

Abstract
2H-Chromenes were synthesized from salicylaldehyde using potassium vinylic borates in the presence of secondary amines. We synthesized these 2H-chromene derivatives as a part of an ongoing project to develop inhibitors for TGF-β receptors. Potassium vinyl trifluoroborates react with salicylaldehydes at 80 °C in the presence of a secondary amine and produced 2-substituted 2H-chromene derivatives with a 70–90% yield.
Keywords: Chromenes, Potassium organic trifluoroborate, TGF-β inhibitors, Signal transduction
We have a long-term goal to develop novel chemical screens capable of analyzing important biological systems by interfacing libraries of small molecules, in the context of developing zebrafish embryos. Toward this end, we were interested in developing potential new inhibitors of TGF-β signaling using 2-substituted 2H-chromene derivatives.
2H-Chromenes are an important class of oxygenated heterocyclic compounds.1 Many biologically active natural products contain a chromene ring system.2 In recent years, there has been increased interest in the synthesis of 2H-chromenes due to the number of compounds that possess this group, and that show a variety of activities including as antidepresssant, antihypertensive, anti-tubulin, antiviral, antioxidative, activator of potassium channels and inhibition of phosphodiesterase IV or dihydrofolate reductase.3 Based on the importance of these compounds, a number of research groups have developed methodologies to synthesize these compounds. The approaches used include intramolecular cyclization of Wittig intermediates,4 microwave-assisted reaction of salicylaldehyde with enamines,5 catalytic Petasis reaction of salicylaldehydes,6 ring-closing olefin metathesis,7 Baylis–Hillman reaction of 2-hydroxybenzaldehydes with methyl vinyl ketones,8 Claisen rearrangement of propargyl phenol ethers,9 Pd-catalyzed ring closure of 2-isoprenyl phenols,10 electrocyclic ring closure of vinylquinone derivatives,11 and the Ylide annulation reaction.12 Despite the availability of these existing methods for the synthesis of chromene derivatives, there remains a demand for general strategies that can more efficiently provide variously substituted chromene systems.
In the context of our goal to generate small molecule probes to study the biology of key signal transduction pathways, including transforming growth factor beta (TGF-β) pathways, we were interested in developing improved methods for the synthesis of a 2H-chromene library.
In this Letter, we report a practical and highly efficient procedure for preparing diverse chromene derivatives using potassium vinyltrifluoroborate as a substrate for the Petasis reaction in the presence of catalytic amounts of dibenzylamine as a secondary base at 80 °C in dimethyl formide. This procedure is complementary to that of Finn and Kabalka, which in both cases uses boronic acid. The utility of organoboronic acids in organic synthesis has flourished in recent years, particularly through developments in Miyaura–Suzuki coupling,13 allyl-boration,14,15 copper catalyzed arylboronic acid–hetero atom coupling16 and Petasis reaction.17,18 However, these organoboron derivatives have many limitations:19 (i) quantitative analysis and stoichiometric reactions using boronic acids are often difficult because of the rapid equilibrium between the boronic acids and the corresponding cyclic, trimeric anhydrides (boroxines), (ii) the diols utilized to generate stable boronate esters such as catechol, pinacol, and diethanolamine add considerable expense to the overall process and they must be separated from the final product, further increasing the cost of synthesis.
(iii) Boronate esters, as Lewis acids and electrophiles, to elaborate the functionalization of multistep synthesis using nucleophiles, hamper the reaction process. To overcome all these difficulties using organoboron derivatives, the use of potassium organotrifluoroborates was developed. These reagents are easily prepared by the addition of inexpensive KHF2 to various organoboron intermediates,20a–e from organostannate20f and by C–H activation method.20g For our reaction, we purchased all alkenyl trifluoroborates from Aldrich chemicals. All these salts have been described as being very stable at elevated temperature. Potassium organotrifluoroborates are more stable and easier to handle than the corresponding organoboronic acids, and they are more reactive due to the higher nucleophilicity of the organic group on the boron atom. Due to these distinctive features (greater nucleophilicity, ready accessibility, and remarkable stability in air), organotrifluoroborate have been used for many organic reactions, as reported by Kabalka, Molander, Genet and others.21
In this Letter, we report a practical and highly efficient procedure for preparing diverse chromene derivatives, and demonstrate the practical utility of potassium organotrifluoroborate in chromene synthesis.
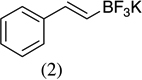
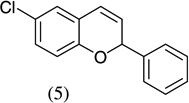
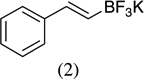
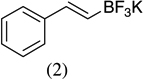
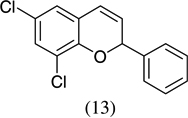
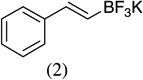
As shown in Scheme 1, salicylaldehyde (1 mmol), vinyl boronic acid (1 mmol) and dibenzylamine (20 mol %) was added to DMF and the solution was stirred at 80 °C for 3 h. The reaction was monitored through thin layer chromatography (solvent system). Unexpectedly we identified a novel spot, and after workup NMR analysis determined it that comprised 2-phenyl chromenes.22 To generalize this methodology, we worked with a variety of substituted salicylaldehydes and boronic acids, as shown in Table 1. We purified all products by column chromatography and the reported yields are the isolated yields. The purity of the products was subsequently established by thin layer chromatography, 1H NMR, 13C NMR and by high resolution mass spectrometry. For all new products, experimental procedures and analytical data are provided in Ref. 22, and for all known compounds we compared the analytical data with our compounds (which were identical).
Scheme 1.

Table 1.
Synthesis of 2-substituted 2H-chromenes from salicylaldehydes in the presence of 20% mol dibenzylamine in DMF at 80 °C
| Entry | Aldehydes | Potassium vinylboronic | Producta | Yieldb (%) |
|---|---|---|---|---|
| 1 | 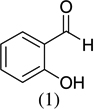 |
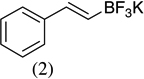 |
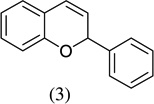 |
51 |
| 2 | 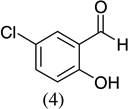 |
 |
 |
89 |
| 3 | 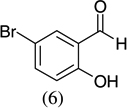 |
 |
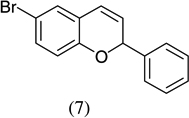 |
71 |
| 4 | 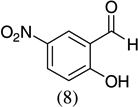 |
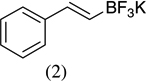 |
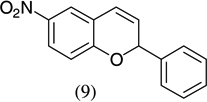 |
90 |
| 5 | 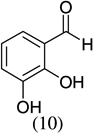 |
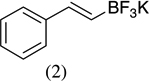 |
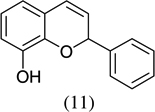 |
54 |
| 6 | 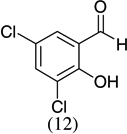 |
 |
 |
83 |
| 7 | 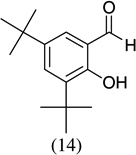 |
 |
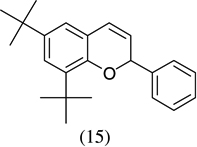 |
78 |
| 8 | 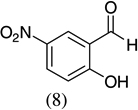 |
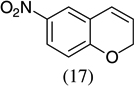 |
65 | |
| 9 | 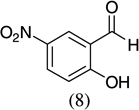 |
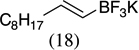 |
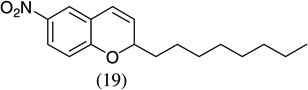 |
85 |
| 10 | 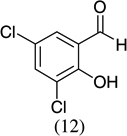 |
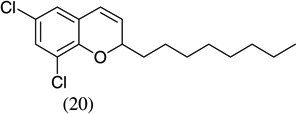 |
87 |
All products exhibited 1H NMR and 13C NMR characteristics in accord with assigned structures and literature values (for known compounds) and high resolution mass spectrometry.
Isolated yields.
A somewhat higher yield is obtained in the case of electron withdrawing groups in the para position of salicylaldehydes, as compared to unsubstituted functional groups. When we changed from potassium trans-styryltrifluoroborate to potassium vinyltrifluoroborate the yields decreased, except in the case of potassium trans-1-decenyltrifluoroborate, where the yield remained similar.
In summary, we report a practical synthesis of chromene derivatives using potassium organotrifluoroborate. Experiments are currently underway to test the biological activity of these 2H derivatives and to determine their utility as inhibitors of TGF-β receptors.
Acknowledgments
The author BCD acknowledges Professor M. Donald Blaufox for his financial support and encouragement and AECOM for startup funding.
References and notes
- 1.(a) Hepworth JD. Pyrans and Fused Pyrans: Synthesis and Applications. In: Katrizky AR, Rees CW, editors. Comprehensive Heterocyclic Chemistry. Vol. 3. Oxford: Pergamon; 1984. p. 737. [Google Scholar]; (b) Milcent R, Chau F. Chimie Organique He’te’rocyclique. Les Ulis, France: EDP Sciences; 2003. p. 434. [Google Scholar]
- 2.(a) Nicolaou KC, Pfefferkorn JA, Cao G-Q. Angew. Chem., Int. Ed. 2000;39:734. [PubMed] [Google Scholar]; (b) Nicolaou KC, Cao G-Q, Pfefferkorn JA. Angew. Chem., Int. Ed. 2000;39:739. [PubMed] [Google Scholar]; (c) Trost BM, Trost FD. J. Am. Chem. Soc. 1998;120:9074. [Google Scholar]; (d) Varma RS, Dahiya R. J. Org. Chem. 1998;63:8038. [Google Scholar]; (e) Miao H, Yang Z. Org. Lett. 2000;2:1765. doi: 10.1021/ol000087t. [DOI] [PubMed] [Google Scholar]; (f) Maggiani A, Tubul A, Brun P. Helv. Chem. Acta. 2000;83:650. [Google Scholar]
- 3.(a) Evans JM, Fake CS, Hamilton TC, Poyser RH, Watts EA. J. Med. Chem. 1983;26:1582. doi: 10.1021/jm00365a007. [DOI] [PubMed] [Google Scholar]; (b) Ashwood VA, Buckingham RE, Cassidy F, Evans JM, Faruk EA, Hamilton TC, Nash DJ, Stemp G, Willcocks K. J. Med. Chem. 1986;29:2194. doi: 10.1021/jm00161a011. [DOI] [PubMed] [Google Scholar]; (c) Bergmann R, Gericke R. J. Med. Chem. 1990;33:492. doi: 10.1021/jm00164a005. [DOI] [PubMed] [Google Scholar]; (d) Gericke R, Harting J, Lues I, Schittenhelm C. J. Med. Chem. 1991;34:3074. doi: 10.1021/jm00114a017. [DOI] [PubMed] [Google Scholar]; (e) Cassidy F, Evans JM, Hadley MS, Haladij AH, Leach PE, Stemp G. J Med. Chem. 1992;35:1623. doi: 10.1021/jm00087a018. [DOI] [PubMed] [Google Scholar]; (f) Pinney D, Kevin G, Arthasary P, Shirali A, Edvardsen K, Chaplin DJ. US2005/024590A1 U.S. Patent. ; (g) Wipf P, Weiner WS. J. Org. Chem. 1999;65:5321. doi: 10.1021/jo990352s. [DOI] [PubMed] [Google Scholar]; (h) Jankun J, Selman SH, Swiercz R, Skrzypczak-Jankun E. Nature. 1997;387:561. doi: 10.1038/42381. [DOI] [PubMed] [Google Scholar]; (i) Van Lommen G, De Bruyn M, Schoven M. J. Pharm. Belg. 1990;45:355–360. [PubMed] [Google Scholar]; (j) Covington AD. Chem. Soc. Rev. 1997:111–126. [Google Scholar]
- 4.Hanamoto T, Shindo K, Matsuoka M, Kiguchi Y, Kondo M. J. Chem. Soc., Perkin Trans. 2000;1:3082. [Google Scholar]
- 5.Varma RS, Dahiya R. J. Org. Chem. 1998;63:8038. [Google Scholar]
- 6.(a) Wang Q, Finn MG. Org. Lett. 2000;2:4063–4065. doi: 10.1021/ol006710r. [DOI] [PubMed] [Google Scholar]; (b) Kabalka GW, Venkataiah B, Das BhaskarC. Synlett. 2004:2194–2196. [Google Scholar]
- 7.Chang S, Grubbs RH. J. Org. Chem. 1998;63:864. doi: 10.1021/jo9712198. [DOI] [PubMed] [Google Scholar]
- 8.Kaye PT, Nocanda XW. J. Chem. Soc., Perkin Trans. 2000;1:1331. [Google Scholar]
- 9.Hlubeck J, Ritchie E, Taylor WC. Tetrahedron Lett. 1969;1369 [Google Scholar]
- 10.Iyer M, Trivedi GR. Synth. Commun. 1990;20:1347. [Google Scholar]
- 11.Parker KA, Mindt TL. Org. Lett. 2001;3(24):3875. doi: 10.1021/ol0167199. [DOI] [PubMed] [Google Scholar]
- 12.Ye LWu, Sun Xiu-L, Zhu Chun-Y, Tang Y. Org. Lett. 2006;8:3853–3856. doi: 10.1021/ol0615174. [DOI] [PubMed] [Google Scholar]
- 13.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. [Google Scholar]
- 14.Roush WR, Adam MA, Walts AE, Harris DJ. J. Am. Chem. Soc. 1986;108:3422–3434. [Google Scholar]
- 15.Flamme EM, Roush WR. J. Am. Chem. Soc. 2002;124:13644–13645. doi: 10.1021/ja028055j. [DOI] [PubMed] [Google Scholar]
- 16.Ley SV, Thomas AW. Angew. Chem., Int. Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 17.Petasis NA, Zavialov IA. J. Am. Chem. Soc. 1997;119:445–446. [Google Scholar]
- 18.Petasis NA, Yudin AK, Zavialov IA, Prakash GKS, Olah GA. Synlett. 1997:606–608. [Google Scholar]
- 19.Molander GA, Ham J. Org. Lett. 2006;8:2031–2034. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For the synthesis of organotrifluoroborates, see: Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J. Org. Chem. 1995;60:3020. Vedejs E, Fields SC, Hayashi R, Hitchcock SR, Powell DR, Schrimpf MR. J. Am. Chem. Soc. 1999;121:2460. Molander GA, Figueroa R. Aldrichim. Acta. 2005;38:49. Darses S, Genêt J-P. Eur. J. Org. Chem. 2003:4313. Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275–286. doi: 10.1021/ar050199q. Chambers RD, Clark HC, Willis CJ. J. Am. Chem. Soc. 1960;82:5298–5301. Takahashi M, Bae C, Hartwig JF. J. Am. Chem. Soc. 2004;126:15334. doi: 10.1021/ja044933x.
- 21.Use of BF3K in organic synthesis. Kabalka GW, Venkataiah B, Dong G. Org. Lett. 2003:3803–3805. doi: 10.1021/ol0351798. Kabalka GW, Reddy M. Organometallics. 2004;23:4519–4521. Kabalka GW, Venkataiah B, Dong G. Tetrahedron Lett. 2004:729–731. Kabalka GW, Dong G, Venkataiah B. Tetrahedron Lett. 2005;46:763–765. Molander GA, Katona BW, Machrouhi FJ. Org. Chem. 2002;67:8416. doi: 10.1021/jo0262356. Darses S, Michaud G, Genet J-P. Eur. J. Org. Chem. 1999:1875. Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MRJ. Org. Chem. 1995;60:3020. Darses S, Michaud G, Genet J-P. Tetrahedron Lett. 1998;39:5045. Molander GA, Biolatto B. Org. Lett. 2002;4:1867. doi: 10.1021/ol025845p. Molander GA, Petrillo DE. J. Am. Chem. Soc. 2006;128:9634–9635. doi: 10.1021/ja062974i. Molander GA, Ribagorda M. J. Am. Chem. Soc. 2003;125:11148–11149. doi: 10.1021/ja0351140. Molander GA, Figueroa R. Org. Lett. 2006;8:75–78. doi: 10.1021/ol052549e. Stefani HA, Cella R, Dörr FA, Pereira CMP, Zeni G, Gomes M., Jr Tetrahedron Lett. 2005;46:563–567. Fang G-H, Yan Z-J, Deng M-Z. Org. Lett. 2004;6:357–360. doi: 10.1021/ol036184e.
- 22.Experimental details: Synthesis of compound 11: A solution of 2,3-dihydroxybenzaldehyde (276.2 mg, 2 mmol), potassium trans-styryltrifluoroborate (420 mg, 2 mmol), was added with dibenzylamine (82 mg, 20 mol %) in anhydrous DMF (10 mL). The reaction mixture was heated at 80 °C overnight. The reaction mixture was quenched with aqueous saturated NH4Cl (8 mL), extracted with CH2Cl2 (10 mL × 3), and combined organic layers were washed with water (10 mL × 3). The water layer was extracted with CH2Cl2 (5 mL × 2). The combined organic layers were dried with Na2SO4, and evaporated. The residue was purified by flash column chromatography using hexane–ethyl acetate (98:2) as the eluent to give a yellow solid, 242.2 mg (54.0%). 1H NMR (300 MHz, CDCl3): δ 7.50–7.39 (m, 5H), 6.87–6.82 (m, 2H), 6.67 (dd, J = 6.9, 2.4 Hz, 1H), 6.60 (dd, J = 9.9, 2.1 Hz, 1H), 5.98 (t, J = 3 Hz, 1H), 5.85 (dd, J = 9.9, 3.3 Hz, 1H), 5.44 (s, 1H). 13C NMR CD3OD: δ 145.04, 141.31, 140.65, 128.49, 126.94, 125.20, 123.97, 122.63, 121.14, 117.80, 116.47, 77.10. ESI MS: calcd for C15H12O2 ([M+H]+) 225.088; found: 225.12.Compound 19: A solution of 2-hydroxy-5-nitrobenzaldehyde (272.92 mg, 1.6 mmol), potassium trans-1-decenyltrifluoroborate (402 mg, 1.6 mmol), was added with dibenzylamine (82 mg, 20 mol %) in anhydrous DMF (8 mL). The reaction mixture was heated at 80 °C overnight. The reaction mixture was quenched with aqueous saturated NH4Cl (8 mL), extracted with CH2Cl2 (10 mL × 3), and the combined organic layers were washed with water (10 mL × 3). The water layer was extracted with CH2Cl2 (5 mL × 2). The combined organic layers were dried with Na2SO4, and evaporated. The residue was purified by flash column chromatography using hexane–ethyl acetate/98:2 as the eluent to give a yellow solid, 402.15 mg (85.1%). 1H NMR(300 MHz, CDCl3): δ 7.99 (dd, J = 8.85, 2.4 Hz, 1H), 7.83 (d, J = 2.7 Hz, 1H), 6.78 (d, J = 8.7 Hz, 1H), 6.40 (dd, J = 10.8, 1.2 Hz, 1H), 5.79 (dd, J = 12, 1.5 Hz, 1H), 5.04–5.01 (m, 1H), 1.82–1.70 (m, 2H), 1.52–1.31 (m, 2H), 1.26 (br s, 10H), 0.875 (t, J = 6.3 Hz, 3H). 13C NMR CD3COCD3: δ 159.56, 141.87, 128.03, 125.64, 122.65, 121.83, 116.48, 77.48, 36.34,32.23, 24.94, 23.04, 14.84. ESI MS calcd for C17H23NO3 ([M▪+−H▪]+): 288.1521; found: 288.120.Compound 20: A solution of 3,5-dichlorosalicyladehyde (382 mg, 2 mmol), potassium trans-1-decenyltrifluoroborate (492 mg, 2 mmol), was added with dibenzylamine (82 mg, 20 mol %) in anhydrous DMF (10 mL). The reaction mixture was heated at 80 °C overnight. The reaction mixture was quenched with aqueous saturated NH4Cl (8 mL), extracted with CH2Cl2 (15 mL × 3), and the combined organic layers were washed with water (10 mL × 3). The water layer was extracted with CH2Cl2 (10 mL × 2). The combined organic layers were dried with Na2SO4, and evaporated. The residue was purified by flash column chromatography using hexane as the eluent to give a yellow solid, 545.00 mg (87.00%). 1H NMR (300 MHz, CDCl3): δ 7.16 (d, J = 2.1 Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 6.33 (dd, J = 9.9, 1.5 Hz, 1H), 5.82 (dd, J = 9.8, 3.6 Hz, 1H), 4.95 (m, 1H), 1.83–1.46 (m, 4H), 1.30 (br s, 10H), 0.93 (t, J = 6.3 Hz, 3H).13C NMR CDCl3: δ 148.22, 129.25, 128.41, 125.73, 124.89, 124.52, 122.80, 122.12, 76.67, 35.71, 32.27, 29.75, 25.24, 23.09, 14.52. ESI MS calcd for C17H22Cl2O ([M▪+−H▪]+): 311.097; found: 311.0903.