

Abstract
Many Archaea and Bacteria isolated from hot, marine environments accumulate di-myo-inositol-phosphate (DIP), primarily in response to heat stress. The biosynthesis of this compatible solute involves the activation of inositol to CDP-inositol via the action of a recently discovered CTP:inositol-1-phosphate cytidylyltransferase (IPCT) activity. In most cases, IPCT is part of a bifunctional enzyme comprising two domains: a cytoplasmic domain with IPCT activity and a membrane domain catalyzing the synthesis of di-myo-inositol-1,3′-phosphate-1′-phosphate from CDP-inositol and l-myo-inositol phosphate. Herein, we describe the first X-ray structure of the IPCT domain of the bifunctional enzyme from the hyperthermophilic archaeon Archaeoglobus fulgidus DSMZ 7324. The structure of the enzyme in the apo form was solved to a 1.9-Å resolution. The enzyme exhibited apparent Km values of 0.9 and 0.6 mM for inositol-1-phosphate and CTP, respectively. The optimal temperature for catalysis was in the range 90 to 95°C, and the Vmax determined at 90°C was 62.9 μmol · min−1 · mg of protein−1. The structure of IPCT is composed of a central seven-stranded mixed β-sheet, of which six β-strands are parallel, surrounded by six α-helices, a fold reminiscent of the dinucleotide-binding Rossmann fold. The enzyme shares structural homology with other pyrophosphorylases showing the canonical motif G-X-G-T-(R/S)-X4-P-K. CTP, l-myo-inositol-1-phosphate, and CDP-inositol were docked into the catalytic site, which provided insights into the binding mode and high specificity of the enzyme for CTP. This work is an important step toward the final goal of understanding the full catalytic route for DIP synthesis in the native, bifunctional enzyme.
INTRODUCTION
Enzymes classified in the nucleoside triphosphate-transferase family (PF00483) typically transfer nucleoside monophosphate (NMP) from nucleoside triphosphates (NTP) to an acceptor phosphoryl group belonging to a small molecule such as phosphocholine, hexose-1-phosphate, or ribitol-5-phosphate (2, 16, 20). This activity leads to release of pyrophosphate and production of a nucleoside diphospho-acceptor that is subsequently utilized by glycosyltransferases in a myriad of reactions of vital importance for the cellular functions.
Recently, we described a novel nucleotidyltransferase that uses CTP and l-myo-inositol-1-phosphate (inositol-1P) as substrates to synthesize a newly identified metabolite, CDP-inositol. Additionally, we showed that the inositol group of this metabolite is subsequently transferred to a second molecule of l-myo-inositol-1-phosphate to yield the phosphorylated form of di-myo-inositol phosphate (DIPP), which is then dephosphorylated to give di-myo-inositol phosphate (DIP) (Fig. 1) (31). This phosphodiester of myo-inositol is a hallmark of marine microorganisms that thrive optimally in very hot environments. Indeed, this unusual compatible solute is highly restricted to hyperthermophilic Archaea and Bacteria and has never been found in mesophilic organisms; furthermore, the DIP pool increases preferentially in response to heat stress, leading to the view that it protects proteins and other cellular macromolecules against the detrimental effects of high temperature (32).
Fig. 1.

Pathway for di-myo-inositol phosphate synthesis. The enzymes shown are CTP:l-myo-inositol-1-phosphate cytidylyltransferase (IPCT), di-myo-inositol-1,3′-phosphate synthase (DIPP synthase), and di-myo-inositol-1,3′-phosphate phosphatase (DIPP phosphatase).
The genes encoding the key activities in DIP synthesis—i.e., CTP:inositol-1-phosphate cytidylyltransferase (IPCT) and the DIPP synthase (DIPPS)—were firmly identified in several microorganisms (Archaeoglobus fulgidus, Pyrococcus furiosus, Thermococcus kodakarensis, Aquifex aeolicus, and Rubrobacter xylanophilus) by functional expression in Escherichia coli (31). In most organisms known to accumulate DIP, the two activities are present in a single polypeptide chain, constituting the bifunctional enzyme IPCT/DIPPS; however, in Thermotoga maritima, the two activities are encoded by separate, consecutive genes and the same gene organization was predicted in other members of hyperthermophilic Bacteria and in a few Archaea (29).
Biochemical studies of the recombinant IPCT/DIPPS from the hyperthermophilic archaeon Archaeoglobus fulgidus have shown that the IPCT domain has absolute specificity for CTP and l-myo-inositol-1-phosphate (31). More importantly, it was predicted that the bifunctional enzyme IPCT/DIPPS combines a cytosolic domain (IPCT) with a membranar domain (DIPPS), expected to have five transmembrane helices. Therefore, the determination of the structure-function relationship of the bifunctional enzyme emerges as an attractive challenge. The attempts to crystallize the whole functional enzyme had been unsuccessful thus far, and hence we set out to determine the structure of IPCT, the cytoplasmic domain, as a first step toward understanding the full catalytic process. IPCT is an intrinsically interesting enzyme, being the sole nucleotidyltransferase known to use l-myo-inositol-1-phosphate as a substrate.
Herein, we describe the first X-ray structure of the IPCT domain of the IPCT/DIPPS bifunctional enzyme from Archaeoglobus fulgidus DSMZ 7324. CTP, l-myo-inositol-1-phosphate, and CDP-inositol were docked to the structure, which gave insight into the binding mode and substrate specificity of the enzyme.
MATERIALS AND METHODS
Materials.
Cytidine-5′-triphosphate (CTP) was purchased from Sigma-Aldrich (St. Louis, MO). l-myo-Inositol-1-phosphate (inositol-1P) was produced enzymatically as previously described (31). Diglycerol phosphate (DGP) was supplied by bitop AG (Witten, Germany).
Protein production and biochemical characterization.
The cytoplasmic domain of the bifunctional IPCT/DIPPS from A. fulgidus was produced in E. coli and purified by chromatographic techniques, as previously described (7). Briefly, the amino acid sequence of AF0263 (436 amino acids) was aligned with the separate IPCT and DIPPS sequences of Hyperthermus butylicus, Aeropyrum pernix, Thermotoga maritima, and Thermotoga petrophila to define the IPCT domain in A. fulgidus. IPCT was identified in the N-terminal region of AF0263, from Met-Ile-Asn-Val-Asp up to residue 232. The gene was amplified by PCR using Pfu polymerase (Fermentas) and cloned in the pET19b plasmid (Novagen) betwen the NdeI and XhoI sites. E. coli BL21DE cells, harboring the construct, were grown at 37°C in LB medium supplemented with ampicillin (100 μg · ml−1) to an absorbance of 0.6 and induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h. After centrifugation, cells were disrupted in a French press. The IPCT was purified from heat-treated cell extracts with a His-Trap column, and the histidine tag was cleaved with enterokinase. A final polishing step was performed by applying IPCT onto a Superdex G75 gel-filtration column. Protein concentration was estimated by the Bradford method (5), and purity was assessed by SDS-PAGE (21). The molecular mass of IPCT was estimated by gel filtration using a Superdex S200 column equilibrated with 20 mM Tris-HCl–150 mM NaCl, pH 7.5. Lysozyme (14.3 kDa), superoxide dismutase (32.6 kDa), albumin (66.8 kDa), and alcohol dehydrogenase (150 kDa) were used as standards.
The IPCT activity was determined in a reaction mixture (final volume of 400 μl), containing 50 mM Bis-Tris-propane, 10 mM MgCl2, 5 mM CTP, and 5 mM inositol-1P. The mixture was preincubated for 2 min at the same temperatures of the assay in 2-ml glass tubes. The reactions were initiated by addition of IPCT (5 μg) and stopped at different time points (0, 15, and 30 s) by immersion in liquid nitrogen. Afterwards, 10 μl of EDTA (0.5 M, pH 8) and 100 μl of 2H2O were added to the reaction mixture. The amount of CDP-inositol produced was quantified by 31P-nuclear magnetic resonance (NMR) spectroscopy using DGP as an internal concentration standard. Spectra were acquired on a Bruker DRX500 spectrometer (Bruker, Rheinstetten, Germany). The effect of Mg2+ (0, 10, and 20 mM) on IPCT activity was studied. The temperature profile for activity was determined between 60°C and 115°C at pH25°C 8.0. (pH25°C indicates that pH values were measured at 25°C and no temperature correction was applied.) The pH profile for activity was determined at 90°C with 50 mM Bis-Tris-propane; pH values were measured at 25°C, but the values displayed (pH90°C) are corrected for the temperature of 90°C by using the conversion factor for Bis-Tris-propane (ΔpKa/ΔT = −0.015). Kinetic parameters (Vmax and Km) were determined at 90°C and a pH90°C of 7.0 in reaction mixtures containing 4 mM CTP and 0 to 7.5 mM inositol-1P or 0 to 4 mM CTP and 7.5 mM inositol-1P. The actual CTP and inositol-1P concentrations of the stock solutions were determined by NMR. Kinetic parameters were obtained by fitting the data to Michaelis-Menten equations with the software Origin 5.0 Professional (Microcal Software, Inc., Northhampton, MA). The data included six concentration values of each substrate.
Crystallization, data collection, structure solution, and refinement.
IPCT was crystallized as described previously (7). In summary, a protein solution at a concentration of 12 mg · ml−1 in 10 mM HEPES, pH 7.5, was used for crystallization trials; cubic crystals were grown at 20°C by the hanging-drop vapor diffusion method by mixing equal volumes of protein and reservoir solution containing 1.2 to 1.4 M sodium citrate, 100 mM HEPES, pH 6.8 to 8.2, and 10 mM MgCl2. Crystals were cryoprotected with the crystallization buffer supplemented with 10% glycerol and flash-cooled in a nitrogen gas stream. An IPCT:HgCl2 derivative crystal was obtained by soaking freshly prepared IPCT crystals in crystallization solution supplemented with 5 mM HgCl2 for 4 h. IPCT was also crystallized by using sodium malonate as the precipitant. Crystals were obtained at 20°C in hanging drops with ∼1.5 μl of protein sample and 1.5 μl of reservoir solution consisting of 1.8 M sodium malonate, pH 6.5 to 7.0. These crystals were flash-cooled directly in liquid nitrogen prior to data collection. X-ray diffraction images of IPCT crystals grown with citrate as precipitant were collected on beamline ID14-2 (ESRF, Grenoble, France) by an ADSC Q4 charge-coupled device (CCD). Data were also acquired for crystals grown in malonate at ESRF beamline ID23-1 using an ADSC Q315r CCD detector. All data were indexed and processed with XDS (15) and scaled with SCALA from the CCP4 program package (13).
Experimental phases were determined by the single isomorphous replacement with anomalous scattering (SIRAS) method, using a mercury derivative. SHELXC and SHELXD (33), both within HKL2MAP (27), were used to prepare the experimental data and to locate the heavy atom sites, respectively. A few positive hits were found out of 1,000 trials, the best solution showing correlation coefficients (CCs) of 17.98% (CCall) and 9.62% (CCweak) and a PATFOM value of 1.37. A total of 10 Hg sites were found with occupancies higher than ca. 0.4, but SHELXE (33) did not produce an interpretable electron density map. This initial set of 10 sites was then refined in SHARP (10), and subsequent analysis of log-likelihood gradient maps revealed 2 additional sites, giving a total of 12 heavy atom positions. The calculated phases had a mean figure of merit of 0.29- to 2.8-Å resolution and were improved through a single cycle of solvent flipping with SOLOMON (1) and extended to the resolution of the native data set. The resulting map was used to build an initial atomic model using the program BUCCANEER (8), which built 1,168 residues in 61 chains (the longest having 104 residues). Upon examination of the model, it became clear that one noncrystallographic symmetry (NCS) operator related two dimers of the enzyme, and this information was used for NCS averaging with DM (9). This further improved the maps, which allowed BUCCANEER to proceed with model building, reducing the number of chains and sequencing more side chains. Iterative model building and crystallographic refinement were performed with the programs COOT (12) and BUSTER-TNT (6). One monomer of the citrate-crystallized IPCT, without solvent and other ligands, was used as a search model for molecular replacement on the malonate-crystallized IPCT by using PHASER (25). The model was built into the electron density maps with COOT (12), and refinement was carried out with BUSTER-TNT (6).
The homologous three-dimensional (3D) structures were superimposed by using the secondary structure-matching algorithm (SSM) (19) in COOT, and CTP, inositol-1P, and CDP-inositol were manually fitted followed by structure idealization with REFMAC5 (26), visualization in COOT, and subsequent analysis in PyMol (11). All structural figures were drawn with PyMol (11).
Protein structure accession numbers.
The coordinates of the malonate-IPCT (apo form) and the citrate-IPCT of A. fulgidus DSMZ 7324 have been deposited in the Protein Data Bank under accession codes 2XME and 2XMH, respectively.
RESULTS AND DISCUSSION
Biochemical characterization of IPCT from A. fulgidus.
The biochemical and kinetic properties of IPCT from A. fulgidus were investigated with CTP and inositol-1P as substrates. The presence of Mg2+ was absolutely required for activity, and maximal activity was obtained with 20 mM Mg2+. The activity of the enzyme was undetectable at temperatures below 60°C, and maximal activity was reached between 90 and 95°C (Fig. 2a). Therefore, the temperature for optimal catalysis is around 10°C above the temperature for optimal growth of the host organism, a feature commonly found in other enzymes from hyperthermophiles (4, 30). The pH profile for A. fulgidus IPCT showed a maximum of activity between 6.5 and 7.5 (Fig. 2b). Kinetic parameters of the enzyme were determined under optimal conditions (Table 1). A typical Michaelis-Menten kinetics was observed for the two substrates. The Vmax (62.9 μmol · min−1 · mg−1) was 2.5-fold lower than that of the T. maritima IPCT (29). With respect to substrate affinity, the truncated A. fulgidus IPCT showed Kms of 0.87 mM and 0.58 mM for inositol-1P and CTP, respectively, which are in the same range of magnitude as those of the homofunctional enzyme from T. maritima (29).
Fig. 2.
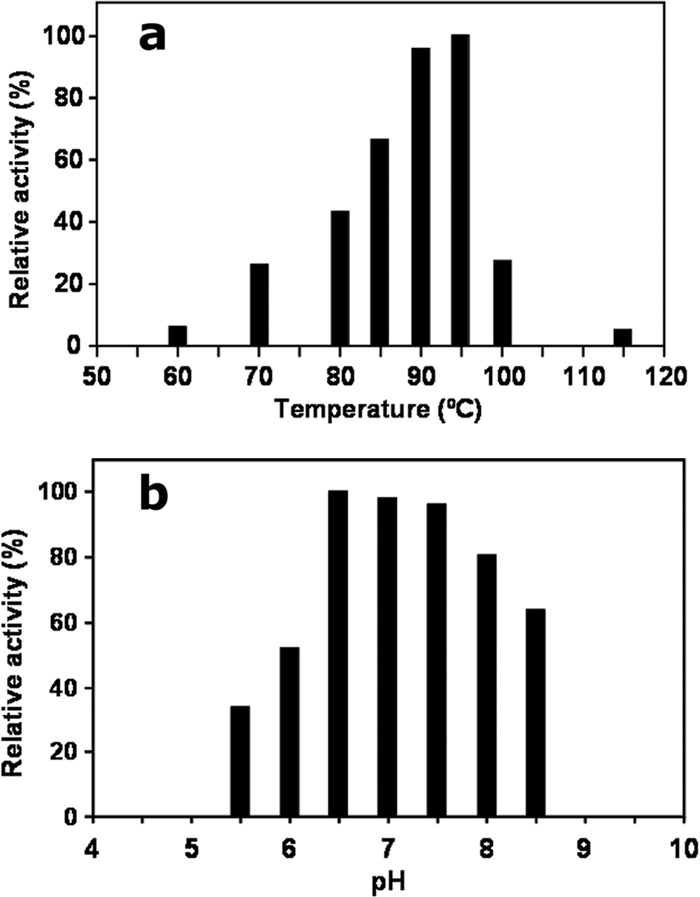
Temperature (a) and pH (b) profiles of IPCT from Archaeoglobus fulgidus. The enzyme activity was determined between 60°C and 115°C and between pH90°C 5.5 and 8.5 in Bis-Tris-propane.
Table 1.
Kinetic properties of the recombinant IPCT of A. fulgidus and effect of Mg2+
| Parametera | IPCT activity |
|---|---|
| Km (mM) | |
| Inositol-1-phosphate | 0.87 ± 0.09 |
| CTP | 0.58 ± 0.08 |
| Vmax (μmol · min−1 · mg protein−1) | 62.9 ± 1.4 |
| % of maximum activity at Mg2+ concn of: | |
| 0 mM | 0 |
| 10 mM | 67 |
| 20 mM | 100 |
Km and Vmax values were determined at 90°C and pH90°C 7.0.
Model quality.
IPCT crystallized in space group P21212 (a = 154.9, b = 83.9, and c = 127.9 Å) with 6 molecules in the asymmetric unit using citrate as precipitant at pH 8.2 (Matthews coefficient [Vm] is 2.6 Å3 · Da−1, and solvent content is ∼53%) (24). The structure was solved by SIRAS based on an Hg derivative and refined to an Rfactor of 23.3% and Rfree of 27.0% at a 2.4-Å resolution. The final IPCT model comprises 1,183 amino acid residues, 204 water molecules, and 4 citrate molecules. Data collection and refinement statistics are shown in Table 2. No clear electron density was visible at both termini (16 to 19 amino acid residues in the N terminus and 10 to 12 residues in the C terminus, depending on the chain, were not built in the model), and for a flexible loop composed of residues 27 to 37. Attempts to soak or cocrystallize IPCT with its substrates were performed, but no well-defined electron density was observed in the calculated maps for either inositol-1P or CTP nearby the active site: instead, a citrate molecule was observed (Fig. 3a).
Table 2.
Data collection and refinement statistics
| Parameter | Result fora: |
|
|---|---|---|
| Citrate-IPCT | Malonate-IPCT (apo) | |
| PDB | ||
| Identification code no. | 2XMH | 2XME |
| Space group | P21212 | P121 |
| Unit cell dimensions | ||
| a, b, c (Å) | 154.7, 83.9, 127.7 | 86.03, 127.55, 141.48 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.56, 90.00 |
| Data collectionb | ||
| Beamline | ESRF ID23-1 | ESRF ID14-2 |
| Wavelength (Å) | 1.067 | 0.933 |
| Resolution range (Å) | 48.28–2.40 (2.53–2.40) | 142.49–1.89 (1.99–1.89) |
| Total no. of observations | 224,153 (32,846) | 941,436 (58,133) |
| No. of unique observations | 64,618 (9,409) | 219,870 (29,484) |
| Rmerge (%) | 5.0 (42.6) | 12.6 (48.9) |
| Rpim (%) | 2.8 (26.2) | 6.6 (45.3) |
| Completeness (%) | 98.8 (99.5) | 90.3 (83.0) |
| Mean 〈I/σ(I)〉 | 13.4 (2.3) | 10.1 (1.5) |
| Refinement statistics | ||
| Resolution range (Å) | 35.14–2.40 (2.46–2.40) | 27.67–1.89 (1.94–1.89) |
| Rwork/Rfree (%) | 23.3/27.5 | 20.6/23.7 |
| Geometry | ||
| RMSDc bonds (Å) | 0.01 | 0.01 |
| RMSD angles (°) | 1.23 | 1.10 |
| Ramachandran statistics | ||
| Residues in most favored regions (%) | 94.9 | 98.3 |
| Residues in allowed regions (%) | 5.0 | 1.6 |
| Residues in disallowed regions (%) | 0.1 | 0.1 |
| Mean B factors | ||
| Protein (Å2) | 73.1 | 45.9 |
| Solvent (Å2) | 60.9 | 41.0 |
Values in parentheses refer to the highest-resolution shell.
RMSD, root mean square deviation.
Fig. 3.

(a) Cartoon representation of IPCT monomer. The color scheme is according to the topology diagram shown in panel b. The active site loop is shown as a thicker purple loop herein and after, and citrate is drawn as sticks (carbon is yellow and oxygen red). (b) TOPS-generated topology diagram of IPCT. Relevant secondary structure elements are numbered, and the location of the active site loop is noted with an asterisk. (c) Cartoon and surface representation of IPCT dimer. Chain F is drawn in yellow with the molecular surface; residues at the interface are colored magenta (chain F) and cyan (chain E). (d) Zoomed view at the dimer interface. Residues establishing side chain interactions are displayed. By the atom color code, carbon is yellow for chain F and cyan for chain E, oxygen is red, and nitrogen is dark blue for both chains.
We then searched for another crystallization condition without citrate and optimized one containing malonate as the precipitant at pH ∼7. Under this condition, IPCT crystals belonged to space group P2, with 12 molecules in the asymmetric unit corresponding to a Vm value of 2.5 Å3 · Da−1 and a solvent content of ∼50% (24). Soaking and cocrystallization experiments were also unsuccessful under this condition since no clear electron density for CTP and/or inositol-1P was visible in the active site. The final model comprises 2,419 amino acid residues, 1,171 water molecules, and 8 glycerol molecules and was refined to a 1.9-Å resolution with an Rfactor of 20.6% (Rfree of 23.7%). The electron density maps are clearly defined, apart from the N and C termini of all subunits (similarly to the citrate-IPCT, chains start at residues 16 to 19 and end up at residues 220 to 222 out of 232) and a loop. This loop (residues 27 to 37) is disordered in most chains and contains gaps in the final model, except in chains E and F. Interestingly, these two loops are quite close to each other in the crystallographic dimer (e.g., the distance between the Cα atoms of Gly34E and Gly33F is only 6.6 Å), similar to the other pairs: A-B, C-D, and so on. The final models of citrate- and malonate-IPCT are almost identical, yielding root mean square deviation (RMSD) values between 0.24 and 0.69 Å for Cα superposition of the different chains (14). Subsequent descriptions will refer to chains E and/or F of the malonate-IPCT structure, unless otherwise stated.
The Ramachandran plot, as assessed with RAMPAGE (23), shows that all nonglycine amino acid residues lie within allowed regions, with the exception of Arg91A and Asp109E for citrate-IPCT. PROCHECK (22) analysis showed no bad contacts and a final model with a good geometry and stereochemistry.
Overall fold of IPCT and related structures.
Each monomer of IPCT has overall dimensions of ∼45 Å by 45 Å by 40 Å and is organized into two domains—a core domain and a sugar-binding domain. The core domain (residues 16 to 135 and 173 to 222), consists of a central seven-stranded mixed β-sheet (β1 to β7), with order 3214657, where β6 is antiparallel to the rest, surrounded by six helices, a fold reminiscent of the dinucleotide-binding Rossmann fold (28). At one of its ends, the central β-sheet is topped by a two-stranded β-sheet (β5a and β5b) that participates in a 30-residue-long stretch connecting strands β5 and β6. One face of the central β-sheet is packed against helices α1, α2, and α4, and the other face, which binds the nucleotide, stacks against helices α3, α5 and α6 (this helix being typical of nucleotide binding proteins). The sugar-binding domain (residues 136 to 172) comprises a short antiparallel three-stranded β-sheet, which sits against the exposed face of the central β-sheet (where the nucleotide binds), with several conserved residues involved in the sugar-phosphate moiety recognition (e.g., Leu24, Gly174, and Trp216; IPCT numbering). A topology diagram drawn with the program TOPS is presented in Fig. 3b (35).
The citrate-IPCT crystallographic structure contains 6 molecules in the asymmetric unit, whereas the malonate one has 12 molecules. Despite the different types of packing in both crystal forms, the dimeric arrangements between two molecules (e.g., chains A-B, C-D, and E-F) are quite similar (Fig. 3c and d). According to PISA (18), a dissociation energy (ΔGdiss) of 6.4 kcal · mol−1 is predicted for this dimeric assembly in solution. The interface is mainly hydrophilic, with an area around 1,110 Å2 corresponding to ca. 11% of the total solvent-accessible area of each monomer. Most residues that contribute to the interface stabilization are located in loops between β1-α1, β5-β5a and at the C-terminal loop. The interface is stabilized by 14 H-bonds, 2 of which are salt bridges. Several hydrogen bonds are established between main chain atoms of both chains, namely, between Arg41-Asp220, Gly43-Asp218, and Asp144-Gly43. Additional stabilization is achieved by side chain-main chain interactions between Arg50-Phe142, Thr221-Leu39, and Asp218-Gly44. Moreover, Arg41E/F (NH2) is H-bonded to Glu147F/E (OE2). Molecular mass determinations by gel filtration indicate that there is a mixture of monomers and dimers in solution, with prevalence of the monomeric form. At this stage, we think that the relevance of the interface interactions in the crystal structure for the physiological assembly of the enzyme cannot be fully assessed before the structure of the whole IPCT/DIPPS bifunctional protein is characterized.
Comparison of one molecule of IPCT (monomer) with other structures in the Protein Data Bank (PDB) by using the DALI server (14), reveals that the highest matches are found with the N-terminal domain of the bifunctional N-acetylglucosamine-1-phosphate uridylyltransferases (GlmU) from different sources (PDB codes 1G97, 2OI6, 1HM9, 1HV9, 1FWY, and 2V0I) and with glucose-1-phosphate thymidylyl/uridylyl-transferases (PDB codes 2PA4, 1FXO, 2E3D, and 2UX8), showing Z scores between 20 and ∼18 and RMSDs ranging from 2.5 to 2.8 Å for ∼190 superimposed Cα atoms. Other cytidylyltransferases (CTP:phosphocholine-, 3-deoxy-manno-octulosonate-, and α-d-glucose-1-phosphate-), with PDB codes 1JYL, 1GQC, and 1TZF, respectively, have Z values from 17.7 down to ∼14.5. Interestingly, GlmU is also a bifunctional enzyme, containing a homologous nucleotidyltransferase domain at the N terminus but with an acetyltransferase domain at the C terminus, which adopts a left-handed parallel β-helix (LβH) structure. The biological assembly of the bifunctional GlmU is a trimer, whereas most members of other nucleotidyltransferases are dimers or tetramers. These structure-related enzymes share between 15% and 22% of sequence identity with IPCT and show conservation in the core region, consisting of a mixed β-sheet of seven strands with the same order and orientation as IPCT, a fold typical of nucleotide-diphospho-sugar transferases (SCOP superfamily 53448).
Interestingly, the 32 best hits retrieved in a BLAST search using the sequence of A. fulgidus IPCT belong to hyperthermophilic or thermophilic organisms, probably reflecting the specialized function of IPCT in DIP synthesis (see Fig. S1 in the supplemental material); the best match relates to proteins of Thermococcus spp. that display sequence identities between 49% and 56%. As expected, the protein from A. fulgidus appears in a cluster dominated by members of the Euryarchaeota; curiously, three representatives of the domain Bacteria also cluster in this group, which is clearly separated from a second major group comprising bacteria of the order Thermotogales. The closest mesophilic homologues share ca. 30% sequence identity with A. fulgidus IPCT and belong to the bacterial genera Gluconacetobacter and Granulibacter. Whether these mesophilic counterparts are able to use inositol-1P and CTP remains elusive.
The active site.
A citrate molecule is observed in the active site pocket in three out of six molecules of IPCT crystallized using citrate as precipitant (chains A, D, and E) (Fig. 3a). The citrate is establishing hydrogen bonds with the main-chain nitrogen atoms of Gly27, Leu28, Lys37, and Arg68. Its presence in only some chains might be related to the position of the disordered loop comprising Gly27-Lys37. This loop is not visible in most chains of both citrate- and malonate-IPCT structures, and, if visible, it adopts different conformations. This loop encompasses the signature sequence Gly-X-Gly-Thr-(Arg/Ser)-X4-Pro-Lys of nucleotidyltransferases, and in IPCT, it starts at Gly27 and has the sequence Gly-Leu-Gly-Thr-Arg-Leu-Gly-Gly-Val-Pro-Lys. In the apo-RmlA structure (PDB code 1FZW), this loop is also disordered but becomes traceable in the thymidine-containing complexes (PDB codes 1G2V and 1G0R) (3).
Attempts to obtain structures of IPCT in complex with substrates CTP, inositol-1P, and CTP:inositol-1P, by either cocrystallization or soaking experiments, did not succeed in both crystal forms. However, due to the core fold conservation among sugar nucleotidyltransferase structures, and based on the DALI results, we were able to identify the catalytic region of IPCT. The most homologous structure to IPCT, besides GlmU, is glucose-1-phosphate thymidylyltransferase (RmlA) from Pseudomonas aeruginosa (RMSD of 2.57 Å for 188 Cα atoms) (3). RmlA is involved in the synthesis of dTDP-l-rhamnose, an important component in the cell wall of many microorganisms. Superposition of various structures of RmlA apo forms and complexes (dTTP, glucose-1-phosphate, and dTDP-d-glucose) shows no relevant structural changes upon ligand binding, as also observed with other nucleotidyltransferase structures (Fig. 4a). We identified the relevant catalytic residues of IPCT based on various structures of RmlA. Moreover, this structural analysis was extended to include other homologous nucleotidyltransferases, such as CTP:phosphocholine cytidylyltransferase from Streptococcus pneumoniae (PDB code 1JYL) (20) and α-d-glucose-1-phosphate cytidylyltransferase from Salmonella enterica serovar Typhi (PDB code 1TZF) (17), leading to similar results. For the sake of simplicity, we will refer to the comparison with RmlA throughout the article.
Fig. 4.

(a) Superposition of IPCT (cartoon in blue) with several structures of RmlA: apo form (PDB code 1FZW; ribbon in dark green), and in complex with different ligands (PDB codes 1FXO, 1G0R, 1G2V, 1G1L, 1G23, and 1G3L; ribbons in different shades of green). dTDP-d-glucose (from 1G1L) is shown in sticks (C, yellow; O, red; N, blue; and P, orange). (b) IPCT active site pocket with fitted CDP-inositol; residues at the entrance are labeled.
The active site of IPCT is thus located in a pocket formed by the sugar- and nucleotide-binding domains of the enzyme (Fig. 4b). The pocket entrance comprises a series of polar or charged residues: Lys37, Arg68, Glu93, Lys163, Glu194, Ser198, and Asp220. The cavity is formed by residues from strands β1, β2, β4, and β6 (namely Leu24, Ala25, Val65, Ala66, Met115-His118, Asp172, Gly174, and Phe176); helices α3 and α6 (residues Gly95, Asn96, and Val201); and residues Pro38, Ser99, and Trp216.
The binding sites of nucleotides are highly conserved, comprising a Rossmann fold motif (αβαβα). In IPCT, the CTP should be nested in a groove composed by the N termini of helices α2 and α5 and the C termini of strands β1, β2, and β4. Based on the structure of RmlA in complex with dTTP (PDB code 1G2V) (3), we have fitted a CTP molecule into the active site of IPCT (Fig. 5a). In RmlA, several conserved amino acid residues, namely, Gly10, Thr14, Arg15, and Lys25 (RmlA numbering), are involved in the stabilization of the nucleotide. In IPCT, the phosphate moiety may be stabilized by Thr30, Arg31, and Lys37 (corresponding to Thr14, Arg15, and Lys25 in RmlA), of which Thr30 and Arg31 are located in the disordered loop. Furthermore, Asp117 and Ala26 (Asp110 and Gly10 in RmlA), may establish several H-bonds to the ribose and pyrimidine ring, respectively. Gln82, a catalytically important residue in RmlA (replaced by His89 in IPCT), along with Gly87 (Gly95 in IPCT) and Gly10 (Ala26 in IPCT), provides specificity for thymidine, while a tight loop formed by Gln82 to Gly87 is responsible for the specificity of pyrimidine over purine bases (3).
Fig. 5.

Structural superposition in stereo view of IPCT fitted with CTP and RmlA with dTTP (1G2V) (a), IPCT fitted with inositol-1-P and RmlA with G-1-P (1G23) (b), and IPCT structure fitted with CDP-inositol and RmlA with dTDP-d-glucose (1G1L) (c). The IPCT ribbon is drawn in yellow and RmlA in gray, with ligands shown as sticks (same atom color code as in Fig. 4, with sulfur atoms colored green and carbon atoms of RmlA colored gray).
IPCT is highly specific for CTP as no activity was detected with UTP, ATP, and GTP as nucleotide donors (31). The structural basis for this substrate preference becomes apparent from our CTP-fitted model: the NH2 amino group of cytidine is able to form an H-bond with the main chain oxygen of Pro92, which is not possible with UTP (NH2 substituted for by a carbonyl group). By analogy with RmlA (2), the tight loop (His89-Gly95 in IPCT) is proposed to be responsible for the specificity of cytosine over purines.
Inositol-1P was docked into IPCT by superposition with the structure of RmlA bound to glucose-1-phosphate (PDB code 1G23) (3), where the ligand is stabilized by several hydrogen bonds and van der Waals interactions (3). The same type of interactions is expected in the inositol-1P fitted structure of IPCT since most of the residues involved in substrate binding are conserved among nucleotidyltransferases (Fig. 5b). The side chains of Thr149, His118, and Asp172 (Tyr145, Asn111, and Val172 in RmlA), can establish H-bonds with the inositol ring, and Asn96 and Lys163 (Leu88 and Lys162 in RmlA), with the phosphate moiety. Additionally, several hydrophobic residues are present in the surroundings of the ligand, such as Met115, Gly174, Leu197, and Trp216 in IPCT (Leu108, Gly174, Ile199, and Trp223 in RmlA). The structure of IPCT fitted with CDP-inositol (Fig. 5c) reveals protein interactions quite similar to those found in inositol-1P- and CTP-docked models and hence will not be the subject of further discussion.
Mg2+ is known to be essential for the catalytic reaction of nucleotidyltransferases but its role is not fully understood. Blakenfeldt et al. (3) proposed that this cation could be coordinating the β- and γ-phosphates of dTTP in a chelated manner, leaving the active site as a complex with pyrophosphate. However, several structures, such as glucose-1-phosphate uridylyltransferase from E. coli (34) and glucose-1-phosphate cytidylyltransferase from Salmonella enterica serovar Typhi (17), show the magnesium bound to the final product of each enzyme, specifically to the α- and β-phosphoryl oxygens of the NDP-sugar product. A more direct role of Mg2+ could be to accelerate catalysis by positioning the phosphate oxygen of the sugar-phosphate adjacent to the α-phosphate of CTP to align the incoming second substrate for in-line nucleophilic attack, as suggested for CTP:phosphocholine cytidylyltransferase of Streptococcus pneumoniae (20).
Concluding remarks.
The first 3D structure of CTP:inositol-1-phosphate cytidylyltransferase (IPCT) revealed an overall architecture similar to the dinucleotide-binding Rossmann fold and a fairly good match with the structures of glucose-1-phosphate thymidylyl/uridylyl-transferases. It is surprising that inositol-1P, a novel substrate for nucleotidyltransferases, does not imply a distinctive structural layout for catalysis. On the other hand, it is curious that IPCT is structurally less related to known cytidyltransferases, namely, those recognizing other polyol-phosphates, such as glycerol-, ribitol-, or methylerythritol-phosphate.
A BLAST search in the genome databases with the A. fulgidus IPCT sequence clearly shows a strong correlation of the closest matches with thermophilic or hyperthermophilic host organisms. This feature underlies the commitment of CDP-inositol to the synthesis of DIP, a compatible solute thus far exclusive to (hyper)thermophiles. This apparently specialized role of CDP-inositol is an intriguing finding that challenges the usual, multitasking character of most metabolites.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Fundação para a Ciência e a Tecnologia (FCT) through projects PTDC/SAU-NEU/103720/2008 (M.A.) and PTDC/BIA-MIC/71146/2006 (H.S.). J.A.B. is the recipient of FCT fellowship BD/30512/2006. We thank the ESRF for financial and technical support for data collection. The NMR spectrometers are part of the National NMR Network (REDE/1517/RMN/2005), supported by the Programa Operacional Ciência e Inovação and FCT.
We acknowledge M. Henriques and I. Pacheco for technical assistance and Pedro Matias for help during data collection.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 4 March 2011.
REFERENCES
- 1. Abrahams J. P., Leslie A. G. 1996. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52:30–42 [DOI] [PubMed] [Google Scholar]
- 2. Baur S., Marles-Wright J., Buckenmaier S., Lewis R. J., Vollmer W. 2009. Synthesis of CDP-activated ribitol for teichoic acid precursors in Streptococcus pneumoniae. J. Bacteriol. 191:1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blankenfeldt W., Asuncion M., Lam J. S., Naismith J. H. 2000. The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). EMBO J. 19:6652–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borges N., Marugg J. D., Empadinhas N., da Costa M. S., Santos H. 2004. Specialized roles of the two pathways for the synthesis of mannosylglycerate in osmoadaptation and thermoadaptation of Rhodothermus marinus. J. Biol. Chem. 279:9892–9898 [DOI] [PubMed] [Google Scholar]
- 5. Bradford M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 6. Bricogne G., et al. 2009. BUSTER, version 2.8.0. Global Phasing, Ltd., Cambridge, United Kingdom [Google Scholar]
- 7. Brito J. A., Borges N., Santos H., Archer M. 2010. Production, crystallization and preliminary X-ray analysis of CTP:inositol-1-phosphate cytidylyltransferase from Archaeoglobus fulgidus. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 66:1463–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cowtan K. 2006. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62:1002–1011 [DOI] [PubMed] [Google Scholar]
- 9. Cowtan K. 1994. ‘dm’: an automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newsl. Protein Crystallogr. 31:34–38 [Google Scholar]
- 10. de la Fortelle E., Bricogne G. 1997. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276:472–494 [DOI] [PubMed] [Google Scholar]
- 11. DeLano W. (ed.). 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- 12. Emsley P., Lohkamp B., Scott W. G., Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evans P. R. 1997. Scaling of MAD data, p. 97–102 In Wilson K. S., Davies G., Ashton A. W., Bailey S. (ed.), Recent advances in phasing: Proceedings of the CCP4 Study Weekend Council for the Central Laboratory of the Research Councils, Daresbury Laboratory, Warrington, United Kingdom [Google Scholar]
- 14. Holm L., Sander C. 1996. Mapping the protein universe. Science 273:595–603 [DOI] [PubMed] [Google Scholar]
- 15. Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26:795–800 [Google Scholar]
- 16. Kim H., et al. 2010. Structural basis for the reaction mechanism of UDP-glucose pyrophosphorylase. Mol. Cells 29:397–405 [DOI] [PubMed] [Google Scholar]
- 17. Koropatkin N. M., Holden H. M. 2004. Molecular structure of alpha-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi. J. Biol. Chem. 279:44023–44029 [DOI] [PubMed] [Google Scholar]
- 18. Krissinel E., Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 19. Krissinel E., Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60:2256–2268 [DOI] [PubMed] [Google Scholar]
- 20. Kwak B. Y., et al. 2002. Structure and mechanism of CTP:phosphocholine cytidylyltransferase (LicC) from Streptococcus pneumoniae. J. Biol. Chem. 277:4343–4350 [DOI] [PubMed] [Google Scholar]
- 21. Laemmli U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 22. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291 [Google Scholar]
- 23. Lovell S. C., et al. 2003. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50:437–450 [DOI] [PubMed] [Google Scholar]
- 24. Matthews B. W. 1968. Solvent content of protein crystals. J. Mol. Biol. 33:491–497 [DOI] [PubMed] [Google Scholar]
- 25. McCoy A. J., et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Vagin A. A., Dodson E. J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 27. Pape T., Schneider T. R. 2004. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37:843–844 [Google Scholar]
- 28. Rao S. T., Rossmann M. G. 1973. Comparison of super-secondary structures in proteins. J. Mol. Biol. 76:241–256 [DOI] [PubMed] [Google Scholar]
- 29. Rodionov D. A., et al. 2007. Genomic identification and in vitro reconstitution of a complete biosynthetic pathway for the osmolyte di-myo-inositol-phosphate. Proc. Natl. Acad. Sci. U. S. A. 104:4279–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodrigues M. V., Borges N., Almeida C. P., Lamosa P., Santos H. 2009. A unique beta-1,2-mannosyltransferase of Thermotoga maritima that uses di-myo-inositol phosphate as the mannosyl acceptor. J. Bacteriol. 191:6105–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodrigues M. V., et al. 2007. Bifunctional CTP:inositol-1-phosphate cytidylyltransferase/CDP-inositol:inositol-1-phosphate transferase, the key enzyme for di-myo-inositol-phosphate synthesis in several (hyper)thermophiles. J. Bacteriol. 189:5405–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Santos H., Lamosa P., Faria T. Q., Borges N., Neves C. 2007. The physiological role, biosynthesis and mode of action of compatible solutes from (hyper)thermophiles. ASM Press, Washington, DC [Google Scholar]
- 33. Sheldrick G. M. 2008. A short history of SHELX. Acta Crystallogr. A Found. Crystallogr. 64:112–122 [DOI] [PubMed] [Google Scholar]
- 34. Thoden J. B., Holden H. M. 2007. Active site geometry of glucose-1-phosphate uridylyltransferase. Protein Sci. 16:1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Westhead D. R., Hatton D. C., Thornton J. M. 1998. An atlas of protein topology cartoons available on the World-Wide Web. Trends Biochem. Sci. 23:35–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.