

Abstract
The mshA::Tn5 mutant of Mycobacterium smegmatis does not produce mycothiol (MSH) and was found to markedly overproduce both ergothioneine and an ∼15-kDa protein determined to be organic hydroperoxide resistance protein (Ohr). An mshA(G32D) mutant lacking MSH overproduced ergothioneine but not Ohr. Comparison of the mutant phenotypes with those of the wild-type strain indicated the following: Ohr protects against organic hydroperoxide toxicity, whereas ergothioneine does not; an additional MSH-dependent organic hydroperoxide peroxidase exists; and elevated isoniazid resistance in the mutant is associated with both Ohr and the absence of MSH. Purified Ohr showed high activity with linoleic acid hydroperoxide, indicating lipid hydroperoxides as the likely physiologic targets. The reduction of oxidized Ohr by NADH was shown to be catalyzed by lipoamide dehydrogenase and either lipoamide or DlaT (SucB). Since free lipoamide and lipoic acid levels were shown to be undetectable in M. smegmatis, the bound lipoyl residues of DlaT are the likely source of the physiological dithiol reductant for Ohr. The pattern of occurrence of homologs of Ohr among bacteria suggests that the ohr gene has been distributed by lateral transfer. The finding of multiple Ohr homologs with various sequence identities in some bacterial genomes indicates that there may be multiple physiologic targets for Ohr proteins.
INTRODUCTION
Mycobacteria, including Mycobacterium tuberculosis, produce mycothiol (MSH) as the major intracellular thiol (39). Like glutathione in eukaryotes and Gram-negative bacteria, MSH has a wide range of functions that include detoxification of electrophilic compounds, conjugation with antibiotics, and maintenance of the redox potential within the cell (30, 42, 53). The biosynthesis of mycothiol requires five enzymes (Fig. 1), MshA, MshA2, MshB, MshC, and MshD (44). Mutants with mutations of mshA and mshC produce undetectable levels of mycothiol, whereas mshB and mshD mutants make ∼10% and 1% of normal levels, respectively, during exponential growth (42). The mshD mutant also produces high levels of the precursor to mycothiol and two related thiols in which the acetyl residue of mycothiol is replaced by formyl or succinyl residues. The decreased levels of mycothiol in mshB and mshD mutants are thought to involve alternative low-level pathways, whereas MshA, a glycosyltransferase, and MshC, a homolog of cysteine tRNA synthetase, do not have compensating enzymes. Several of the mycothiol mutants of Mycobacterium smegmatis have been characterized and exhibit increased sensitivity to peroxides, alkylating agents, and antibiotics (53).
Fig. 1.

Biosynthesis of mycothiol from 1L-myo-inositol-1-phosphate (Ins-P) via 1-O-(2-acetamido-2-deoxy-α-d-glucopyranosyl)-d-myo-inositol-3-phosphate (GlcNAc-Ins-P), 1-O-(2-acetamido-2-deoxy-α-d-glucopyranosyl)-d-myo-inositol (GlcNAc-Ins), 1-O-(2-amino-2-deoxy-α-d-glucopyranosyl)-d-myo-inositol (GlcN-Ins), and 1-O-[2-[[(2R)-2-amino-3-mercapto-1-oxopropyl]amino]-2-deoxy-α-d-glucopyranosyl]-d-myo-inositol (Cys-GlcN-Ins). AcOH, acetic acid.
Mycothiol has been of special interest due to its apparent requirement for the growth of M. tuberculosis (5, 58). Like the M. smegmatis mutants, M. tuberculosis mutants bearing mutations of the mshB (7) and mshD (6) genes have decreased levels of mycothiol and increased sensitivity to peroxides. Attempts to generate viable mutants with mutations of the native mshA (5) and mshC (58) genes in M. tuberculosis were unsuccessful unless a second functional copy of the gene was present. This suggested that the mshA and mshC genes of mycothiol biosynthesis, like the gene encoding the mycothiol disulfide reductase (mtr, Rv2855), are essential in M. tuberculosis, in accordance with results from transposon site hybridization studies (60). More recently, M. tuberculosis mutants lacking mshA have been isolated (65), indicating that under appropriate conditions, M. tuberculosis can adapt to the loss of mycothiol. Mycothiol is not required for the growth of M. smegmatis, which made this bacterium highly useful in identifying mycothiol biosynthesis genes. Curiously, M. smegmatis mutants deficient in mycothiol synthesis are sensitized to many toxins and antibiotics but are resistant to isoniazid (INH) (55).
A surprising observation with the M. smegmatis mshA transposon mutant used to identify the mshA gene (mshA::Tn5 strain) (43) seemed potentially relevant for its ability to grow without mycothiol. As shown in this study, this mutant dramatically overproduces an ∼15-kDa protein compared to its level in the wild-type strain mc2155. The expression of the unknown protein was even more pronounced when the cells were grown on agar at room temperature for periods longer than a week, which allowed them to enter stationary phase while under constant exposure to oxygen. It was thought that the overexpressed protein might compensate for the lack of mycothiol. In the studies presented here, we identified this protein as organic hydroperoxide reductase (Ohr), further characterized the mutant, purified Ohr, and characterized the substrate specificity, peroxide sensitivity, and reducing systems for Ohr. We also showed that ΔmshA mutants markedly increase their production of ergothioneine.
MATERIALS AND METHODS
Reagents.
5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB), cumene hydroperoxide (CuOOH), tert-butylhydroperoxide (t-BuOOH), lipoamide, NADH, soybean lipoxygenase, insulin, and linoleic acid were from Sigma. Porcine heart lipoamide dehydrogenase (Lpd) was from Calzyme, and M. tuberculosis Lpd and dihydrolipoamide acetyltransferase (DlaT) were kindly provided courtesy of Carl F. Nathan and Ruslana Bryk. All other reagents were from Fisher.
Bacterial strains and culture conditions.
The M. smegmatis strains used are described in Table S1 in the supplemental material. M. smegmatis mc2155 was grown in Middlebrook 7H9 broth (Difco) with 0.05% Tween 80 and supplemented with either OADC (oleic acid, albumin, glucose, and catalase supplement) or 1% glucose. M. smegmatis mc2155 was also grown on Middlebrook 7H9 solid medium (1.8% Difco agar), with 0.5% glycerol supplemented with OADC or 1% glucose. Escherichia coli DH5α was used as the host strain for cloning experiments. Antibiotics were added as needed; 25 μg ml−1 kanamycin and 75 μg ml−1 hygromycin were used for M. smegmatis strains, and 100 μg ml−1 ampicillin, 100 μg ml−1 hygromycin for E. coli strains. Unless otherwise indicated, cultures were incubated at 37°C. The mshA::Tn5 strain is difficult to initiate in culture and requires the presence of OADC. Growth in liquid culture from 5 to 10% inoculums occurs at the same rate as that of the mc2155 strain, but the mshA::Tn5 strain often fails to grow from 1% inoculums. Other growth conditions are detailed for the specific experiments.
Cell extracts for SDS-PAGE.
M. smegmatis strains were grown for 11 days on 100-mm plates containing Middlebrook 7H9 agar and 1.0% glucose at 23°C. The mshA::Tn5 and mshD::Tn5 strains were grown in the presence of 25 μg/ml kanamycin; mshA::Tn5 mshA and mshA::Tn5(pcv125) strains were grown in the presence of streptomycin at 30 μg per ml. Cells were scraped off the plates with a small volume of Middlebrook 7H9 medium and centrifuged. The pellet was extracted by sonication at 5°C with extraction buffer (25 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM 2-mercaptoethanol, and 35 μM each N-α-p-tosyl-l-phenylalanyl chloromethyl ketone and N-α-p-tosyl-l-lysine chloromethyl ketone protease inhibitors). The extract was centrifuged at 16,100 × g for 15 min at 4°C, and the soluble fraction used for protein analysis by SDS-polyacrylamide gel electrophoresis (PAGE).
Analysis of thiols.
Thiols were analyzed by monobromobimane (mBBr) labeling and high-performance liquid chromatography (HPLC) as previously described (7, 17), except for ergothioneine, lipoic acid, and lipoamide. The bimane derivative of ergothioneine has a low fluorescence with mBBr and was incompletely resolved from other bimane-labeled peaks using method 1, a reversed-phase chromatography using sodium acetate buffer (17). The asymmetric peak at 14.8 min was purified and rechromatographed using method 2 chromatography, an ion-pairing program used for coenzyme A (CoA) analysis (17). The 14.8-min peak generated 2 to 5 peaks in method 2, one of which coeluted with authentic ergothioneine-methylbimane (ergothioneine-mB) standard at 7.1 min. The ergothioneine content of the cells was calculated from the fraction of total peak fluorescence (∼25 to 50%) that coeluted with ergothioneine-mB in method 2. The ergothioneine-mB peak was verified by mass spectrometry, showing an MH+ 420 m/z ion (electrospray-positive mode) as previously described (14).
Enzymatically reduced lipoamide and lipoic acid were derivatized at pH 10, due to their high thiol pKa (10.7 for both [29]), to generate their authentic bis-bimane derivatives. Lipoamide-(mB)2 and lipoate-(mB)2 were made by reacting 1 mM reduced lipoamide or lipoic acid with 4 mM mBBr in a solution of 20 mM sodium borate, pH 10, in 50% acetonitrile–water. The reaction mixture was incubated for 10 min at 60°C and acidified with 10 mM methanesulfonic acid (Fluka). Lipoamide-(mB)2 and lipoate-(mB)2 were analyzed in method 1 chromatography (17) with buffer A (0.25% acetic acid) adjusted to pH 4.0 with NaOH and acetonitrile substituted for methanol as buffer B; lipoate-(mB)2 and lipoamide-(mB)2 eluted at 32 and 38.5 min, respectively.
A concentrated cell extract was made to assay lipoic acid and lipoamide in M. smegmatis exponential-phase cultures. M. smegmatis mutants and mc2155 were cultured to an A600 value of 1.4 to 2 in Middlebrook 7H9 medium with 0.4% glucose and 0.05% Tween 80. The cells were pelleted by centrifugation of 20 ml of culture at 4°C and 4,000 × g. The cells were extracted in 1 ml of 50% acetonitrile–water without buffer at 60°C for 15 min. The extract was cleared by centrifugation at 13,000 × g for 5 min, and the supernatant was dried in a SpeedVac. The pellet was dissolved in 200 μl of lipoamide dehydrogenase assay buffer (100 mM NaCl, 1 mM EDTA, 50 mM NaPO4, pH 7.0), and the insoluble material was removed by centrifugation at 13,000 × g for 5 min. A 100-μl aliquot of extract was reduced with 25 μg of lipoamide dehydrogenase in the presence of 1 mM NADH at room temperature. The lipoic acid and lipoamide contents were assayed by mixing a 10-μl aliquot with 2 μl of 100 mM Na borate, pH 10, and 1 μl of 20 mM mBBr (in dimethyl sulfoxide [DMSO]) and incubating in the dark at 23°C for 15 min. This sample was diluted into 10 mM methanesulfonic acid for HPLC assay. Control experiments showed that the reduction of lipoic acid and lipoamide was complete after 30 min. The recovery of lipoic acid or lipoamide spiked at 5 μM into the 50% warm acetonitrile–water extract was ∼80%.
Identification of Ohr.
The M. smegmatis mshA mutant with the mshA::Tn5 mutation was grown on Middlebrook 7H9 agar with 1% glucose as described above for SDS-PAGE analysis, and the overexpressed protein was purified to ∼85% purity using stepwise ammonium sulfate precipitation and gel filtration with Sephadex G100 as described below. The purified protein was separated on a 15% Tris–HCl gradient SDS-PAGE gel and electroblotted onto a polyvinylidene difluoride membrane according to the manufacturer's instructions (Bio-Rad). Staining with Coomassie blue revealed two bands of approximately equal intensity at ∼15 kDa. Both bands were submitted to the UCSD Division of Biological Sciences Protein Sequencing Facility.
Assay of MSH biosynthesis enzymes and Mca.
MshA (44), MshB (40), MshC (59), MshD (31), and Mca (41) activities were assayed using cell extracts of mid-log phase M. smegmatis mc2155 and the mshA::Tn5 and mshA(G32D) strains as described in the referenced publications.
Generation of mshA(G32D) mutant.
We produced a mutant with an unmarked single-base-change mutation in the mshA gene in M. smegmatis mc2155 that reproduced the mutation observed (G32D) in mycothiol chemical mutant strain 49 (43). A 1,366-bp fragment, including 765 bp upstream and 588 bp downstream of the desired mutation, was amplified from M. smegmatis genomic DNA using primers smegAreg up (5′-GGATCCGTCTCGATGGCCTCGCACGT-3′) and smegAreg down (5′-AAGCTTGGGAAGCCCGAACACCGCGC-3′), containing BamHI and HindIII restriction sites, respectively, for cloning into p2NIL (49). The fragment was amplified from M. smegmatis chromosomal DNA using Platinum Taq and cloned into TOPO pCR4 (Invitrogen). After verification of the sequence with the universal primers T3 and T7, the construct was amplified with site-directed (underlining) mutagenesis primers mutmshA1 (5′-GGCACCGGCGACGCGGACGGCATGAACG-3′) and mutmshA2 (5′-CCGTGGCCGCTGCGCCTGCCGTACTTGC-3′) using Pfu Turbo DNA polymerase. The amplified plasmids were digested with DpnI to remove the parent template DNA, and the remaining mutant construct was transformed into E. coli DHAα. Kanamycin-resistant transformants were selected and sequenced with T3 and T7 primers to confirm mutation. Then, the mutated construct was digested with BamHI and HindIII and the insert was ligated to p2NIL (49), also digested with BamHI and HindIII. After cloning of the mutated insert into p2NIL was confirmed, the hyg, lacZ, and sacB fragment from pGOAL19 (49) was cloned into the p2NIL PacI restriction site. The resulting construct was transformed into M. smegmatis mc2155 by electroporation, and transformants were selected on Middlebrook 7H9 agar with kan/hyg and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (49). Single-crossover mutant colonies (blue) were grown and plated on sucrose-X-Gal plates to select double-crossover mutants (white). The mutants were confirmed to be kanamycin sensitive and isoniazid resistant. The M. smegmatis colonies were further confirmed to be mutants by amplifying the genomic DNA with primers smegAreg up and smegAreg down and sequencing the fragment.
Sensitivity to peroxide.
M. smegmatis mc2155 and mycothiol mutants were cultured in Middlebrook 7H9 medium with 0.05% Tween 80 and 0.4% glucose at 37°C for 17 days. This culture condition generates overexpressed levels of Ohr in the mshA::Tn5 mutant. The cells were harvested by centrifugation and were resuspended at an A600 of 0.1. The cells (2 ml) were aliquoted into 4-ml culture tubes containing dilutions of cumene hydroperoxide (80%; Aldrich) and incubated at 37°C for 2 h. The cells were immediately diluted into ice-cold phosphate-buffered saline, and serial dilutions were plated on Middlebrook 7H9 agar containing 0.05% Tween 80 and 10% OADC (Difco). The colonies on the plates (n = 3) were counted after 3 to 5 days of incubation at 37°C.
Sensitivity to isoniazid.
The sensitivity of M. smegmatis mycothiol mutants to isoniazid (INH) was assayed on agar plates containing various levels of isoniazid. Overnight cultures (5 ml) of M. smegmatis strains mc2155 and Tn1 (MshC−) (55) and the mshD::Tn5 (31, 45), Myco504 (mshB::aph) (54), mshA(G32D) (this study), and mshA::Tn5 (43) mutants were grown to an A600 of ∼1 in Middlebrook 7H9 medium containing 0.4% glucose and 0.4% glycerol in 15-ml polypropylene tubes. The cultures were diluted to an A600 of 0.06 in Middlebrook 7H9 medium without a carbon source, and 10-μl aliquots of serial dilutions were spotted on plates containing Middlebrook 7H9 medium with 1.5% Bacto agar, 10% OADC with INH from 0 to 600 μg per ml. Colonies on plates with 0 to 30 μg per ml INH were counted after 2 to 3 days of incubation at 37°C. Plates with 600 μg per ml INH contained smaller colonies that were counted on days 7 and 8, and those with 30 to 300 μg per ml were counted on days 4 and 6.
Purification of Ohr.
The M. smegmatis mshA mutant mshA::Tn5 was grown for 15 days on 40 100-mm plates containing 7H9 agar, 0.05% Tween 80, 1% glucose, and 25 μg per ml kanamycin at 23°C. Fresh 7H9 medium was added to the plates, the cells scraped off, and the suspension centrifuged to generate ∼18 g (wet weight) of cells. The pellet was frozen at −70°C until used for extraction and purification by ammonium sulfate fractionation, gel filtration chromatography, and reversed-phase high-performance liquid chromatography as described in the supplemental material.
Assay of Ohr activity.
Assays of Ohr activity were performed in a 100-μl quartz cuvette with a 1-cm path length, and absorbance measured in a Beckman DU640 spectrophotometer. The buffer was 50 mM sodium phosphate, pH 7, 100 mM NaCl, and 1 mM EDTA. Lipoamide and NADH stock solutions were made at 10 mM in DMSO and water, respectively. Porcine heart lipoamide dehydrogenase stock solution (9 μM in 50 mM sodium phosphate, pH 7, 100 mM NaCl) was stored at −70°C. To 90 μl of buffer, 1.6 μl (160 μM final concentration) lipoamide and 3.2 μl (320 μM) NADH were added. Lipoamide dehydrogenase (6 μl) was then added to convert lipoamide to dihydrolipoamide using one equivalent of NADH, and the reaction was completed instantaneously based on the decrease in A340. After 2 min, 2 μl of Ohr from a stock solution (or 50 mM HEPES, pH 7.5, 100 mM NaCl as a control) was added. After five additional minutes to allow for reduction of Ohr, the peroxide substrate was added to start the reaction (see the supplemental material for additional detail). Absorbance was recorded periodically to follow the loss of NADH. The initial reaction rates were calculated as the rate with Ohr minus the control rate.
Reduction of CuOOH by Ohr/DlaT/Lpd.
Assays of the reduction of CuOOH by Ohr, DlaT, and Lpd were performed similarly to Ohr activity assays in a 100-μl cuvette. The buffer contained 50 mM sodium phosphate, pH 7, 100 mM NaCl, and 1 mM EDTA. The enzyme stock solution concentrations were as follows: Ohr, 700 μM; M. tuberculosis DlaT, 65 μM; and M. tuberculosis Lpd, 99 μM. Final concentrations of 160 μM NADH, 2 μM Lpd, 2 μM DlaT, and 100 μM CuOOH were added to the buffer. Ohr at 40 nM was added last to start the reaction. The controls lacked DlaT or Lpd, and a comparison assay contained 320 μM NADH, 160 μM lipoamide, 5 μM Lpd, 100 μM CuOOH, and 40 nM Ohr. The reaction was measured by following the decrease in A340. Negative results for the reduction of Ohr by MSH and thioredoxin are described in the supplemental material.
Phylogenetic analysis.
GenBank was searched with Blastp 2.2.23 (1) for homologs of M. smegmatis Ohr. The phylogenetic tree for selected Ohr sequences was generated using SeaView 4.2.6 (21) for alignment with ClustalW2 (32) and implementing PhyML version 3.0.1 (23) using the Dayhoff evolutionary model (12) and default parameters.
RESULTS
The mshA::Tn5 mutant of M. smegmatis overexpresses a 15-kDa protein.
When the mshA::Tn5 mutant was grown on agar at 23°C for over 1 week or in liquid culture at 37°C for over 21 days and the cells harvested, lysed, and analyzed for protein by SDS-PAGE, an ∼15-kDa protein band constituted ∼30% of the total protein (Fig. 2A, lane 2). This protein band was not prevalent in the wild-type strain mc2155 grown under the same conditions (Fig. 2A, lane 1). Another Tn5 transposon mutant, mshD::Tn5, that produces low levels of mycothiol and a mycothiol homolog in which the acetyl group is replaced by the formyl group, also failed to overproduce the 15-kDa protein (Fig. 2A, lane 5). Complementation of mshA::Tn5 with M. tuberculosis mshA in the pcv125 vector to generate strain mshA::Tn5c eliminated the prominent 15-kDa band on a protein gel of the cell extract (Fig. 2B, lane 3) and restored MSH to the wild-type level. However, when only the empty vector was used for complementation, overproduction of the 15-kDa protein persisted (Fig. 2B, lane 4). This indicates that either MSH or MshA functions to repress the expression of the gene encoding the 15-kDa protein. The mshA chemical mutant strain 49 lacks MSH (46) and contains a G32D mutation in the gene encoding MshA but does not overproduce the 15-kDa protein (Fig. 2A, lane 3). To demonstrate that chemical mutant strain 49 does not contain additional mutations responsible for its phenotype, we produced an mshA(G32D) strain by site-directed mutagenesis to replace the MshA protein with the G32D MshA protein. The mshA(G32D) strain also did not overexpress the 15-kDa protein (Fig. 2A, lane 4) and did not produce MSH or its precursors. This indicates that it is not MSH or its precursors but the MshA protein and its G32D mutant that function to repress the expression of the 15-kDa protein.
Fig. 2.

One-dimensional SDS-PAGE analysis. (A) Overexpression of ∼15-kDa protein in M. smegmatis. Std, molecular mass standards (kDa); lane 1, wild-type mc2155; lane 2, mshA::Tn5 strain; lane 3, strain 49 [mshA(G32D) chemical mutant]; lane 4, mshA(G32D) strain with site-directed mutation; lane 5, mshD::Tn5 strain (MshD-deficient transposon mutant). (B) Regulation of ohr expression by mshA. Std, molecular mass standards (kDa); lane 1, wild-type mc2155; lane 2, mshA::Tn5 strain; lane 3, mshA::Tn5c (mshA::Tn5 mutant complemented with pcv125 containing M. tuberculosis mshA); lane 4, mshA::Tn5(pcv125) (empty vector) strain. (C) Purification of Ohr: Std, molecular mass standards (kDa); lane 1, crude extract run with 2-mercaptoethanol; lane 2, purified Ohr run with 2-mercaptoethanol; lane 3, purified Ohr run without 2-mercaptoethanol; lane 4, Ohr treated with diamide; lane 5, Ohr treated with dithiothreitol and then with N-ethylmaleimide. For the assays whose results are shown in panels A and B, cells were grown on Middlebrook 7H9 agar with 1% glucose and 0.05% Tween 80 for 1 day at 37°C followed by 10 days at 23°C.
Overexpressed protein identified as Ohr.
The 15-kDa protein was purified by stepwise ammonium sulfate precipitation and gel filtration over Sephadex G100 to ∼85% purity. On gel electrophoresis, there were two bands of approximately equal amounts at 15 kDa (see below). The N-terminal sequence of the upper band was determined to be TTSTDNVLYTAKTHT. The lower band sequence was identical to the upper band to 14 amino acids, TTSTDNVLYTAKTH, with the 15th amino acid unable to be called. The sequence corresponds to that encoded by the open reading frame MSMEG_0447, annotated as an organic hydroperoxide resistance protein (Ohr).
Activities of mycothiol enzymes other than MshA were little influenced by loss of MSH or overproduction of Ohr in M. smegmatis mshA mutants.
The enzyme activities in extracts of exponential-phase cells of strain mc2155 and the mshA::Tn5 and mshA(G32D) mutants were measured (Table 1). MshA activity was undetectable in the two mshA mutant strains. No marked differences in the activities of the other mycothiol biosynthesis enzymes (MshB, MshC, and MshD) compared to the levels in the parental strain (mc2155) were seen. Also, no major differences were seen in the activity of mycothiol S-conjugate amidase (Mca), an enzyme involved in MSH catabolism (8) and MSH-dependent detoxification reactions (56). The results indicate that the activities of these proteins are not controlled by MSH or the regulatory system involved in ohr expression.
Table 1.
Activity of mycothiol biosynthesis enzymes and mycothiol S-conjugate amidase in M. smegmatis mshA mutants
| Strain or genotypea | Mean activityb ± SD (nmole/min/mg protein, n = 3) |
||||
|---|---|---|---|---|---|
| MshA | MshB | MshC | MshD | Mca | |
| mc2155 | 0.40 ± 0.05 | 1.9 ± 0.3 | 1.6 ± 0.1 | 8.0 ± 0.3 | 37 ± 6 |
| mshA::Tn5 | ≤0.06 | 2.0 ± 0.3 | 2.7 ± 0.3 | 4.6 ± 1.6 | 42 ± 6 |
| mshA(G32D) | ≤0.01 | 1.2 ± 0.05 | 2.4 ± 0.1 | 4.9 ± 0.7 | 45 ± 3 |
M. smegmatis strains were cultured for 16 h (A600 ∼ 1) in Middlebrook 7H9 medium supplemented with 1% glucose and 0.05% Tween 80.
MshA saturating conditions, 0.5 mM 1L-Ins-1-P and 0.5 mM UDP-GlcNAc, 17% maximum conversion; MshB saturating conditions, 1 mM GlcNAc-Ins, 3% maximum conversion; MshC saturating conditions, 1 mM GlcN-Ins, 0.5 mM Cys, 1 mM ATP, 6% maximum conversion based on GlcN-Ins or 11% based on Cys; MshD nonsaturating conditions, 40 μM Cys-GlcN-Ins, 250 μM acetyl-CoA, 63% maximum conversion; Mca saturating conditions, 0.5 mM mycothiol-methylbimane, 12% maximum conversion. See Fig. 1 legend for abbreviations.
Marked elevation of ergothioneine production correlates with lack of MSH but not with overproduction of Ohr.
Thiol levels were measured in the mshA mutants to ascertain what changes in other thiols accompanied the loss of MSH and enhanced production of Ohr (Table 2). No significant differences were seen between mc2155 and the mshA mutants in cysteine or coenzyme A content. Unbound lipoic acid and lipoamide levels were also measured since these represent possible reductants for Ohr; both were below measureable levels. Prior determinations of lipoic acid in bacteria have measured the amount released by acid hydrolysis of cell pellets and reflect protein-bound lipoyl residues in amounts as high as ∼0.2 μmol per g (dry weight) (27).
Table 2.
Thiol levels in wild-type M. smegmatis mc2155 and mshA mutants
| Strain or genotype and growth phasea | Mean level ± SD (μmol/g [dry wt], n = 3) |
|||||
|---|---|---|---|---|---|---|
| MSH | Cysteine | Coenzyme A | Lipoic acidb | Lipoamideb | Ergothioneine | |
| mc2155 | ||||||
| Exponential | 8.8 ± 0.2 | 0.080 ± 0.005 | 1.8 ± 0.1 | <0.001 | <0.001 | 0.053 ± 0.01 |
| Stationary | 2.4 ± 0.3 | 0.043 ± 0.004 | 1.2 ± 0.1 | ND | ND | 0.11 ± 0.002 |
| mshA::Tn5 | ||||||
| Exponential | <0.003 | 0.082 ± 0.004 | 1.4 ± 0.1 | <0.001 | <0.001 | 1.4 ± 0.1 |
| Stationary | <0.003 | 0.027 ± 0.001 | 1.0 ± 0.3 | ND | ND | 0.52 ± 0.02 |
| mshA(G32D) | ||||||
| Exponential | <0.003 | 0.095 ± 0.004 | 1.8 ± 0.2 | <0.003 | <0.003 | 1.85 ± 0.020 |
| Stationary | <0.003 | 0.057 ± 0.007 | 1.2 ± 0.2 | ND | ND | 0.85 ± 0.06 |
In the exponential phase, growth was at 37°C in liquid Middlebrook 7H9 medium, 1% glucose, 0.05% Tween 80 for 16 h (A600 ∼ 1). In the stationary phase, growth was as for exponential growth but for 48 h (A600 > 2.5).
Unbound forms. ND, not determined.
The unexpected result was that ergothioneine increased 26-fold in exponential-phase cells and 5-fold in stationary-phase cells of the mshA::Tn5 mutant relative to the levels in mc2155 (Table 2). Ergothioneine was thought to be produced only by mycobacteria and fungi (19, 20) but is imported in eucaryotes via the OCTN1 transporter (22). It has long been speculated to function as an antioxidant (26), although recent studies have led to contradictory conclusions (16, 52). The function of ergothioneine in mycobacteria has been little studied, but the recent identification of the biosynthetic genes in M. smegmatis paves the way for more detailed studies and suggests that ergothioneine production in bacteria may be more widespread than previously recognized (61). The enhanced production of ergothioneine in mshA::Tn5 is not correlated with Ohr upregulation, since the mshA(G32D) strain shows even greater elevations, 35-fold for exponential-phase cells and 8-fold for stationary-phase cells (Table 2). The mshC::Tn5 transposon mutant strain, generated and isolated like the mshA::Tn5 mutant (55), was also shown to produce undetectable levels of MSH (<0.003 μmol per g [dry weight]) but to have ∼20-fold-elevated ergothioneine levels during exponential growth compared with the levels in the wild-type strain. Upregulation of ergothioneine production thus appears to be associated with loss of MSH.
Ohr overexpression confers increased resistance to CuOOH and enhanced resistance to isoniazid.
When cells were grown for 18 days in liquid culture and treated with 5 mM CuOOH, the wild-type strain showed ∼25% loss of viability (Fig. 3A), whereas the mshA(G32D) mutant strain showed ∼98% loss of viability. The Ohr-overproducing mshA::Tn5 mutant displayed an intermediate level of loss (∼72%). The overproduction of Ohr provides enhanced protection against CuOOH but does not provide protection equivalent to that in the wild-type strain with its full complement of MSH but normal level of Ohr. This suggests that there is an additional MSH-dependent process which contributes to the protection against CuOOH in the wild-type strain.
Fig. 3.

(A) Resistance of M. smegmatis to CuOOH is dependent in part upon Ohr. Cells were cultured to late stationary phase (17 days) in Middlebrook 7H9 medium. Cells were then suspended in the same medium at an A600 of 0.1 and incubated with CuOOH for 2 h at 37°C without shaking, after which they were plated on Middlebrook 7H9 agar with OADC supplement. The mshA(G32D) strain lacking MSH and not overproducing Ohr is most sensitive to CuOOH. The mshA::Tn5 strain that lacks MSH but overproduces Ohr is more resistant to CuOOH but not as resistant as the wild-type strain mc2155, which produces MSH but does not overproduce Ohr. (B) Sensitivity to INH requires a low level of MSH and is decreased by overproduction of Ohr. The M. smegmatis strains were cultured overnight at 37°C in tubes containing Middlebrook 7H9 medium. The exponential-phase cells were plated on Middlebrook 7H9 agar supplemented with OADC and various levels of isoniazid. Wild-type M. smegmatis mc2155 (100% MSH production) and MshB-defective strain Myco504 (mshB::aph, 10% MSH) are maximally sensitive to INH, and MshC-deficient strain I64 [mshC chemical mutant, mshC(L205P), 1% MSH] is sensitive to INH, whereas strain Tn1 (MshC-deficient Tn611 mutant, 0.0% MSH) and site-directed MshA-deficient mutant mshA(G32D) (0.0% MSH) are INH resistant, and Ohr-overproducing mshA::Tn5 transposon mutant (0.0% MSH) is highly resistant to INH.
When various cell strains were grown in liquid medium overnight and plated on isoniazid-containing agar, their survival was dependent upon both the MSH content and the Ohr content (Fig. 3B). Maximum sensitivity to killing by INH was seen in the wild-type mc2155 and Myco504 (mshB::aph) (54) strains producing ≥10% of the normal MSH level. Even the mshC chemical mutant strain I64 (55) generating 1% of normal MSH exhibited only slightly enhanced resistance. However, the MshC-deficient strain Tn1 (55) and the mshA(G32D) strain that produce no detectable MSH are an order of magnitude more resistant to INH. Most resistant of all was the mshA::Tn5 mutant, lacking MSH and having markedly elevated Ohr, which was more than two orders of magnitude more resistant to INH than the parent strain. These results can be understood in terms of current ideas regarding the activation of the prodrug INH. INH is converted to an active INH adduct with NAD (57) by KatG (67), and the development of cellular resistance to INH has been the subject of extensive studies (68). Studies of KatG-mediated production of the adduct (IN-NAD) in vitro are consistent with an oxidative step promoted by oxygen, H2O2, or t-BuOOH leading to loss of HN NH and generation of the isonicotinyl radical IN· that adds to NAD+ to generate IN-NAD+· (66). In the in vitro aerobic process, the latter undergoes a one-electron reduction by superoxide anion to generate the IN-NAD adduct. The results for the cellular process in M. smegmatis are generally consistent with this mechanism since overproduction of Ohr would reduce peroxide levels in the cell and inhibit the first step. The requirement for MSH to maximize inhibition by INH suggests that MSH or its thiolate ion may be the cellular reductant for the second step of activation in M. smegmatis. The in vitro studies have been generally conducted in the absence of added thiols. In the absence of MSH [in the mshA(G32D) (this study) and mshA::Tn5 (43) mutants] (Fig. 3), another less available or less reactive reductant may replace MSH, but the elevated resistance to peroxide in the mshA::Tn5 strain appears to enhance INH resistance by limiting peroxide availability.
NH and generation of the isonicotinyl radical IN· that adds to NAD+ to generate IN-NAD+· (66). In the in vitro aerobic process, the latter undergoes a one-electron reduction by superoxide anion to generate the IN-NAD adduct. The results for the cellular process in M. smegmatis are generally consistent with this mechanism since overproduction of Ohr would reduce peroxide levels in the cell and inhibit the first step. The requirement for MSH to maximize inhibition by INH suggests that MSH or its thiolate ion may be the cellular reductant for the second step of activation in M. smegmatis. The in vitro studies have been generally conducted in the absence of added thiols. In the absence of MSH [in the mshA(G32D) (this study) and mshA::Tn5 (43) mutants] (Fig. 3), another less available or less reactive reductant may replace MSH, but the elevated resistance to peroxide in the mshA::Tn5 strain appears to enhance INH resistance by limiting peroxide availability.
Purification of Ohr.
Ohr was purified from cells harvested from plates. Following ammonium sulfate fractionation, the protein eluted on Sepharose G100 at a volume corresponding to a native molecular weight of 34 ± 3 kDa (mean ± standard deviation), indicating that the native protein is a dimer. The central peak of activity from the G100 column contained minor impurities and was purified to homogeneity by preparative HPLC on a reversed-phase C8 column. The purified protein gave a single band on SDS-PAGE without treatment with 2-mercaptoethanol (Fig. 2C, lane 3) but yielded two bands (Fig. 2C, lane 2) when 0.7 M 2-mercaptoethanol was included in the SDS sample buffer; these corresponded to the two strong bands evident in the crude extract that was similarly treated (Fig. 2C, lane 1) and are presumed to reflect partial conversion of Ohr to mixed disulfides with 2-mercaptoethanol under aerobic oxidation. When the native enzyme was titrated with DTNB at pH 8, it showed little or no reaction (0.02 equivalents thiol per mol Ohr), but in the presence of 6 M guanidine hydrochloride, the titration gave 1.60 ± 0.01 (n = 3) equivalents of thiol per mol Ohr. Thus, the two cysteine residues (C58 and C122) are predominately in the reduced state in the isolated protein but are unreactive toward DTNB. Treatment of the purified Ohr with diamide prior to SDS-PAGE produced no change (Fig. 2C, lane 4) from the purified protein (Fig. 2C, lane 3), whereas treatment with dithiothreitol followed by N-ethylmaleimide generated product eluting at an apparently higher molecular weight (Fig. 2C, lane 5). Diamide is ineffective in converting Ohr to the disulfide form, causing <5% change in thiol content, as detected by DTNB titration in 6 M guanidine hydrochloride on samples taken during a 2-h incubation of 694 μM Ohr with 770 μM diamide in 25 mM HEPES, pH 7.5, and 100 mM NaCl at 23°C. It appears that small neutral reactants (2-mercaptoethanol/oxygen and N-ethylmaleimide) can access the buried thiols of Ohr more readily than larger or charged reactants (diamide and DTNB), although inherent reactivity toward thiols is likely also important.
Assay of Ohr and kinetics of peroxide reductions.
Ohr activity with peroxides was measured by following the change in A340 in an assay employing dihydrolipoamide to reduce Ohr and dihydrolipoamide dehydrogenase plus NADH to reduce lipoamide (48) (see Fig. S1A in the supplemental material), analogous to the assay used with the AhpC/AhpD-dependent peroxidase system (4). The reduction of H2O2 by Ohr followed simple Michaelis-Menten kinetics (see Fig. S1B in the supplemental material), with a kcat value of 21 ± 1 s−1 and a Km value of 235 ± 7 mM, generating the kcat/Km value given in Table 3. The reduction of cumene hydroperoxide (CuOOH), tert-butyl hydroperoxide (t-BuOOH), and linoleic acid hydroperoxide (LaOOH) (see Fig. S2 in the supplemental material) could not be fit to simple Michaelis-Menten kinetics owing to inactivation of Ohr at high peroxide concentrations, but values of kcat/Km could be calculated from the linear increase in rate with peroxide at low peroxide concentrations (Table 3). The three organic hydroperoxides have comparable activities and are more than three orders of magnitude more reactive than H2O2. Ohr has not been previously tested as a reducing agent for disulfides, but no activity was found with the disulfides of glutathione, mycothiol, or coenzyme A (see the supplemental material).
Table 3.
Summary of rates of reduction by Ohr
| Substrate | Mean kcat/Km ± SD (M−1 s−1) | Relative rate |
|---|---|---|
| H2O2 | 89 ± 5 | 0.02 |
| CuOOH | 4.3 ± 0.5 × 105 | 100 |
| t-BuOOH | 1.6 ± 0.1 × 105 | 37 |
| LaOOH | 3.0 ± 0.5 × 105 | 70 |
DlaT plus Lpd provides a reducing system for Ohr.
Since lipoic acid and lipoamide are not present in M. smegmatis at significant levels (Table 2), there must be another physiologic reductant for Ohr. Several possible systems were tested. Mycothiol (320 μM) together with mycothiol disulfide reductase (Mtr; 0.4 μg) (50, 51) and NADPH (160 μM) exhibited no activity with Ohr (100 μM) following the addition of 100 μM CuOOH in 100 μl of assay buffer. An analogous assay with 6 μM M. tuberculosis TrxB or TrxC in the presence of thioredoxin reductase TrxB2 (340 μM) and NADPH (200 μM) also showed no activity. However, positive activity (Fig. 4) was obtained with M. tuberculosis DlaT (SucB, Rv2215; 2 μM), containing two covalently linked lipoamide residues, together with M. tuberculosis Lpd (Rv0462; 2 μM) and NADH (160 μM), a reducing system previously shown to support peroxidase activity of AhpD with AhpC toward H2O2 in vitro (4). Ohr plus DlaT showed no activity and Lpd plus Ohr showed only low activity, whereas DlaT in combination with Lpd and Ohr showed substantial peroxidase activity toward CuOOH (100 μM), comparable to that measured with lipoamide in combination with Lpd and Ohr (Fig. 4). The DlaT and Lpd proteins used were from M. tuberculosis (4). Since the M. smegmatis genome contains single strong homologs of Lpd (YP_885306, 85% identity) and of DlaT (YP_888560, 72% identity), it is likely that the M. tuberculosis proteins provide good models for their M. smegmatis counterparts. A rapid second-order rate of ∼1.3 × 105 M−1 s−1 was calculated from the initial rate and the CuOOH and Ohr concentrations for the DlaT reaction whose results are shown in Fig. 4. This is severalfold lower than the value (4.3 × 105 M−1 s−1) obtained with CuOOH and lipoamide as reductant (Table 3) but likely represents a lower limit, since the substrate concentrations were not fully optimized. These rates are two orders of magnitude lower than the value (4 × 107 M−1 s−1) for catalase with H2O2 that is considered to be close to diffusion controlled (18, 47).
Fig. 4.
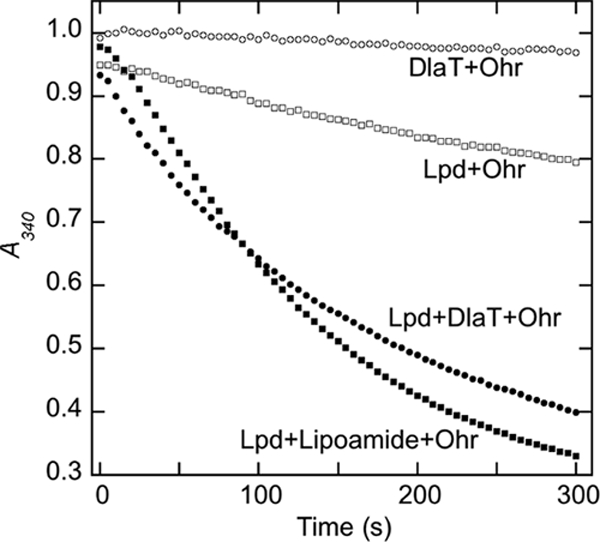
DlaT-dependent reducing system for Ohr. Reduction of CuOOH (0.1 mM) by Ohr (40 nM) supported by M. tuberculosis lipoamide dehydrogenase (Lpd, 2 μM), NADH (160 μM), and M. tuberculosis dihydrolipoamide acetyltransferase (DlaT, 2 μM) or lipoamide (3 μM), followed by decreasing absorbance of NADH at 340 nM.
M. smegmatis Ohr homologs are widely but not universally distributed in bacteria.
It was established a decade ago that the OsmC family of proteins involves two distinct branches, one designated OsmC and the other Ohr (2, 38). Since then, the bacterial genomic database has markedly expanded, and a number of interesting features became evident during a search for homologs of M. smegmatis Ohr. Many bacteria appear to lack the gene for this protein. Not surprisingly, it is absent in the anaerobic Clostridia; no sequences with a E-value of ≤0.1 were found in 40 species examined. Among the Archaea, only 10% of the 65 species tested had distant homologs of M. smegmatis Ohr. Of the 76 completed genomes of Actinobacteria available at GenBank, 39% had no significant homologs of M. smegmatis Ohr, 36% had 1 homolog, 16% 2 homologs, 7% 3 homologs, and 3% 4 homologs. Among mycobacteria, there were no significant homologs (E-value < 5) in Mycobacterium bovis, Mycobacterium kansasii, Mycobacterium leprae, Mycobacterium marinum, M. tuberculosis, or Mycobacterium ulcerans. The gammaproteobacterium Pseudomonas fluorescens Pf0-1 produced 7 homologs of M. smegmatis Ohr, the most of any of the bacteria examined, whereas in the Gammaproteobacteria generally, 37% of the 142 species had no homologs of M. smegmatis Ohr. Thus, the general occurrence of M. smegmatis Ohr homologs in bacteria varies markedly, with a substantial fraction having no significant homologs and the number of homologs within a given species varying markedly.
A closer examination of the homologs in a selected group of bacteria provides some important insights, as illustrated by a phylogenomic tree (Fig. 5). We include those proteins with a known crystal structure (Fig. 5), i.e., Bacillus subtilis Ohr-1 (9), Deinococcus radiodurans Ohr-1 (37), Escherichia coli (36), Pseudomonas aeruginosa Ohr-1 (35), and Xylella fastidiosa (48). These studies established that all of these proteins have very similar structures, with two cysteines in the active site. Representatives of different bacterial phyla and bacteria with multiple M. smegmatis Ohr homologs were also chosen to illustrate several points. First, some organisms have only one homolog, either from the Ohr branch (Corynebacterium glutamicum, M. smegmatis, and Myxococcus xanthus) or the OsmC branch (E. coli and Thermus thermophilus), whereas others have homologs in both branches (Flavobacterium johnsoniae, D. radiodurans, Burkholderia phymatum, Rhizobium leguminosarum, P. aeruginosa, Pseudomonas fluorescens, B. subtilis, and Staphylococcus aureus). Second, the closest homologs to M. smegmatis Ohr come not from the Actinobacteria but from the Alphaproteobacteria (Oligotropha carboxidovorans Ohr-1 [67% identity] and R. leguminosarum Ohr-1 and -2), Betaproteobacteria (B. phymatum Ohr-1 and -4), Gammaproteobacteria (P. fluorescens Ohr-1, -3, -4, and -5), and Bacteroidetes (F. johnsoniae Ohr-2). All of these close homologs are found in soil or aqueous environments where lateral gene transfer is viable, and it therefore seems likely that this is how M. smegmatis acquired the ohr gene and why many of the pathogenic mycobacteria that survive in an animal habitat have not acquired this gene. The plasmid pMAC, identified in Acinetobacter baumannii by Dorsey et al. (13), carries 11 open reading frames that include both a gene encoding an Ohr homolog with 43% identity to M. smegmatis Ohr and a gene encoding an OhrR homolog. These authors propose that pMAC could facilitate the spread of these genes among bacteria.
Fig. 5.

Phylogenetic tree for selected homologs of M. smegmatis Ohr (numbers indicate one of multiple homologs in the genome). Sequences with known crystal structures are denoted with an asterisk. Sequence identities and GenBank sequence accession numbers are as follows: Bacillus subtilis subsp. subtilis strain 168 (1, NP_389199; 2, NP_389197; and 3, NP_389610), Burkholderia phymatum STM815 (1, YP_001863266; 2, YP_001861723; 3, YP_001861801; 4, YP_001862707; 5, YP_001862485; and 6, YP_001861352), Corynebacterium glutamicum ATCC 13032 (NP_599276), Deinococcus radiodurans R1 (1, NP_295580, and 2, NP_295261), Escherichia coli strain K-12 substrain MG1655 (NP_415999), Flavobacterium johnsoniae UW101 (1, YP_001194932; 2, YP_001196333; and 3, YP_001196355), M. smegmatis mc2155 (YP_884860), Myxococcus xanthus DK 1622 (Mx YP_628923), Oligotropha carboxidovorans OM5 (1, YP_002288723; 2, YP_002287603; and 3, YP_002287723), Pseudomonas aeruginosa PAO1 (1, NP_251540, and 2, NP_248749), Pseudomonas fluorescens Pf0-1 (1, YP_348195; 2, YP_347938; 3, YP_347995; 4, YP_351428; 5, YP_351444; 6, YP_349725; and 7, YP_345830), Rhizobium leguminosarum bv. viciae 3841 (1, YP_768321; 2, YP_768512; 3, YP_766908; 4, YP_769801; and 5,YP_765600), Staphylococcus aureus subsp. aureus D139 (1, ZP_06323910, and 2, ZP_06324740), Streptomyces coelicolor A3(2) (1, NP_626642; 2, NP_627209; and 3, NP_631172), Thermus thermophilus HB8 (YP_144891), and Xylella fastidiosa 9a5c (NP_299113). Scale defines changes per site. The tree was generated as described in Materials and Methods.
Among those species with multiple homologs of Ohr, there is a wide variation in the degree of identity. Thus, B. phymatum Ohr-2 and -3 are 82% identical, whereas B. phymatum Ohr-4 shares only 25% identity with B. phymatum Ohr-5. The closest homolog of P. fluorescens Ohr-1 in the P. fluorescens genome is P. fluorescens Ohr-2, with which it shares 68% identity, but P. fluorescens Ohr-1 is also 65% identical to M. smegmatis Ohr. The identity of P. fluorescens Ohr-1 to other Ohr members ranges from 51% for P. fluorescens Ohr-5 to 60% for P. fluorescens Ohr-4. The maximal identity is between P. fluorescens Ohr-3 and P. fluorescens Ohr-5, at 66%. The occurrence of multiple homologs with substantial variation in primary sequence within a given genome suggests that these proteins may have significantly different physiological substrates.
DISCUSSION
We originally thought that identification of the overexpressed protein in the mshA::Tn5 mutant might provide insight concerning how M. smegmatis is able to survive and grow without MSH. The identification of this protein as M. smegmatis Ohr, with its ability to destroy organic hydroperoxides, might well explain how it could partially compensate for loss of the protective action of MSH. However, while the overproduction of Ohr in the mshA::Tn5 mutant increases protection against CuOOH relative to the sensitivity of the mshA(G32D) strain lacking excess Ohr, it does not increase it to the level of the wild-type mc2155 strain with normal levels of Ohr and MSH (Fig. 3A). Thus, in addition to Ohr, there appears to be an MSH-dependent peroxidase active against CuOOH in M. smegmatis, and this protein remains to be identified.
Another significant finding is the marked increase in ergopthioneine in MSH-deficient strains, ∼30-fold for the mshA::Tn5 and mshA(G32D) strains and ∼20-fold for the mshC::Tn5 strain. Ergothioneine is thought to provide protection against oxidative stress, and the genes for its biosynthesis have been recently identified (61). However, the mshA(G32D) mutant with highly elevated ergothioneine but lacking MSH was quite sensitive to CuOOH (Fig. 3A), indicating that ergothioneine provides little protection against this toxin. The ohr gene is not present in the M. tuberculosis genome, and its absence may be one reason why M. tuberculosis is more sensitive than M. smegmatis to loss of MSH. The genes for ergothioneine biosynthesis are present in the M. tuberculosis genome, but whether M. tuberculosis mutants deficient in MSH biosynthesis increase ergothioneine production is unknown (65).
The chemistry of ergothioneine is substantially different from that of cysteine-containing thiols, such as GSH and MSH, and may influence the role that ergothioneine plays in mycothiol-deficient mutants. Although ergothioneine at physiological pH exists in the thione rather than thiol form, is a much poorer reductant, and is less subject to autoxidation than cysteine or its derivatives, it is nevertheless considered to be an effective antioxidant through quenching of singlet oxygen, scavenging of hydroxyl radicals, and inhibition of heavy metal-catalyzed autoxidation (24). Despite hundreds of publications dealing with ergothioneine, mostly involving eucaryotes, its function in bacteria has been little studied and no enzymes utilizing ergothioneine as a cofactor have been reported. However, as pointed out by Seebeck (61), close homologs of the M. smegmatis EgtB and EgtD ergothioneine biosynthesis proteins are encoded in most cyanobacterial genomes (24 out of 28 completed genomes in our recent count), suggesting that protection of DNA against visible and UV radiation damage may be an important function of ergothioneine in cyanobacteria, as has been reported for human keratinocytes (3). Soil organisms such as M. smegmatis could also benefit from such protection when subject to surface exposure. Identification of the ergothioneine biosynthesis genes (61) should allow the generation of mutants deficient in ergothioneine production whose phenotype should serve to clarify the functions of ergothioneine in bacteria.
We originally thought that upregulation of the ohr gene (MSMEG_0447) in the mshA::Tn5 strain should involve OhrR (MSMEG_0448), the product of a neighboring gene, as found in many other bacteria (64), via a thiol-mediated redox mechanism (33, 63). However, the present results indicate that MshA and its G32D mutant function as repressors of ohr expression. Whether OhrR can also serve as a repressor of ohr in M. smegmatis is unclear.
A cellular reducing system for Ohr based upon Lpd and DlaT (SucB) was identified recently in Xylella fastidiosa (10) and is confirmed in M. smegmatis in the present study (Fig. 4). Lipoamide itself is sufficient for the in vitro assay, but neither free lipoamide nor lipoic acid can function as the cellular reductant, because they are expected to rapidly diffuse out of the cell (25, 62) and their cellular levels are undetectable (Table 2). Protein-bound lipoyl residues have been measured at levels as high as ∼0.2 μmol per gram (27). Both Lpd and DlaT are more widely distributed in bacteria than Ohr, so a reduction system for Ohr might be available if the ohr gene were acquired by lateral gene transfer. This assumes that the dihydrolipoamide form of DlaT is sufficiently available in vivo to function as the reductant for Ohr. In exponentially growing E. coli, the lipoyl residues of the pyruvate dehydrogenase complex were found to be predominately in the oxidized form (34), so whether sufficient reduced DlaT is available in M. smegmatis to reduce oxidized Ohr in vivo is unclear.
With regard to the cellular function of Ohr, the present results accord with those of earlier qualitative studies establishing a preference for organic hydroperoxides over hydrogen peroxide in Ohr from P. aeruginosa (35), X. fastidiosa (11), and Mycoplasma gallisepticum (28) and identify lipid hydroperoxides as possible physiological targets. Quantitative kinetic data on the destruction of peroxides by Ohr with which to compare the rate data of Table 3 were not available until the study of X. fastidiosa Ohr by Cussiol et al. appeared (10). Their results for H2O2 (kcat/Km = 230 M−1 s−1) with lipoamide and Lpd as reductants are comparable to those reported here for M. smegmatis Ohr (kcat/Km = 89 M−1 s−1). The value of kcat/Km obtained here for M. smegmatis Ohr with t-BuOOH (1.6 × 105 M−1 s−1) is an order of magnitude lower than the kcat/Km value (2.06 × 106 M−1 s−1) for X. fastidiosa Ohr (10). Although we were unable to determine precise Km values owing to the inactivation of Ohr at high alkyl hydroperoxide concentrations, it is apparent from our results shown in Fig. S2B in the supplemental material that the Km value for t-BuOOH must be ≥200 μM, more than an order of magnitude greater than the value (14.5 μM) determined for X. fastidiosa Ohr, which may contribute to the difference in measured rates.
M. smegmatis Ohr was resistant to inactivation by H2O2 but sensitive to inactivation by CuOOH and LaOOH and, to a lesser extent, by t-BuOOH (see Fig. S2 in the supplemental material). X. fastidiosa Ohr showed no loss of activity during assay at t-BuOOH concentrations up to 10 mM but lost about 85% of its activity during incubation in 5.1 mM t-BuOOH for 1 week at 4°C (48). M. smegmatis Ohr was most sensitive to inactivation by LaOOH, the most physiological substrate tested. It might seem strange that a peroxidase is sensitive to inactivation by the peroxide substrates it is designed to control but, in the cellular environment, with adequate Ohr and reductive capacity available, the level of peroxide is not likely to accumulate to damaging levels.
Although a good deal is known about the primary, secondary, tertiary, and quaternary structure of Ohr from numerous sources, many questions remain about the functions of this group of enzymes. The high activity with LaOOH is indicative that lipid hydroperoxides are important substrates. However, in many bacteria, there are multiple Ohr homologs, the extreme case being P. fluorescens, with 6 homologs in the Ohr branch (Fig. 5) plus an additional one associated with the OsmC branch (P. fluorescens Ohr-7). Since there is substantial variation in sequence among these homologs, it seems likely that they function with different hydroperoxide substrates. Are all of these substrates lipid hydroperoxides or could other classes of hydroperoxides, such as protein hydroperoxides (15), be the preferred targets of some Ohr homologs? The Ohr proteins of P. fluorescens represent an important resource to provide answers to this question.
In conclusion, disruption of the mshA gene in M. smegmatis leads to upregulation of the ohr gene that is reversed by complementation. Overproduction of Ohr and loss of MSH influences organic hydroperoxide sensitivity and results in marked resistance to INH, consistent with the proposed requirement for both an oxidative and a reductive step in the activation of INH. M. smegmatis Ohr is highly active with linoleyl hydroperoxide, indicating that lipid hydroperoxides may be the natural substrates. Oxidized M. smegmatis Ohr is efficiently reduced in vitro by the widely distributed NADH-dependent Lpd/DlaT-reducing system. If this system functions in vivo, it could have facilitated the acquisition of the ohr gene through lateral gene transfer in diverse bacteria by providing an established system for the reduction of Ohr. The occurrence in P. fluorescens and other bacteria of multiple ohr genes that encode proteins with markedly varying sequences indicates that there may be multiple cellular functions for Ohr proteins. Finally, the dramatic upregulation of ergothioneine production in mshA- and mshC-deficient mutants suggests a compensating role of ergothioneine for the loss of MSH, but the results indicate that ergothioneine has little influence on resistance to simple organic hydroperoxides.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants AI072133 to R.C.F. and GM061223 to M.R. and by a University of Pittsburgh School of Medicine Dean's Summer Research Grant to P.T.
We thank Carl F. Nathan and Ruslana Bryk for the gift of the M. tuberculosis Lpd and DlaT used in these studies.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 18 February 2011.
REFERENCES
- 1. Altschul S. F., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atichartpongkul S., et al. 2001. Bacterial Ohr and OsmC paralogues define two protein families with distinct functions and patterns of expression. Microbiology 147:1775–1782 [DOI] [PubMed] [Google Scholar]
- 3. Botta C., Di Giorgio C., Sabatier A. S., De Meo M. 2008. Genotoxicity of visible light (400-800 nm) and photoprotection assessment of ectoin, l-ergothioneine and mannitol and four sunscreens. J. Photochem. Photobiol. B 91:24–34 [DOI] [PubMed] [Google Scholar]
- 4. Bryk R., Lima C. D., Erdjument-Bromage H., Tempst P., Nathan C. 2002. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295:1073–1077 [DOI] [PubMed] [Google Scholar]
- 5. Buchmeier N., Fahey R. C. 2006. The mshA gene encoding the glycosyltransferase of mycothiol biosynthesis is essential in Mycobacterium tuberculosis Erdman. FEMS Microbiol. Lett. 264:74–79 [DOI] [PubMed] [Google Scholar]
- 6. Buchmeier N. A., Newton G. L., Fahey R. C. 2006. A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J. Bacteriol. 188:6245–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchmeier N. A., Newton G. L., Koledin T., Fahey R. C. 2003. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol. Microbiol. 47:1723–1732 [DOI] [PubMed] [Google Scholar]
- 8. Bzymek K. P., Newton G. L., Ta P., Fahey R. C. 2007. Mycothiol import by Mycobacterium smegmatis and function as a resource for metabolic precursors and energy production. J. Bacteriol. 189:6796–6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cooper D. R., Surendranath Y., Devedjiev Y., Bielnicki J., Derewenda Z. S. 2007. Structure of the Bacillus subtilis OhrB hydroperoxide-resistance protein in a fully oxidized state. Acta Crystallogr. D Biol. Crystallogr. 63:1269–1273 [DOI] [PubMed] [Google Scholar]
- 10. Cussiol J. R., Alegria T. G., Szweda L. I., Netto L. E. 2010. Ohr (organic hydroperoxide resistance protein) possesses a previously undescribed activity, lipoyl-dependent peroxidase. J. Biol. Chem. 285:21943–21950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cussiol J. R., Alves S. V., de Oliveira M. A., Netto L. E. 2003. Organic hydroperoxide resistance gene encodes a thiol-dependent peroxidase. J. Biol. Chem. 278:11570–11578 [DOI] [PubMed] [Google Scholar]
- 12. Dayhoff M., Schwartz R., Orcutt B. 1978. A model of evolutionary change in proteins. In Dayhoff M. (ed.), Atlas of protein sequence and structure, vol. 5 National Biomedical Research Foundation, Washington, DC [Google Scholar]
- 13. Dorsey C. W., Tomaras A. P., Actis L. A. 2006. Sequence and organization of pMAC, an Acinetobacter baumannii plasmid harboring genes involved in organic peroxide resistance. Plasmid 56:112–123 [DOI] [PubMed] [Google Scholar]
- 14. Dosanjh M., Newton G. L., Davies J. 2008. Characterization of a mycothiol ligase mutant of Rhodococcus jostii RHA1. Res. Microbiol. 159:643–650 [DOI] [PubMed] [Google Scholar]
- 15. Du J., Gebicki J. M. 2004. Proteins are major initial cell targets of hydroxyl free radicals. Int. J. Biochem. Cell Biol. 36:2334–2343 [DOI] [PubMed] [Google Scholar]
- 16. Ey J., Schomig E., Taubert D. 2007. Dietary sources and antioxidant effects of ergothioneine. J. Agric. Food Chem. 55:6466–6474 [DOI] [PubMed] [Google Scholar]
- 17. Fahey R. C., Newton G. L. 1987. Determination of low-molecular-weight thiols using monobromobimane fluorescent labeling and high-performance liquid chromatography. Methods Enzymol. 143:85–96 [DOI] [PubMed] [Google Scholar]
- 18. Fersht A. 1999. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W. H. Freeman and Company, New York, NY [Google Scholar]
- 19. Genghof D. S. 1970. Biosynthesis of ergothioneine and hercynine by fungi and Actinomycetales. J. Bacteriol. 103:475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Genghof D. S., Vandamme O. 1964. Biosynthesis of ergothioneine and hercynine by mycobacteria. J. Bacteriol. 87:852–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gouy M., Guindon S., Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27:221–224 [DOI] [PubMed] [Google Scholar]
- 22. Grundemann D., et al. 2005. Discovery of the ergothioneine transporter. Proc. Natl. Acad. Sci. U. S. A. 102:5256–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guindon S., Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 24. Hand C. E., Honek J. F. 2005. Biological chemistry of naturally occurring thiols of microbial and marine origin. J. Nat. Prod. 68:293–308 [DOI] [PubMed] [Google Scholar]
- 25. Handelman G. J., Han D., Tritschler H., Packer L. 1994. Alpha-lipoic acid reduction by mammalian cells to the dithiol form, and release into the culture medium. Biochem. Pharmacol. 47:1725–1730 [DOI] [PubMed] [Google Scholar]
- 26. Hartman P. E. 1990. Ergothioneine as antioxidant. Methods Enzymol. 186:310–318 [DOI] [PubMed] [Google Scholar]
- 27. Herbert A. A., Guest J. R. 1975. Lipoic acid content of Escherichia coli and other microorganisms. Arch. Microbiol. 106:259–266 [DOI] [PubMed] [Google Scholar]
- 28. Jenkins C., Samudrala R., Geary S. J., Djordjevic S. P. 2008. Structural and functional characterization of an organic hydroperoxide resistance protein from Mycoplasma gallisepticum. J. Bacteriol. 190:2206–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jocelyn P. C. 1972. Biochemistry of the SH group. Academic Press, New York, NY [Google Scholar]
- 30. Jothivasan V. K., Hamilton C. J. 2008. Mycothiol: synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 25:1091–1117 [DOI] [PubMed] [Google Scholar]
- 31. Koledin T., Newton G. L., Fahey R. C. 2002. Identification of the mycothiol synthase gene (mshD) encoding the acetyltransferase producing mycothiol in actinomycetes. Arch. Microbiol. 178:331–337 [DOI] [PubMed] [Google Scholar]
- 32. Larkin M. A., et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 33. Lee J. W., Soonsanga S., Helmann J. D. 2007. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc. Natl. Acad. Sci. U. S. A. 104:8743–8748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leichert L. I., Jakob U. 2004. Protein thiol modifications visualized in vivo. PLoS Biol. 2:e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lesniak J., Barton W. A., Nikolov D. B. 2002. Structural and functional characterization of the Pseudomonas hydroperoxide resistance protein Ohr. EMBO J. 21:6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lesniak J., Barton W. A., Nikolov D. B. 2003. Structural and functional features of the Escherichia coli hydroperoxide resistance protein OsmC. Protein Sci. 12:2838–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meunier-Jamin C., Kapp U., Leonard G. A., McSweeney S. 2004. The structure of the organic hydroperoxide resistance protein from Deinococcus radiodurans. Do conformational changes facilitate recycling of the redox disulfide? J. Biol. Chem. 279:25830–25837 [DOI] [PubMed] [Google Scholar]
- 38. Mongkolsuk S., Praituan W., Loprasert S., Fuangthong M., Chamnongpol S. 1998. Identification and characterization of a new organic hydroperoxide resistance (ohr) gene with a novel pattern of oxidative stress regulation from Xanthomonas campestris pv. phaseoli. J. Bacteriol. 180:2636–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newton G. L., et al. 1996. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 178:1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newton G. L., Av-Gay Y., Fahey R. C. 2000. N-Acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) is a key enzyme in mycothiol biosynthesis. J. Bacteriol. 182:6958–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Newton G. L., Av-Gay Y., Fahey R. C. 2000. A novel mycothiol-dependent detoxification pathway in mycobacteria involving mycothiol S-conjugate amidase. Biochemistry 39:10739–10746 [DOI] [PubMed] [Google Scholar]
- 42. Newton G. L., Buchmeier N., Fahey R. C. 2008. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 72:471–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Newton G. L., et al. 2003. The glycosyltransferase gene encoding the enzyme catalyzing the first step of mycothiol biosynthesis (mshA). J. Bacteriol. 185:3476–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Newton G. L., Ta P., Bzymek K. P., Fahey R. C. 2006. Biochemistry of the initial steps of mycothiol biosynthesis. J. Biol. Chem. 281:33910–33920 [DOI] [PubMed] [Google Scholar]
- 45. Newton G. L., Ta P., Fahey R. C. 2005. A mycothiol synthase mutant of Mycobacterium smegmatis produces novel thiols and has an altered thiol redox status. J. Bacteriol. 187:7309–7316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Newton G. L., et al. 1999. Characterization of a Mycobacterium smegmatis mutant defective in 1-d-myo-inosityl-2-amino-2-deoxy-alpha-d-glucopyranoside and mycothiol biosynthesis. Biochem. Biophys. Res. Commun. 255:239–244 [DOI] [PubMed] [Google Scholar]
- 47. Ogura Y. 1955. Catalase activity at high concentration of hydrogen peroxide. Arch. Biochem. Biophys. 57:288–300 [DOI] [PubMed] [Google Scholar]
- 48. Oliveira M. A., et al. 2006. Structural insights into enzyme-substrate interaction and characterization of enzymatic intermediates of organic hydroperoxide resistance protein from Xylella fastidiosa. J. Mol. Biol. 359:433–445 [DOI] [PubMed] [Google Scholar]
- 49. Parish T., Stoker N. G. 2000. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146:1969–1975 [DOI] [PubMed] [Google Scholar]
- 50. Patel M. P., Blanchard J. S. 1999. Expression, purification, and characterization of Mycobacterium tuberculosis mycothione reductase. Biochemistry 38:11827–11833 [DOI] [PubMed] [Google Scholar]
- 51. Patel M. P., Blanchard J. S. 2001. Mycobacterium tuberculosis mycothione reductase: pH dependence of the kinetic parameters and kinetic isotope effects. Biochemistry 40:3119–3126 [DOI] [PubMed] [Google Scholar]
- 52. Paul B. D., Snyder S. H. 2010. The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ. 17:1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rawat M., Johnson C., Cadiz V., Av-Gay Y. 2007. Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 363:71–76 [DOI] [PubMed] [Google Scholar]
- 54. Rawat M., Kovacevic S., Billman-Jacobe H., Av-Gay Y. 2003. Inactivation of mshB, a key gene in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Microbiology 149:1341–1349 [DOI] [PubMed] [Google Scholar]
- 55. Rawat M., et al. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals and antibiotics. Antimicrob. Agents Chemother. 46:3348–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rawat M., et al. 2004. Targeted mutagenesis of the Mycobacterium smegmatis mca gene, encoding a mycothiol-dependent detoxification protein. J. Bacteriol. 186:6050–6058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rozwarski D. A., Grant G. A., Barton D. H., Jacobs W. R., Jr., Sacchettini J. C. 1998. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279:98–102 [DOI] [PubMed] [Google Scholar]
- 58. Sareen D., Newton G. L., Fahey R. C., Buchmeier N. A. 2003. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J. Bacteriol. 185:6736–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sareen D., Steffek M., Newton G. L., Fahey R. C. 2002. ATP-dependent L-cysteine:1D-myo-inosityl 2-amino-2-deoxy-alpha-d-glucopyranoside ligase, mycothiol biosynthesis enzyme MshC, is related to class I cysteinyl-tRNA synthetases. Biochemistry 41:6885–6890 [DOI] [PubMed] [Google Scholar]
- 60. Sassetti C. M., Boyd D. H., Rubin E. J. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 61. Seebeck F. P. 2010. In vitro reconstitution of mycobacterial ergothioneine biosynthesis. J. Am. Chem. Soc. 132:6632–6633 [DOI] [PubMed] [Google Scholar]
- 62. Sen C. K., Roy S., Han D., Packer L. 1997. Regulation of cellular thiols in human lymphocytes by alpha-lipoic acid: a flow cytometric analysis. Free Radic. Biol. Med. 22:1241–1257 [DOI] [PubMed] [Google Scholar]
- 63. Soonsanga S., Lee J. W., Helmann J. D. 2008. Oxidant-dependent switching between reversible and sacrificial oxidation pathways for Bacillus subtilis OhrR. Mol. Microbiol. 68:978–986 [DOI] [PubMed] [Google Scholar]
- 64. Sukchawalit R., Loprasert S., Atichartpongkul S., Mongkolsuk S. 2001. Complex regulation of the organic hydroperoxide resistance gene (ohr) from Xanthomonas involves OhrR, a novel organic peroxide-inducible negative regulator, and posttranscriptional modifications. J. Bacteriol. 183:4405–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vilcheze C., et al. 2008. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 69:1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wiseman B., et al. 2010. Isonicotinic acid hydrazide conversion to isonicotinyl-NAD by catalase-peroxidases. J. Biol. Chem. 285:26662–26673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Y., Heym B., Allen B., Young D., Cole S. 1992. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358:591–593 [DOI] [PubMed] [Google Scholar]
- 68. Zhang Y., Vilchéze C., Jacobs W. R., Jr 2005. Mechanisms of drug resistance in Mycobacterium tuberculosis, p. 115–140 In Cole S. T., Eisenach K. D., McMurray D. N., Jacobs W. R., Jr (ed.), Tuberculosis and the tubercle bacillus. ASM Press, Washington, DC [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.