of unbound EF-G with the 70S ribosome-bound form, is observed along the MD simulation samples in a smaller range.
Putting the structural information together, it becomes clear that the function of EF-G in the translocation process relies on its structural flexibility, as seen from two conformations of the ribosome-bound EF-G. EF-G binds to the ribosome in two different conformations: EF-G•GDPNP binds to the ribosome in the state with the inter-subunit rotation; in contrast, EF-G•GDP binds with the ribosome in the state without the rotation. Comparing these two states, the conformational changes in the ribosome are substantial, as summarized by Frank and coworkers.13 Some of these changes are associated with the binding of EF-G. One special binding site in the ribosome with numerous ribosomal factors, including EF-Tu, EF-G and RF3, is the L11 lobe. As a previous study shows,31 when bound to EF-Tu, the L11 lobe has been found in a “closed” position with respect to the main body of the 70S ribosome, compared with its “open” position seen in the pre-translocational ribosome. The position of the L11 lobe, moving from the closed to the open state, is in a range of 10 Å. In addition, there are also some relatively minor, yet crucial, conformational changes within the lobe, namely a substantial rotation of the N-terminal domain of L11, and the flipping of base A1067.22 These local changes are directly involved in the binding between the lobe and the EF-Tu•tRNA complex. One direct binding site is the flexible base of A1067 in H43, which interacts with the T-loop of the aa-tRNA bound with EF-Tu.22,32 When these two components make contact, the base of A1067 is flipped outward from the helix. The base, otherwise, is in the position of stacking with its neighboring nucleotide when the lobe is unbound. Similar to this inter-molecular interaction, we observe the loose involvement of the base of A1067 in the binding to Domain V of EF-G•GDP in the ribosome-bound complex, but a substantially stronger binding in the 70S ribosome•EF-G•GDPNP complex when the ribosome is in the inter-subunit rotated state. In this strong binding situation, protein L11 forms part of the binding network: together with helices H43–H44 they completely wrap around Domain V of EF-G, seemingly with the purpose to lock Domain V in its extensively rotated position. These multiple binding interactions between the L11 lobe and EF-G are obviously stronger than the single binding site in the case of binding of tRNA•EF-Tu, showing a rotation of super-domain III–V by about 14 degrees from the other ribosome-bound position seen in the 70S•EF-G•GDP•fusidic acid crystal structure.12 The strong involvement of the L11 lobe is indicated by the fact that the rotation of super-domain II is affected by its contact with the lobe. This situation changes when EF-G is to be released from the ribosome after the GTP hydrolysis. The loose contact between the L11 lobe and EF-G enables the outgoing EF-G to leave the ribosome. Thus, our observations on the structural differences between the two ribosome-EF-G binding states imply that the GTPase-associated center plays a key role in locking Domain V of EF-G to the position in the ribosome entrance when the ribosome undergoes the inter-subunit rotation. The intrinsic flexibility of EF-G allows these conformational changes to take place, and the interactions with the ribosome are likely to push EF-G to change its conformation to a larger extent than that occurring spontaneously.
Interestingly, we observed that the 70S ribosome-bound EF-G8 shows a conformation close to that of the ternary complex including the A/T aa-tRNA bound with EF-Tu•GTP•kirromycin.31,32 In that cryo-EM study, as well as more recent studies,33,34 the A/T tRNA is found to have a large structural distortion in its D stem and anticodon-stem loop. The A/T tRNA is in a position equivalent to super-domain II of EF-G, and the anticodon-stem loop is at the position of Domain IV of EF-G. The distortion in the A/T-tRNA is similar to the conformational change of EF-G due to the rotation of Domain IV relative to III and V. Thus, it seems that the ternary complex and EF-G share a common mechanism of conformational changes as they bind to the ribosome.
Acknowledgments
The authors thank the National Cancer Institute and the Advanced Biological Computer Center in the NCI for support with their supercomputer resources. They thank Melissa Thomas for her assistance with the illustrations. L.G.T. is a recipient of the European Molecular Biology Organization fellowship. Computer time for MDFF simulations were provided through the National Resources Allocation Committee (MCA93S028).
Grant sponsor: Joachim Frank; Grant numbers: HHMI and NIH R01 GM 55440; Grant sponsor: Klaus Schulten; Grant numbers: NIH P41-RR005969 and NSF PHY0822613.
References
- 1.Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 2.Kim HD, Puglisi JD, Chu S. Fluctuations of transfer RNAs between classical and hybrid states. Biophys J. 2007;93:3575–3582. doi: 10.1529/biophysj.107.109884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munro JB, Altman RB, O’Connor N, Blanchard SC. Identification of two distinct hybrid state intermediates on the ribosome. Mol Cell. 2007;25:505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ermolenko DN, Spiegel PC, Majumdar ZK, Hickerson RP, Clegg RM, Noller HF. The antibiotic viomycin traps the ribosome in an intermediate state of translocation. Nat Struct Mol Biol. 2007;14:493–497. doi: 10.1038/nsmb1243. [DOI] [PubMed] [Google Scholar]
- 5.Fei J, Bronson JE, Hofman JM, Srinivas RL, Wiggins CH, Gonzalez RL., Jr Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc Natl Acad Sci USA. 2009;106:15702–15707. doi: 10.1073/pnas.0908077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank J, Gonzalez RL. Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu Rev Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor DJ, Nilsson J, Merrill AR, Andersen GR, Nissen P, Frank J. Structures of modified eEF2 80S ribosome complexes reveal the role of GTP hydrolysis in translocation. EMBO J. 2007;26:2421–2431. doi: 10.1038/sj.emboj.7601677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J. Locking and unlocking of ribosomal motions. Cell. 2003;114:123–134. doi: 10.1016/s0092-8674(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 9.Cornish PV, Ermolenko DN, Noller HF, Ha T. Spontaneous intersubunit rotation in single ribosomes. Mol Cell. 2008;30:578–588. doi: 10.1016/j.molcel.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agirrezabala X, Frank J. Elongation in translation as a dybamic intergction among the ribosome, tRNA, and elongation factors EF-G and EF-Tu. Q Rev Biophys. 2009;42:159–200. doi: 10.1017/S0033583509990060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Julián P, Konevega AL, Scheres SH, Lázaro M, Gil D, Wintermeyer W, Rodnina MV, Valle M. Structure of ratcheted ribosome’s with tRNAs in hybrid states. Proc Natl Acad Sci USA. 2008;105:16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao YG, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, Ramakrishnan V. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science. 2009;326:694–699. doi: 10.1126/science.1179709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frank J, Gao H, Sengupta J, Gao N, Taylor DF. The process of mRNA-tRNA translocation. Proc Natl Acad Sci USA. 2007;104:19671–19678. doi: 10.1073/pnas.0708517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agrawal RK, Heagle AB, Penczek P, Grassucci RA, Frank J. EF-G-dependent GTP hydrolysis induces translocation accompanied by large conformational changes in the 70S ribosome. Nat Struct Biol. 1999;6:643–647. doi: 10.1038/10695. [DOI] [PubMed] [Google Scholar]
- 15.Agrawal RK, Penczek P, Grassucci RA, Frank J. Visualization of elongation factor G on the Escherichia coli 70S ribosome: the mechanism of translocation. Proc Natl Acad Sci USA. 1998;95:6134–6138. doi: 10.1073/pnas.95.11.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munro JB, Altman RB, Tung CS, Sanbonmatsu KY, Blanchard SC. A fast dynamic mode of the EF-G-bound ribosome. EMBO J. 2010;29:770–781. doi: 10.1038/emboj.2009.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornell WD, Cieplak P, Bayly CL, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins and nucleic acids. J Am Chem Soc. 1995;177:5179–5197. [Google Scholar]
- 19.Laurberg M, Kristensen O, Martemyanov K, Gudkov AT, Nagaev I, Hughes D, Liljas A. Structure of a mutant EF-G reveals Domain III and possibly the fusidic acid binding site. J Mol Biol. 2000;303:593–603. doi: 10.1006/jmbi.2000.4168. [DOI] [PubMed] [Google Scholar]
- 20.Wriggers W, Agrawal RK, Drew DL, McCammon A, Frank J. Domain motions of EF-G bound to the 70S ribosome: insights from a hand-shaking between multi-resolution structures. Biophys J. 2000;79:1670–1678. doi: 10.1016/S0006-3495(00)76416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jorgensen WL, Chandreskhar J, Madura JD, Imprey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 22.Li W, Sengupta J, Rath BK, Frank J. Functional conformations of the L11-ribosomal RNA complex revealed by correlative analysis of cryo-EM and molecular dynamics simulations. RNA. 2006;12:1240–1253. doi: 10.1261/rna.2294806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Gunsteren WF, Berendsen HJC. Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys. 1977;34:1311–1327. [Google Scholar]
- 24.Petersen HG. Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys. 1995;103:3668–3679. [Google Scholar]
- 25.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 26.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trabuco LG, Schreiner E, Eargle J, Cornish P, Ha T, Luthey-Schulten Z, Schulten K. The role of L1 stalk:tRNA interaction in the ribosome elongation cycle. J Mol Biol. 2010;402:741–760. doi: 10.1016/j.jmb.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ævarsson A, Brazhnikov E, Garber M, Zheltonosova J, Chirgadze Y, al-Karadaghi S, Svensson LA, Liljas A. Three-dimensional structure of the ribosomal translocase: elongation factor G from Thermus thermophilus. EMBO J. 1994;13:3669–3677. doi: 10.1002/j.1460-2075.1994.tb06676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czworkowski J, Wang J, Steitz TA, Moore PB. The crystal structure of elongation factor G complexed with GDP, at 2.7 Å resolution. EMBO J. 1994;13:3661–3668. doi: 10.1002/j.1460-2075.1994.tb06675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. Incorporation of amino-acyl-tRNA into the ribosome as seen by cryo-electron microscopy. J Nat Struct Biol. 2003;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- 32.Villa E, Sengupta J, Trabuco LG, LeBarron J, Baxter WT, Shaikh TR, Grassucci RA, Nissen P, Ehrenberg M, Schulten K, Frank J. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc Natl Acad Sci USA. 2009;106:1063–1068. doi: 10.1073/pnas.0811370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FV, IV, Weir JR, Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuette JC, Murphy FV, IV, Kelley AC, Weir JR, Giesebrecht J, Connell SR, Loerke J, Mielke T, Zhang W, Penczek PA, Ramakrishnan V, Spahn CM. GTPase activation of elongation factor EF-Tu by the ribosome during decoding. EMBO J. 2009;28:755–765. doi: 10.1038/emboj.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]