

Abstract
CpxP is a novel bacterial periplasmic protein with no homologues of known function. In Gram-negative enteric bacteria, CpxP is thought to interact with the two-component sensor kinase, CpxA, to inhibit induction of the Cpx envelope stress response in the absence of protein misfolding. CpxP has also been shown to facilitate DegP-mediated proteolysis of misfolded proteins. Six mutations that negate the ability of CpxP to function as a signaling protein are localized in or near two conserved LTXXQ motifs that define a class of proteins with similarity to CpxP, Pfam PF07813. To gain insight into how these mutations might affect CpxP signaling and/or proteolytic adaptor functions, the crystal structure of CpxP from Escherichia coli was determined to 2.85-Å resolution. The structure revealed an antiparallel dimer of intertwined α-helices with a highly basic concave surface. Each protomer consists of a long, hooked and bent hairpin fold, with the conserved LTXXQ motifs forming two diverging turns at one end. Biochemical studies demonstrated that CpxP maintains a dimeric state but may undergo a slight structural adjustment in response to the inducing cue, alkaline pH. Three of the six previously characterized cpxP loss-of-function mutations, M59T, Q55P, and Q128H, likely result from a destabilization of the protein fold, whereas the R60Q, D61E, and D61V mutations may alter intermolecular interactions.
INTRODUCTION
CpxP is a small, 147-amino-acid, periplasmic protein with no homologues of known function. The cpxP gene was originally identified as an upregulated product of the Cpx envelope stress response pathway in Escherichia coli (8). The Cpx response senses and mediates adaptation to a variety of stresses that are thought to result in periplasmic protein misfolding (for recent reviews see references 36 and 51). These include the overexpression of pilus subunits in the absence of their cognate chaperones or assembly machineries (24, 40), as well as alkaline pH (8). A two-component sensor histidine kinase, CpxA, detects inducing signals and conveys this information by means of phosphorylation to the cognate response regulator, CpxR, which directs altered transcription of the Cpx regulon (44, 50). A number of Cpx-regulated genes are directly involved in protein folding or degradation in the bacterial envelope (inner membrane, periplasm, and outer membrane), including the chaperone/protease DegP (9, 10, 36, 44, 47, 51, 63).
CpxP functions as both a signaling protein to control the activity of CpxA and as a proteolytic adaptor to facilitate degradation of some misfolded proteins (11, 23, 48, 49). Overexpression of CpxP leads to downregulation of the Cpx response by inhibition of the autokinase activity of CpxA (11, 15, 48, 49), likely through direct interaction with the periplasmic sensing domain of CpxA. Inhibition of CpxA activity is relieved in the presence of inducing cues through DegP-mediated degradation of CpxP (5, 23). This step is likely preceded by titration of CpxP from the CpxA sensing domain by either or both misfolded proteins and DegP, as degradation of some misfolded pilus subunits by DegP is dependent upon CpxP (23).
In an effort to understand the molecular mechanism(s) by which CpxP carries out its signaling function, we performed a genetic screen for mutations that negate the ability of CpxP to inhibit CpxA (5). Mutations identified from the screen affect residues in or near two conserved LTXXQ motifs, a sequence for which no function has been associated that defines a large family of proteins (Pfam protein family database accession number PF07813 [http://pfam.sanger.ac.uk/family?entry=PF07813&type=Family]). Many of these proteins share similarity with CpxP and Spy, the only known homologue of CpxP. In this study, we determined the crystal structure of CpxP to 2.85-Å resolution, performed additional biochemical studies using purified protein, and further characterized the previously identified mutants using in vivo techniques. Our results define a role for the conserved LTXXQ motif in the large PF07813 family of proteins and identify several sites on the surface of CpxP that are likely involved in intermolecular interactions.
MATERIALS AND METHODS
Strains and plasmids.
Bacterial strains and plasmids used in this study are described in Table 1.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| MC4100 | F−araD139 Δ(argF-lac)U169 rpsL150 (Strr) relA1 flbB5301 decC1 ptsF25 rbsR | 6 |
| TR50 | MC4100 λRS88 (cpxP-lacZ+) | 50 |
| TR61 | MC4100 λRS88 (cpxP-lacZ+) arar | This study |
| TR757 | MC4100 λRS88 (cpxP-lacZ+) degP::Tn10 | 2 |
| TR930 | TR50 (pBBR1MCS) | 5 |
| TR932 | TR50 (pPB) | 5 |
| DB4 | TR757 (pPB) | 5 |
| DB5 | TR757 (pBBR1MCS) | This study |
| DB31 | TR50 (pPB2) | 5 |
| DB40 | TR50 (pPB6) | 5 |
| DB71 | TR757 (pPB2) | 5 |
| DB75 | TR757 (pPB6) | 5 |
| SS1 | TR757 (pMCP) | This study |
| Plasmids | ||
| pCpxP | cpxP overexpression vector (Ampr) | 49 |
| pCpxPQ55P | cpxP(Q55P) overexpression vector (Ampr) | 5 |
| PCpxPQ128H | cpxP(Q128H) overexpression vector (Ampr) | 5 |
| pMCP | Overexpresses a functional MBP-CpxP fusion protein (Ampr) | 49 |
| pPB | CpxP-Bla translational fusion overexpression vector; Ampr Camr | 5 |
| pPB2 | pPB encoding a CpxP(Q55P)-Bla mutation Ampr Camr | 5 |
| pPB6 | pPB encoding a CpxP(Q128H)-Bla mutation; Ampr Camr | 5 |
| ptrc99A | High expression vector with a multiple cloning site following an IPTG inducible trc promoter; Ampr | Pharmacia |
| pCpxP(40–151) | CpxP40–151 was amplified from pMCP and subcloned into pGEX-6P-1 using BamHI and EcoRI restriction sites | This study |
Media, antibiotics, and growth conditions.
Strains were grown on Luria-Bertani (LB) agar (52) at 30°C or 37°C. All liquid cultures were grown in LB broth with aeration at either 30°C or 37°C. Strains were maintained with the appropriate antibiotic selection, 100 μg/ml ampicillin (Amp), 25 μg/ml chloramphenicol (Cam), 25 μg/ml kanamycin (Kn), or 25 μg/ml tetracycline (Tet). All antibiotics were purchased from Sigma.
Expression and purification of wild-type CpxP for biochemical assays.
Wild-type CpxP was overexpressed as a maltose-binding protein (MBP) fusion from pMCP in TR757, a degP null strain (SS1) (Table 1). Cells containing the pMCP plasmid were grown at 30°C in LB medium with 2 g/liter glucose and ampicillin until they reached an optical density at 600 nm (OD600) of 0.7, at which point they were induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown overnight at 22°C. Cells were then harvested, resuspended, and osmotically shocked to release the periplasmic fusion protein as per the pMAL Protein Fusion and Purification System manual (41). The shock fluid was then applied to amylose resin (New England BioLabs) which was preequilibrated with wash buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT]). The MBP-CpxP fusion protein was eluted with elution buffer (wash buffer supplemented with 10 mM maltose [final concentration]); the buffer was exchanged into 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1 mM CaCl2 and concentrated to approximately 2 ml using a 10,000-molecular-weight-cutoff (MWCO) spin concentrator (Millipore, Fisher Scientific). The concentrated sample was quantitated using the theoretical extinction coefficient at 280 nm of 78,840 M−1 cm−1 calculated using ProtParam (16) from the primary sequence of MBP-CpxP. Factor Xa protease (GE Healthcare) was then added to a 2.5% (wt/wt) ratio of Factor Xa/MBP-CpxP and incubated for 16 h at 4°C. The cleavage reaction was monitored by SDS-PAGE, and the reaction was stopped with phenylmethylsulfonyl fluoride (PMSF) (Sigma). The cleavage reaction was diluted in wash buffer and reflowed over the amylose resin to separate CpxP from MBP and residual fusion protein. The CpxP-containing flowthrough was concentrated using a 5,000-MWCO spin concentrator (Millipore, Fisher Scientific) and applied to a Superdex 75 26/60 gel filtration column (GE Healthcare) which was equilibrated in buffer containing 250 mM NaCl, 1 mM EDTA, and 50 mM sodium phosphate (pH 5.8 or 8.0). The purified CpxP was subjected to matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (University of Alberta Chemistry Mass Spectrometry Facility), where it was found to have an average m/z of 17,336 ± 5, which corresponded to the protein having its last two C-terminal amino acids removed. Amino acid analysis (Alberta Peptide Institute) produced an experimental extinction coefficient at 280 nm of 12,214 M−1 cm−1, which was subsequently used for protein quantitations.
Size exclusion chromatography with MALLS.
Fifty microliters of a 2 mg/ml CpxP sample was injected at 0.5 ml/min onto a Superose 6 HR 10/300 gel filtration column (GE Healthcare) preequilibrated with 50 mM sodium phosphate (pH 5.8 or 8.0), 250 mM NaCl, and 1 mM EDTA. After flowing over the column, the effluent was directly passed over an in-line Dawn EOS multiangle laser light-scattering (MALLS) apparatus and an Optilab rEX differential refractive index detector (Wyatt Technologies, Santa Barbara, CA). Light scattering data were processed using Astra, version 4.90, software. Average molecular weights were calculated from the elution peak (35). A minimum of two runs were collected at each pH, from which the average and standard deviation were determined.
Formaldehyde-mediated in vivo cross-linking and Western blot analysis.
Cross-linking experiments were performed as described in Larsen et al. (30), with minimal changes. Cells were subcultured at 1:50 in 5 ml of fresh medium with the corresponding antibiotics and grown until mid-log phase (OD600 of 0.6). Cells were harvested and resuspended in the original volume of 0.1 M sodium phosphate buffer (pH 6.8). Fifty microliters of formaldehyde (37%, wt/wt) (Fisher) was added to 1-ml sample aliquots. Samples were mixed at room temperature using a Labquake mixer (Fisher) for either 10 or 15 min, centrifuged, and resuspended in 50 μl of 2× SDS-PAGE loading buffer. Samples were analyzed via Western blot analysis. Strains were grown overnight at 30°C with aeration. The next day, overnight cultures were diluted 1:50 into 5 ml of LB medium with the appropriate antibiotic. Cultures were then grown at 37°C for 4 h, and the OD600s of the cultures were measured. Whole-cell extracts were prepared by pelleting volumes that were adjusted to correspond to 1 ml of the culture that reached the lowest OD600 and resuspended in 50 μl of 2× SDS-PAGE loading dye. The membrane was labeled with polyclonal CpxP antibody (1:25,000), maltose-binding protein antibody (1:50,000), or β-lactamase monoclonal antibody (1:1,000 dilution) (QED BioScience Inc.). A secondary anti-mouse antibody conjugated to alkaline phosphatase (Sigma) was used at a 1:25,000 dilution to detect the CpxP-Bla fusion, and a secondary anti-rabbit antibody conjugated to alkaline phosphatase (Sigma) was used at a 1:50,000 dilution to detect the CpxP-MBP fusion and native CpxP protein. Proteins were visualized using the Immun-Star AP substrate pack chemiluminescent kit (Bio-Rad).
SAXS analysis.
Small-angle X-ray scattering (SAXS) curves were collected on beam line 12.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory (Berkeley, CA). Data were collected on wild-type CpxP at 7.5 mg/ml dialyzed against 250 mM NaCl, 50 mM sodium phosphate (at either pH 5.8 or 8.0), 1 mM EDTA, and 10% glycerol. CpxP exhibited no radiation-induced damage, as determined by comparison of a sequential series of exposure times, two 6-s, one 60-s, and a final 6-s exposure (data not shown). Guinier plots (data not shown) plotted in PRIMUS (27) were linear in the range of s · Rg < 1.3, where s is momentum transfer and Rg is the radius of gyration, indicative of nonaggregated samples. The radii of gyration calculated from the scattering data using the Guinier plots were 23.3 ± 0.1 and 22.3 ± 0.1 Å for the protein at pH 5.8 and 8.0, respectively. The distance distribution function, a transformation from reciprocal to real space, calculated using the program GNOM (53), gave mean estimates of the maximum particle dimension (Dmax) of 66 and 60 Å for all four exposures in the series for the pH 5.8 and 8.0 samples, with corresponding Rg of 22.8 and 21.9 Å, respectively. The program GASBOR (54) was used to generate an ab initio low-resolution envelope of wild-type CpxP at pH 5.8 (see Fig. S3B and C in the supplemental material). The envelope was constructed from the 60-s exposure using 15 models from independent GASBOR simulations having P2 symmetry imposed. The resulting models were superimposed and averaged with DAMAVER and filtered with DAMFILT (55). The X-ray model was docked into the SAXS envelope at pH 5.8 using SITUS (66).
CD.
Circular dichroism (CD) experiments were performed on a Jasco J720 spectropolarimeter. Near-UV (320 to 255 nm) experiments used 1 mg/ml samples of CpxP in a 1-cm path length thermostated fused silica cell at 24°C. CpxP samples were in 50 mM sodium phosphate (pH 5.8 or 8.0), 250 mM NaCl, 1 mM EDTA. Raw ellipticities were subtracted from the buffer and converted into mean residue ellipticities using the average amino acid weight of 117.1 Da, calculated from the CpxP primary sequence. Near-UV spectra were an average of 12 scans. For melting temperature (Tm) experiments, ellipticity was recorded at 220 nm for 5 min at each temperature, measuring a reading every second. From the data, an average ellipticity at each temperature was calculated. CpxP samples were at a concentration of 0.25 mg/ml in 250 mM NaCl, 1 mM EDTA, and 50 mM sodium phosphate (pH 5.8 or 8.0) in a 0.05-cm silica cell. The samples were equilibrated for 5 min at each temperature before data collection. Temperatures measured ranged from 24 to 62°C, employing a Lauda water bath (Brinkmann Instruments) to control the temperature of the cell. The loss of ellipticity at 220 nm indicated unfolding of CpxP, and the Tm is defined as the temperature at which the ellipticity at 220 nm decreases by half. Melting temperatures were calculated using the SigmaPlot 2001 software (SPSS Inc.). Tm values represent an average of two experiments.
Limited proteolysis of wild-type CpxP.
Five micrograms of purified wild-type CpxP in 50 mM sodium phosphate (pH 7.0), 250 mM NaCl, and 1 mM EDTA was digested with 0, 5 × 10−4, 0.001, 0.005, 0.01, 0.05, and 0.1 μg of trypsin (Sigma-Aldrich) for 60 min at 37°C. Reactions were stopped with PMSF, and the reaction products were separated on an 18% SDS-PAGE gel. MALDI-TOF mass spectrometry (University of Alberta Chemistry Mass Spectrometry Facility) was used to identify the molecular mass of the two digestion products from a parallel trypsin digest of CpxP at a 1/100 (wt/wt) ratio of trypsin/CpxP. The two fragments had average m/z of 15,990 and 13,336 ± 5. Analysis using the program MS-Digest (in the ProteinProspector suite; Mass Spectrometry Facility, University of California, San Francisco) showed that the fragments corresponded to residues 20 to 151 and 40 to 151, respectively.
Cloning and purification of recombinant CpxP for structure determination.
The sequence corresponding to residues 40 to 151 of CpxP (CpxP40–151) was amplified from full-length cpxP in the pMCP vector (Table 1) using the PCR primers 5′ (5′-GGCGGCGGATCCAGTACGCAGAGCCATATGTTCGACGGCATAAGTTTA) and 3′ (5′-GGCGGCGAATTCCTATTTTTGCCATTGCGTCACGTCACG). The fragment was ligated into the BamHI and EcoRI restriction sites of the pGEX-6P-1 expression vector (GE Healthcare), which contains N-terminal glutathione S-transferase (GST) tag and PreScission Protease cleavage site sequences. The subclones were confirmed by DNA sequencing (The Applied Genomics Centre, University of Alberta). The CpxP40-151-containing plasmid was transformed into Escherichia coli BL21(DE3) cells. Cells were cultured at 37°C in Luria-Bertani medium with 100 μg/ml ampicillin. Upon reaching an optical density at 600 nm of 0.8 to 0.9, CpxP overexpression was induced with 0.05 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and the cells were grown for a further 20 h at 30°C. CpxP40-151 was purified using a protocol modified from the GST Gene Fusion System Handbook (17). Cells were harvested by centrifugation, and the cell pellet was resuspended in buffer composed of 50 mM 2-(N-morpholino)ethanesulfonic acid (MES), pH 5.8, and 150 mM NaCl, plus protease inhibitors. Cells were lysed by chicken egg white lysozyme and sonication, and the lysate was fractionated by centrifugation. The clarified lysate was applied to glutathione Sepharose 4B resin (GE Healthcare), and the captured GST fusion protein was eluted using resuspension buffer containing 20 mM reduced glutathione. The elutions were pooled, and buffer was exchanged into resuspension buffer and concentrated to a small volume (∼2 ml) using a 10,000-MWCO centrifugal filter device (Millipore/Fisher Scientific). The GST tag was removed by PreScission protease (GE Healthcare) over 16 h at 4°C, and the cleavage reaction mixture was applied to a second GST affinity column to remove proteolyzed GST and any remaining fusion protein. The flowthrough was concentrated using a 5,000-MWCO spin concentrator (Millipore/Fisher Scientific). Size exclusion chromatography was performed as a final purification step using a HiLoad Superdex 75 26/60 gel filtration column equilibrated in resuspension buffer on an ÄKTApurifier (GE Healthcare). Fractions were pooled, and the protein was dialyzed into storage buffer containing 50 mM MES, pH 5.8, and 50 mM NaCl and concentrated by ultrafiltration using a 5,000-MWCO spin concentrator. The protein concentration was estimated from the absorbance at 280 nm using the theoretical extinction coefficient of 6,990 M−1 cm−1, which was derived from the amino acid sequence of CpxP40-151 using ProtParam (16). Selenomethionine (Se-Met)-substituted CpxP40-151 was produced in cells grown in M9 minimal medium supplemented with amino acids and seleno-l-methionine (Sigma-Aldrich). The Se-Met-labeled protein was purified using the same methods as above, with resuspension buffer composed of 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, and 0.1% β-mercaptoethanol and storage buffer consisting of 50 mM HEPES, pH 7.0, 50 mM NaCl, and 1 mM DTT.
Crystallization and data collection.
Initial crystallization conditions for CpxP40–151 were identified from the Emerald Wizard II suite, N11 (Emerald Biosystems) and were further optimized. Se-Met-substituted CpxP40–151 was crystallized by hanging-drop vapor diffusion at room temperature (∼21°C) by mixing 1 μl of CpxP at 24 mg/ml in the final storage buffer with 1 μl of reservoir solution containing 9% (vol/vol) 2-propanol, 0.1 M sodium cacodylate, pH 5.4, 0.7 M Zn(OAc)2, and 1 mM DTT. Native protein was crystallized under similar conditions by mixing 2 μl of CpxP at 37 mg/ml in the final storage buffer with 1.5 μl of reservoir solution containing 9% (vol/vol) 2-propanol, 0.1 M sodium cacodylate, pH 5.6, and 0.7 M Zn(OAc)2). No additional cryoprotectant was required, and the crystals were flash frozen in liquid nitrogen. All diffraction data were collected at the Canadian Light Source, beam line CMCF 08ID-1 (Saskatoon, Canada). A multiwavelength anomalous dispersion (MAD) data set was collected from the Se-Met-substituted crystal at three wavelengths (peak, 0.97945 Å; inflection, 0.97961 Å; and remote, 0.97754 Å). Zinc single-wavelength anomalous dispersion (SAD) and native data sets were collected from the native crystal at the zinc peak (1.28197 Å) and remote (0.97949 Å) wavelengths, respectively.
Structure solution and refinement.
All diffraction data sets were processed and integrated with DENZO and scaled in the space group P6122 using SCALEPACK (HKL2000 suite) (43). The structure of CpxP was solved by the MAD method using the selenomethionine derivative data sets processed to 2.9-Å resolution. The heavy atom positions for 15 of the 16 expected selenium atoms (given two molecules in the asymmetric unit) were determined and used to obtain preliminary phases in SOLVE (59). Density modification of phases and automatic model building into the calculated electron density map were performed by the program RESOLVE (57, 58). However, the partial models obtained from automatic building needed to be rebuilt manually using both 2.9 Å- and 2.6 Å-resolution remote 2Fo − Fc and Fo − Fc (where Fo and Fc are observed and calculated structure factor amplitudes, respectively) electron density maps and the known locations of the selenium atoms to place the CpxP sequence into the maps in COOT (14). The starting model was completed by iterative rounds of model building and refinement using REFMAC5 in the CCP4 suite (7, 39, 46, 65). Although it was speculated from large positive difference densities remaining in the maps and peaks in the difference Fourier maps that zinc ions could be present, placing these in the model did not aid the refinement. Therefore, additional data sets were collected from a single native crystal at both the zinc peak and at a remote wavelength (Table 2). SAD phases were determined for the zinc peak data using SOLVE (59), and zinc sites were located. Further model building and refinement were carried out using COOT and PHASER against the 2.85-Å data collected on the native CpxP crystal at the remote wavelength (14, 37). Nine zinc sites identified from the zinc SAD solution were added to the model. Geometry restraints were applied to zinc sites where possible using ideal values obtained from Tamames et al. (56). One translation-libration-screw (TLS) group was refined for each of the two chains in the later stages of refinement using PHENIX (1). Although CpxP is a dimer, the application of noncrystallographic symmetry (NCS) was not beneficial at any stage of refinement. The final model consisted of amino acid residues 44 to 151 of protomer A and of residues 40 to 151 of protomer B. The four N-terminal residues of protomer A were not observed in the electron density map as a result of disorder. The Rwork and Rfree of the final model were 24.7% and 29.8%, respectively. Analysis of the final model with PROCHECK (31) revealed 89.4% of residues in the most favored regions of the Ramachandran plot and 9.6% in additional allowed regions, with no residues in the generously allowed region. Two residues, corresponding to Leu51 from each protomer, were found to be in disallowed regions. However, these residues form a critical part of the conserved LTXXQ motif and pack in a hydrophobic region under the turn, for which there is good electron density. The data collection and refinement statistics are summarized in Table 2.
Table 2.
Data collection and refinement statistics
| Parameter | Value for the CpxP40–151 native crystal at: |
|
|---|---|---|
| Remote wavelength | Zinc peak | |
| Data collectiona | ||
| Space group | P6122 | P6122 |
| Unit cell parameters | ||
| a = b, c (Å) | 88.03, 134.93 | 88.10, 135.05 |
| α = β, γ (°) | 90.0, 120.0 | 90.0, 120.0 |
| Wavelength (Å) | 0.97949 | 1.28197 |
| Resolution (Å) | 41.85–2.85 (2.95–2.85) | 44.05–3.20 (3.31–3.20) |
| Rsymb | 0.042 (0.489) | 0.045 (0.510) |
| I/σ(I) | 34.2 (3.1) | 27.7 (3.2) |
| Completeness (%) | 99.8 (100.0) | 100.0 (100.0) |
| Redundancy | 9.9 (5.9) | 8.9 (9.0) |
| Refinement | ||
| Resolution (Å) | 44.0–2.85 | |
| No. of reflectionsc | 13,092 (588) | |
| Rwork/Rfreed | 0.247/0.298 | |
| No. of atoms | ||
| Protein | 1,834 | |
| Zinc ions | 9 | |
| Water | 15 | |
| B factors | ||
| Protein | 90 | |
| Zinc ions | 104 | |
| Water | 62 | |
| RMSD | ||
| Bond length (Å) | 0.015 | |
| Bond angle (°) | 1.183 | |
| Ramachandran statistics (%) | ||
| Most favored regions | 89.4 | |
| Additional allowed regions | 9.6 | |
| Generously allowed regions | 0.0 | |
| Disallowed regions | 1.0 | |
Data collection statistics for the highest resolution shell are shown in parentheses.
Rsym = Σ|I − <I>|2/Σ|I2|.
Value in parentheses represents the number of reflections used in the calculation of Rfree.
R = Σ||Fo| − |Fc||/Σ|Fo|.
The dimeric interface of CpxP was analyzed using PISA (28). The electrostatic surface potential was computed and visualized using the PDB2PQR server (12, 13) in conjunction with the APBS plug-in in PyMOL (4; PyMOL molecular graphics system, version 1.2, Schrödinger LLC, New York, NY; APBS [adaptive Poisson-Boltzmann solver] plugin, M. G. Lerner and H. A. Carlson, University of Michigan, Ann Arbor, MI). The two CpxP protomers were superpositioned, and the root mean square deviation (RMSD) between them was calculated using the maximum-likelihood alignment method in THESEUS (60). The secondary structure in the atomic model was delineated by DSSP (3, 25). The structural coordinates of CpxP were compared to those of all structures in the Protein Data Bank (PDB) in a search performed by the Dali server (20, 21). Figures were generated using PyMol (PyMOL molecular graphics system, version 1.2; Schrödinger LLC, New York, NY).
β-Galactosidase assays.
β-Galactosidase assays were performed in triplicate as described previously by Buelow and Raivio (5).
Protein structure accession number.
The atomic coordinates and structure factors for CpxP40–151 have been deposited in the RCSB Protein Data Bank (PDB) under accession number 3QZC.
RESULTS
CpxP forms a dimer that undergoes a subtle structural rearrangement in response to alkaline pH.
In order to determine the oligomeric state of wild-type E. coli CpxP, size exclusion chromatography was performed in conjunction with multiangle laser light scattering (MALLS) at pH 8.0, a condition where the Cpx pathway is activated, and pH 5.8, where the pathway is repressed (8). The elution volume of CpxP from the gel filtration column was found to be independent of pH (see Fig. S1A in the supplemental material). Additionally, light scattering data established that at both pHs CpxP formed a dimer (pH 5.8, MW of 34,400 ± 500; pH 8.0, MW of 33,800 ± 1,000), which indicates that CpxP does not undergo a change in oligomeric state in response to the inducing cue of alkaline pH in vitro. To establish the oligomeric state of CpxP in vivo, wild-type CpxP or various CpxP fusion proteins were overexpressed and cross-linked with formaldehyde. In the absence of cross-linker, monomers of the expected molecular weights for the MBP-CpxP and CpxP-Bla fusion proteins and CpxP were observed (see Fig. S1B, lanes 1). Within 10 min of incubation with formaldehyde, major cross-reactive protein species were present at 114 kDa, 98 kDa, and 34 kDa, which are consistent with double the monomeric weight of MBP-CpxP, CpxP-Bla, and CpxP, respectively (see Fig. S1B, lanes 2). This experiment suggests that in vivo CpxP exists as a dimer.
Since size exclusion chromatography and MALLS experiments did not reveal a change in the oligomeric state of full-length CpxP in response to the pathway-inducing cue of alkaline pH (see Fig. S1A in the supplemental material), small-angle X-ray scattering (SAXS) and circular dichroism (CD) spectroscopy methods were used to investigate whether the structure of CpxP was altered under the same conditions (Fig. 1A to D). The values for the radius of gyration (Rg) and maximum particle dimension (Dmax) from SAXS revealed a small but reproducible compression of the structure at pH 8.0 compared to pH 5.8 (Fig. 1B). A small increase in the protein fold stability was also detected through a slight rise in the melting temperature (Tm) of CpxP at the higher pH as measured by CD (Fig. 1C). Near-UV CD experiments (320 to 255 nm) identified variation in the chemical environment of one or both tryptophan residues in CpxP (W30 and W149) with a change in pH from 5.8 to 8.0 (Fig. 1D). Cumulatively, these results suggest that CpxP undergoes slight conformational adjustment to a more compact and stable form upon a shift from pH 5.8 to 8.0.
Fig. 1.
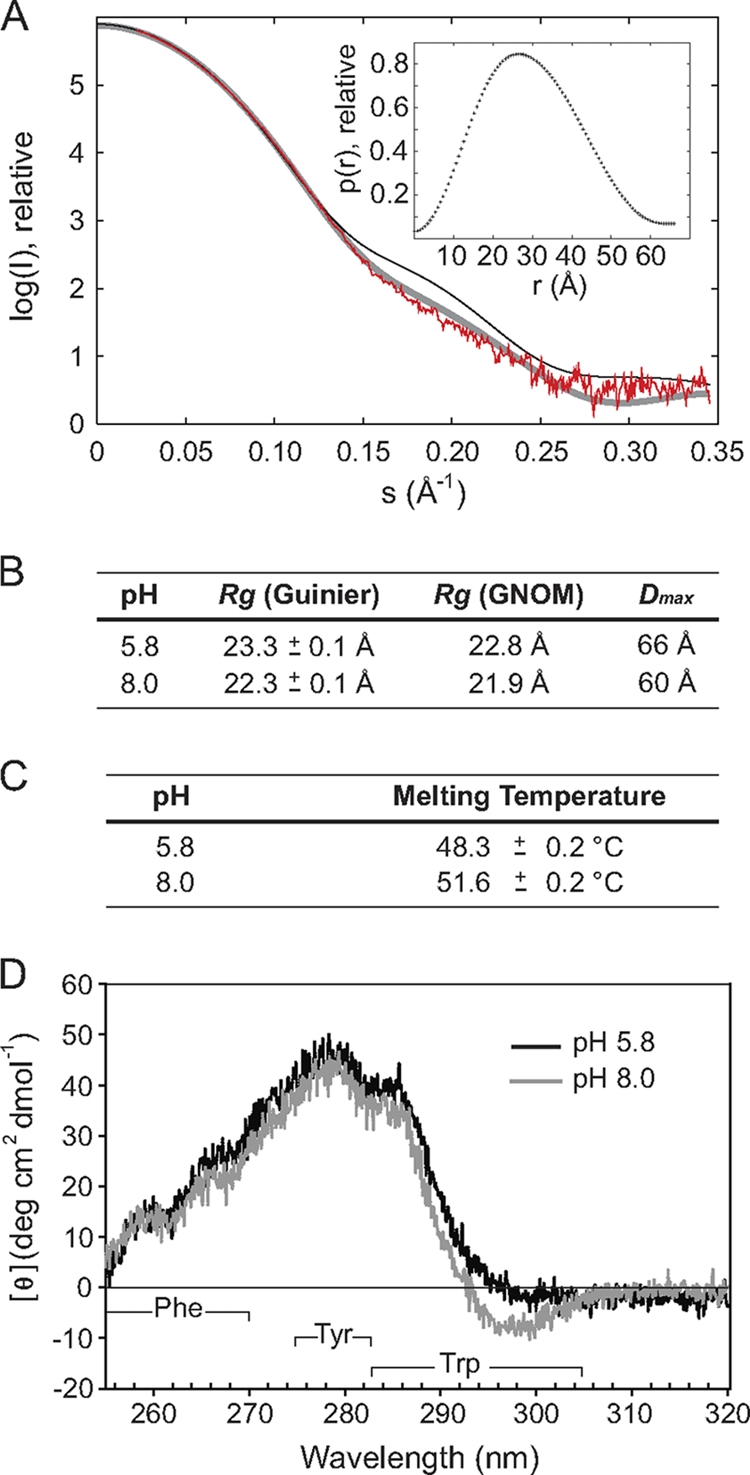
A small structural rearrangement occurs in CpxP in response to the inducing cue of alkaline pH. (A) Small-angle X-ray scattering curve of wild-type CpxP at pH 5.8 with a 60-s exposure (red). Overlaid are the back-calculated theoretical scattering curves of the CpxP X-ray crystal structure (thin black line) and a model CpxP dimer constructed from chain B of the X-ray structure (thick gray line). The chain B-chain B dimer model provides a better fit (χ2 = 1.8) to the solution SAXS data than the X-ray crystal structure (χ2 = 4.2) by effectively removing the crystal-pack-constrained orientation of α4 from chain A. The inset shows the distance distribution function, p(r), calculated using GNOM. The symmetrical bell-shaped curve is typical of a folded, globular particle. (B) Summary of values for the radius of gyration (Rg) calculated using both the Guinier approximation and GNOM and for the maximum dimension of the particle (Dmax) for CpxP at pH 5.8 and pH 8.0. (C) Summary of the melting temperatures (Tm) for CpxP at pH 5.8 and 8.0, calculated from the temperature at which the average ellipticity at 220 nm is decreased by half. (D) Near-UV CD spectra of CpxP at pH 5.8 (black) and 8.0 (gray). The Phe, Tyr, and Trp signal wavelength ranges are indicated. Each of the CD experiments was repeated in duplicate, and representative spectra are shown.
CpxP is a curved α-helical dimer with a highly basic concave face.
Wild-type CpxP was subjected to limited proteolysis to identify an ordered protein domain suitable for crystallization (see Fig. S2 in the supplemental material). Mass spectrometry revealed that the smallest stable domain corresponded to residues 40 to 151, which was sufficient to inhibit the Cpx response (data not shown). CpxP40–151 was purified and crystallized, and the structure was determined to 2.85-Å resolution using a combination of selenomethionine multiwavelength anomalous dispersion and zinc single-wavelength anomalous dispersion methods (Table 2).
The crystal structure revealed that CpxP is a largely helical dimer (Fig. 2A). The two protomers are intertwined in an antiparallel arrangement with an extensive, buried interface of ∼1,600 Å2. The dimer adopts an elongated, bowl-shaped structure, with a highly basic interior concave surface and a rather acidic convex outer surface (Fig. 2B). Each protomer can be described as a long, bent, and hooked hairpin made up of four α-helices (Fig. 2A). Helices α1 and α2 combine to span the length of the protein and are separated by a short region of random coil that permits a kink between the two helices. After a hairpin turn, helix α3 returns along the length of the protein, approximately parallel to α1 and α2 and with a bend opposite the coil region between α1 and α2. Together, these three helices form the bent hairpin fold. After another short turn, α4 runs alongside the N terminus and completes the protein, with the N terminus and α4 comprising the hooks on the end of the hairpin.
Fig. 2.

The structure of CpxP reveals an α-helical dimer with a highly basic surface. (A) Cartoon representation of the structure of CpxP40–151, shown in two orientations that are rotated by 90° about the vertical axis. Chain A is depicted in blue, while chain B is green. The conserved N- and C-terminal LTXXQ motifs are displayed in gold and purple, respectively. Helices α1 to α4 and the N and C termini are labeled. (B) Electrostatic potential distribution on the molecular surface of the CpxP dimer. The concave surface is highly basic (dark blue), while the convex surface (180° rotation around the vertical axis) is mainly acidic (red). Hydrophobic regions are shown in white.
The two protomers are similar to each other in overall structure yet are distinct, with an overall root mean square deviation (RMSD) of 3.1 Å between the Cα atoms of the two chains. Most of the variation occurs in the coil region between helices α1 and α2, the angle of offset of α4, and the N and C termini of the protein (see Fig. S3A in the supplemental material). The divergence between the protomers in all of these regions is likely due to distortion caused by the close packing of a symmetry-related molecule to the coil region and N terminus of chain B and to helix α4 and the C terminus of chain A. The deformation of these segments is correlated with high B factors for the affected atoms, which are indicative of high thermal motion or flexibility. The crystal structure was found to be in good agreement with data and an ab initio molecular envelope generated from SAXS experiments of CpxP in solution, taking into account the distortion due to the crystal pack (Fig. 1A; see also Fig. S3B and C in the supplemental material). The consistency between the X-ray and SAXS structures demonstrates that the crystal structure is representative of the CpxP dimer in solution.
Mutations in and near the conserved LTXXQ motifs disrupt CpxP regulatory function.
The crystal structure of CpxP revealed that the LTXXQ motifs are situated proximal to each other at one end of the monomer, with the beginning of the first motif (residues 51 to 55) positioned before α1 and the second motif (residues 124 to 128) between α3 and α4 (Fig. 2A and 3A). Each LTXXQ motif forms a diverging turn, stabilized by the conserved leucine (L51 or L124) that packs in a hydrophobic region under the turn, and the conserved threonine (T52 or T125), that caps the N terminus of the following α helix (Fig. 3B and C). Hydrogen bond interactions between the conserved glutamine residues of the N- and C-terminal LTXXQ motifs (Q55 and Q128) link the two motifs to one another and likely stabilize the overall structure of the protomer (Fig. 3B and D).
Fig. 3.

Loss-of-function mutations are located in and near the highly conserved LTXXQ motifs of CpxP. (A) Structure-based sequence alignment of CpxP with Spy, ZraP, and CnrX, generated using Dali (20, 21) with numbering from the full protein sequences (62). The conserved N- and C-terminal LTXXQ motifs, located at residues 51 to 55 and 124 to 128 of CpxP, are shown in gold and purple, respectively. The LTXXQ motifs of Spy and ZraP are shown in brown. Residues of chain A that contribute to the dimerization interface of CpxP are highlighted in blue, while the locations of helices α1 to α4 are illustrated below the alignment. Three of the six previously identified cpxP loss-of-function mutations (5), Q55P, M59T, and Q128H, which are predicted to impact the structure or stability of CpxP, are listed in orange above the CpxP sequence. The three remaining mutations, R60Q, D61E, and D61V, that are predicted to have functional implications on CpxP signaling or chaperone activity are indicated above the CpxP sequence in gray. (B to D) Cartoon representation of the CpxP structure illustrating the locations of the conserved LTXXQ motifs and residues conveying loss-of-function mutations. (C) Hydrophobic region including L51 and M59. (D) Hydrogen bonds between the conserved glutamine residues of the N- and C-terminal LTXXQ motifs (Q55 and Q128). Color-coding is the same as for panel A, with chain A of CpxP in blue and chain B in green. Residues of the LTXXQ motifs, mutated residues, and residues of the hydrophobic region surrounding L51 and M59 are shown as sticks. Prominent residues, helices α1 to α4, and the N and C termini are labeled.
All six of the cpxP loss-of-function mutations previously identified (5) cluster to the N- and C-terminal LTXXQ motifs and a segment of α1 adjacent to the N-terminal LTXXQ motif (Fig. 3A to D). Three of these mutations, M59T, Q55P, and Q128H, resulted in reduced levels of CpxP, which were returned to near normal levels in a degP-null strain (5). M59 resides in the α1 helix and forms part of a hydrophobic protein core beneath the N-terminal LTXXQ motif (Fig. 3A to C). The introduction of the smaller, hydrophilic threonine side chain at this position in the M59T mutant would likely disrupt the hydrophobic packing of the α1 and α3 helices, thereby destabilizing the protein fold. Mutation of the conserved glutamine residue within each LTXXQ motif (Fig. 3A, B, and D) to Q55P or Q128H would eliminate the double hydrogen bond that links the two motifs and would therefore reduce the stability of the mutant protein. Additionally, the substitution to P55 may impose a conformational constraint on the protein backbone that might disrupt the α1 helix. Thus, it is likely that each of these three mutations lead to a loss of function through a reduction in protein stability.
Since protein levels of CpxP with the Q55P and Q128H mutations [CpxP(Q55P) and CpxP(Q128H), respectively] were restored in the absence of DegP (5), we tested whether these mutations affected CpxP function in a degP mutant background. The ability of CpxP(Q55P) and CpxP(Q128H) to inhibit CpxA in both wild-type and degP mutant strains was examined. Plasmids overexpressing C-terminal β-lactamase fusions (previously shown not to affect CpxP function [5]) to either the wild-type, CpxP(Q55P), or CpxP(Q128H) proteins were transformed into wild-type and degP-null strains bearing a Cpx-regulated lacZ reporter gene. β-Galactosidase activity was measured as an indicator of Cpx transcriptional activation. As previously observed, overexpression of wild-type CpxP leads to a dramatic decrease in Cpx pathway activity in both strains (Fig. 4, compare lanes 1 and 2 to 3 and 4). In contrast, the Q55P mutant protein was unable to inhibit Cpx pathway activity in either the wild-type strain background, where the Q55P mutant protein is degraded by DegP, or the degP mutant strain, where the mutant CpxP protein level is higher (Fig. 4, lanes 5 and 6) (5). This result suggests that, even in the absence of proteolysis, the structural perturbation predicted to be a consequence of the Q55P mutation disrupts protein function. The Q128H mutant was similarly unable to inhibit the Cpx response in a wild-type strain; however, in contrast to the Q55P mutant, it partially regained the ability to inhibit Cpx pathway activity in the degP mutant strain (Fig. 4, lanes 7 and 8) (5). The Q128H mutation may allow for a weak interaction between the two LTXXQ motifs, via a single hydrogen bond between the substituted H128 and Q55 that could not be formed by the Q55P mutant, thus explaining the milder phenotype associated with the Q128H substitution. These data support the premise that the Q55P and Q128H mutations disrupt CpxP regulatory function by destabilizing critical structural elements of the conserved LTXXQ motifs.
Fig. 4.
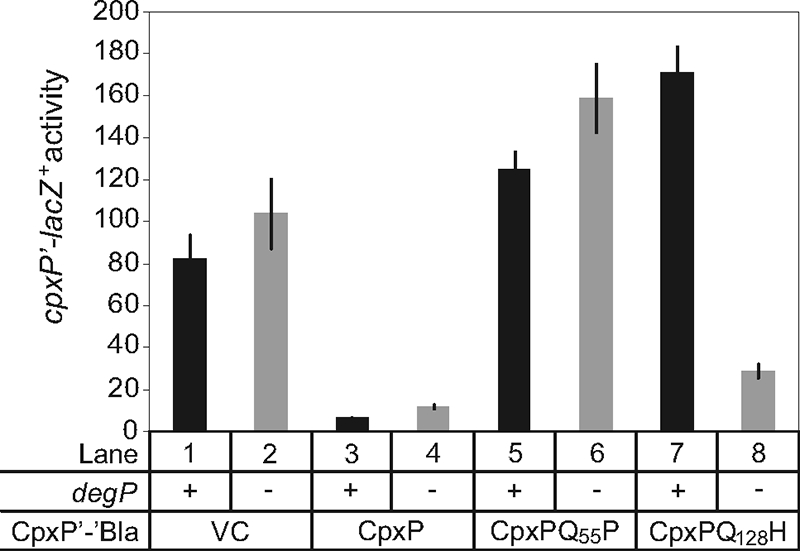
A CpxP(Q128H) mutant regains signaling activity in a degP mutant background. A chromosomal cpxP-lacZ+ fusion was used to determine the effect of overproduction of CpxP-Bla fusion proteins on the Cpx pathway in a degP-null background. β-Galactosidase levels were measured from wild-type (odd lanes) and degP-null strains (even lanes) carrying wild-type and mutant CpxP-Bla overexpression plasmids. Strains used in this experiment are as follows TR930 (lane 1), DB5 (lane 2), TR932 (lane 3), DB4 (lane 4), DB30 (lane 5), DB71 (lane 6), DB40 (lane 7), and DB75 (lane 8). VC, vector control.
DISCUSSION
The molecular mechanisms by which the periplasmic protein CpxP acts to control signaling by the sensor kinase CpxA and facilitate DegP-mediated proteolysis of misfolded pilus proteins are not known. The crystal structure of truncated E. coli CpxP, determined to 2.85-Å resolution, demonstrates that CpxP forms an antiparallel dimer of interconnected α-helices, with each protomer assuming a bent, hooked hairpin fold. SAXS studies illustrated that the crystal structure is a good representation of the structure of CpxP in solution. Residues Q55, Q128, and M59 of CpxP are specifically required to maintain protein structural integrity, while residues R60 and D61 are solvent exposed and could mediate interactions with other proteins.
CpxP has several potential protein binding partners that may interact with either of the discrete, charged surfaces. The highly basic concave cavity strongly suggests a role as a binding site for a negatively charged surface (Fig. 2B). It is also possible that molecules could recognize the acidic and hydrophobic patches on the convex surface of the CpxP structure. A large body of evidence suggests that CpxP interacts with the histidine kinase, CpxA (15, 48–50). Previous studies have also proposed that CpxP could act as an adaptor or chaperone, associating with misfolded envelope proteins to escort them to DegP for degradation or refolding (22, 23, 64).
Three mutations in two neighboring residues of CpxP, R60Q, D61E, and D61V, which are known to disrupt CpxP regulatory function but do not affect mutant protein levels (5), were found to be localized to a region of α1 close to the N-terminal LTXXQ motif (Fig. 3A to C). These mutations are not predicted to alter CpxP structural stability; therefore, residues R60 and D61 may be essential for interaction with the sensing domain of CpxA or might play a role in recognition of other binding partners such as DegP or misfolded proteins. Given that the highly conservative substitution of aspartic acid for glutamic acid at residue 61 obliterates signaling activity, the role of this residue must be highly specific. Notably, the homologous protein, Spy, contains a glutamic acid residue at this position (Fig. 3A) and does not function as an inhibitor of the Cpx response (48). Further investigation into the identity of any binding partners and the elucidation of their mechanism of recognition of CpxP will lead us closer to understanding the signaling, chaperone, and other possible activities of CpxP.
Searches of sequence and protein structure databases uncovered several proteins with a similar core fold to CpxP. Although the function of E. coli Spy, the closest homologue of CpxP with 26.5% sequence identity, remains unknown, its crystal structure reveals striking similarities. Spy (PDB code 3OEO) forms a nearly identical dimer comprised of antiparallel α-helical hairpins, similar positioning of the LTXXQ motifs, and a basic concave face (RMSD of 1.4 Å over 91 aligned Cα atoms) (Fig. 3A; see also Fig. S4C and D in the supplemental material) (29). A thorough examination of the differences in the structures of CpxP and Spy may reveal clues to their distinct roles in bacterial stress response pathways.
Another dimeric periplasmic protein, CnrX (PDB code 3EPV) (45), also bears strong structural resemblance to CpxP (RMSD of 3.0 Å over 101 aligned Cα atoms); however, they share only 8% sequence identity (Fig. 3A; see also Fig. S4E and F in the supplemental material). CnrX does not contain canonical LTXXQ motifs but forms comparable turns to those seen in CpxP and Spy. Instead, CnrX retains only the conserved leucines, which also pack under the turn, suggesting a critical role for this residue in the LTXXQ-like diverging turn.
The structure of the zinc resistance-associated protein ZraP/STM4172 (PDB code 3LAY) also adopts the bent hairpin fold common to CpxP and Spy (RMSD of 1.6 Å over 74 aligned Cα atoms). This periplasmic protein assembles into a decameric α-helical barrel, with hairpins forming antiparallel arrangements analogous to the CpxP dimer (Fig. 3A; see also Fig. S4G and H in the supplemental material). However, ZraP contains only one N-terminal LTXXQ motif, and the structure does not reveal the presence of the hooks in the hooked hairpin fold found in CpxP, Spy, or CnrX.
Analysis of the structures of CpxP, Spy, and ZraP reveals that the LTXXQ motif forms a structural element which incorporates a diverging turn in periplasmic stress response proteins with disparate functions, and it is likely that this role will be extended to the remaining members of this Pfam PF07813 family (http://pfam.sanger.ac.uk/family?entry=PF07813&type=Family). Furthermore, the family may be expanded to include LTXXQ-like turns, such as those observed in CnrX.
Both CnrX and ZraP function in bacterial stress response pathways that are responsible for the efflux of toxic metals from the cell (19, 33, 34, 42, 61). ZraP is a metal-binding protein that is induced by a two-component system that responds to zinc or lead (33, 42), while CnrX is thought to act as a metal sensor that binds cobalt, nickel, and potentially copper in an extracytoplasmic function (ECF)-sigma factor signaling pathway (18, 19, 34, 38, 61). Interestingly, copper ions have been implicated as an inducer of the Cpx pathway (26, 67–69). In addition, the mRNA levels of cpxP, zraP, and spy were found to increase by 11-, 47-, and 21-fold, respectively, in response to zinc ions (32). It is unknown whether the upregulation of CpxP stems from a genuine function related to these metals or if it is a consequence of general protein misfolding as a result of their presence. It is interesting, however, that CpxP crystallized in the presence of high zinc concentrations, and several zinc ions were found to be bound in the structure. While the biological significance of the candidate zinc sites in CpxP remains to be explored, the structural similarities between CpxP, CnrX, and ZraP may point to common periplasmic functions.
Two hypotheses regarding the fate of CpxP upon induction of the Cpx response are as follows: (i) the inducing cue might cause CpxP itself to become misfolded, leading to its proteolysis by DegP, (ii) alternatively, CpxP could interact with misfolded proteins to target them to DegP, where they would jointly be degraded (5, 23). Size exclusion chromatography, MALLS, SAXS, and CD experiments indicate that the shift to alkaline pH that triggers the Cpx response does not initiate a change in oligomeric state or large conformational rearrangements in CpxP in vitro. These biophysical techniques demonstrate that CpxP does not become a misfolded protein substrate suitable for DegP in the presence of natural Cpx pathway-inducing conditions in vitro. The absence of a structural change in CpxP is analogous to CnrX, for which no conformational changes were correlated with copper-binding, a pathway activating signal (38, 45). It is therefore likely that these antiparallel hooked-hairpin proteins sense or react to stress response cues through an alternative mechanism, perhaps by influencing the conformation of a binding partner such as DegP or the ECF sigma factor, CnrH, in the presence of an inducing ligand.
Here, we outline several sites, including the charged surfaces of CpxP and residues R60 and D61, which are likely involved in interactions with potential CpxP binding partners including CpxA, DegP, or misfolded proteins. We suggest that the conserved LTXXQ motifs in periplasmic stress response proteins have a structural role in the formation of diverging turns. And finally, we identify two additional periplasmic stress proteins that share common structural elements and similar core folds with CpxP and Spy; however, differences remain in the overall bent and hooked hairpin fold of CpxP and Spy that render them distinct.
Supplementary Material
ACKNOWLEDGMENTS
We thank James Gorin and Pawel Grochulski at beam line CMCF 1 at the Canadian Light Source, Saskatoon, Canada, for their support during X-ray crystallographic data collection and the staff at beam line 12.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA, for providing SAXS support. We thank Paul Scott for assistance with the MALLS experiments.
This work was supported by grants from the Natural Sciences and Engineering Research Council (T.L.R.) and Canadian Institutes of Health Research (T.L.R. and J.N.M.G), as well as the support of the Howard Hughes International Scholar Program (J.N.M.G) and the Alberta Heritage Foundation for Medical Research Senior Scholar Award (T.L.R.). G.L.T. is supported by an AHFMR Studentship Award. J.L.W. and G.L.T. are recipients of the CIHR Frederick Banting and Charles Best CGS Masters, and Doctoral Awards, respectively.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 11 February 2011.
ADDENDUM IN PROOF
While this paper was under review, a paper presenting a similar structure of CpxP was published. Their results are broadly in agreement with ours (X. Zhou et al., J. Biol. Chem., in press).
REFERENCES
- 1. Adams P. D., et al. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948–1954 [DOI] [PubMed] [Google Scholar]
- 2. Ades S. E., Grigorova I. L., Gross C. A. 2003. Regulation of the alternative sigma factor σE during initiation, adaptation, and shutoff of the extracytoplasmic heat shock response in Escherichia coli. J. Bacteriol. 185:2512–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andersen C. A., Palmer A. G., Brunak S., Rost B. 2002. Continuum secondary structure captures protein flexibility. Structure 10:175–184 [DOI] [PubMed] [Google Scholar]
- 4. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 98:10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buelow D. R., Raivio T. L. 2005. Cpx signal transduction is influenced by a conserved N-terminal domain in the novel inhibitor CpxP and the periplasmic protease DegP. J. Bacteriol. 187:6622–6630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Casadaban M. J. 1976. Transposition and fusion of lac genes to selected promoters in Escherichia coli using bacteriophages lambda and Mu. J. Mol. Biol. 104:541–555 [DOI] [PubMed] [Google Scholar]
- 7. Collaborative Computational Project, Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 8. Danese P. N., Silhavy T. J. 1998. CpxP, a stress-combative member of the Cpx regulon. J. Bacteriol. 180:831–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danese P. N., Silhavy T. J. 1997. The σE and the Cpx signal transduction systems control the synthesis of periplasmic protein-folding enzymes in Escherichia coli. Genes Dev. 11:1183–1193 [DOI] [PubMed] [Google Scholar]
- 10. Danese P. N., Snyder W. B., Cosma C. L., Davis L. J., Silhavy T. J. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 9:387–398 [DOI] [PubMed] [Google Scholar]
- 11. DiGiuseppe P. A., Silhavy T. J. 2003. Signal detection and target gene induction by the CpxRA two-component system. J. Bacteriol. 185:2432–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dolinsky T. J., et al. 2007. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35:W522–W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dolinsky T. J., Nielsen J. E., McCammon J. A., Baker N. A. 2004. PDB2PQR: an automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32:W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 15. Fleischer R., Heermann R., Jung K., Hunke S. 2007. Purification, reconstitution, and characterization of the CpxRAP envelope stress system of Escherichia coli. J. Biol. Chem. 282:8583–8593 [DOI] [PubMed] [Google Scholar]
- 16. Gasteiger E., et al. 2005. Protein identification and analysis tools on the ExPASy server, p. 571–607 In Walker J. M. (ed.), The proteomics protocols handbook. Humana Press, New York, NY [Google Scholar]
- 17. GE Healthcare UK, Ltd 2010. GST gene fusion system handbook. GE Healthcare UK, Ltd., Little Chalfont, England [Google Scholar]
- 18. Grass G., Fricke B., Nies D. H. 2005. Control of expression of a periplasmic nickel efflux pump by periplasmic nickel concentrations. Biometals 18:437–448 [DOI] [PubMed] [Google Scholar]
- 19. Grass G., Grosse C., Nies D. H. 2000. Regulation of the cnr cobalt and nickel resistance determinant from Ralstonia sp. strain CH34. J. Bacteriol. 182:1390–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holm L., Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38(Suppl.):W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holm L., Sander C. 1996. Mapping the protein universe. Science 273:595–603 [DOI] [PubMed] [Google Scholar]
- 22. Humphries R. M., et al. 2010. N-Acetyllactosamine-induced retraction of bundle-forming pili regulates virulence-associated gene expression in enteropathogenic Escherichia coli. Mol. Microbiol. 76:1111–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Isaac D. D., Pinkner J. S., Hultgren S. J., Silhavy T. J. 2005. The extracytoplasmic adaptor protein CpxP is degraded with substrate by DegP. Proc. Natl. Acad. Sci. U. S. A. 102:17775–17779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones C. H., Danese P. N., Pinkner J. S., Silhavy T. J., Hultgren S. J. 1997. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 16:6394–6406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kabsch W., Sander C. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637 [DOI] [PubMed] [Google Scholar]
- 26. Kershaw C. J., Brown N. L., Constantinidou C., Patel M. D., Hobman J. L. 2005. The expression profile of Escherichia coli K-12 in response to minimal, optimal and excess copper concentrations. Microbiology 151:1187–1198 [DOI] [PubMed] [Google Scholar]
- 27. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36:1277–1282 [Google Scholar]
- 28. Krissinel E., Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 29. Kwon E., Kim D. Y., Gross C. A., Gross J. D., Kim K. K. 2010. The crystal structure Escherichia coli Spy. Protein Sci. 19:2252–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Larsen R. A., Thomas M. G., Wood G. E., Postle K. 1994. Partial supression of an Escherichia coli TonB transmembrane domain mutation (DV17) by a missense mutation in ExbB. Mol. Microbiol. 13:627–640 [DOI] [PubMed] [Google Scholar]
- 31. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291 [Google Scholar]
- 32. Lee L. J., Barrett J. A., Poole R. K. 2005. Genome-wide transcriptional response of chemostat-cultured Escherichia coli to zinc. J. Bacteriol. 187:1124–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leonhartsberger S., Huber A., Lottspeich F., Bock A. 2001. The hydH/G Genes from Escherichia coli code for a zinc and lead responsive two-component regulatory system. J. Mol. Biol. 307:93–105 [DOI] [PubMed] [Google Scholar]
- 34. Liesegang H., Lemke K., Siddiqui R. A., Schlegel H. G. 1993. Characterization of the inducible nickel and cobalt resistance determinant cnr from pMOL28 of Alcaligenes eutrophus CH34. J. Bacteriol. 175:767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu J., et al. 2006. Protonation-mediated structural flexibility in the F conjugation regulatory protein, TraM. EMBO J. 25:2930–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. MacRitchie D. M., Buelow D. R., Price N. L., Raivio T. L. 2007. Two-component signaling and gram negative envelope stress response systems. In Utsumi R. (ed.), Bacterial signal transduction: network and drug targets. Landes Bioscience, Austin, TX: [DOI] [PubMed] [Google Scholar]
- 37. McCoy A. J., et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monchy S., et al. 2007. Plasmids pMOL28 and pMOL30 of Cupriavidus metallidurans are specialized in the maximal viable response to heavy metals. J. Bacteriol. 189:7417–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murshudov G. N., Vagin A. A., Dodson E. J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 40. Nevesinjac A. Z., Raivio T. L. 2005. The Cpx envelope stress response affects expression of the type IV bundle-forming pili of enteropathogenic Escherichia coli. J. Bacteriol. 187:672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. New England BioLabs 2007. pMAL protein fusion and purification system instruction manual. New England BioLabs, Ipswich, MA [Google Scholar]
- 42. Noll M., Petrukhin K., Lutsenko S. 1998. Identification of a novel transcription regulator from Proteus mirabilis, PMTR, revealed a possible role of YJAI protein in balancing zinc in Escherichia coli. J. Biol. Chem. 273:21393–21401 [DOI] [PubMed] [Google Scholar]
- 43. Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 44. Pogliano J. A., Lynch S., Belin D., Lin E. C. C., Beckwith J. 1997. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev. 11:1169–1182 [DOI] [PubMed] [Google Scholar]
- 45. Pompidor G., et al. 2008. X-ray structure of the metal-sensor CnrX in both the apo- and copper-bound forms. FEBS Lett. 582:3954–3958 [DOI] [PubMed] [Google Scholar]
- 46. Potterton E., Briggs P., Turkenburg M., Dodson E. 2003. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59:1131–1137 [DOI] [PubMed] [Google Scholar]
- 47. Price N. L., Raivio T. L. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J. Bacteriol. 191:1798–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raivio T. L., Laird M. W., Joly J. C., Silhavy T. J. 2000. Tethering of CpxP to the inner membrane prevents spheroplast induction of the Cpx envelope stress response. Mol. Microbiol. 37:1186–1197 [DOI] [PubMed] [Google Scholar]
- 49. Raivio T. L., Popkin D. L., Silhavy T. J. 1999. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J. Bacteriol. 181:5263–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Raivio T. L., Silhavy T. J. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruiz N., Silhavy T. J. 2005. Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr. Opin. Microbiol. 8:122–126 [DOI] [PubMed] [Google Scholar]
- 52. Silhavy T. J., Berman M. L., Enquist L. W. 1984. Experiments in gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 53. Svergun D. I. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503 [Google Scholar]
- 54. Svergun D. I., Petoukhov M. V., Koch M. H. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80:2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Svergun D. I., Volkov V. V. 2003. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36:860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tamames B., Sousa S. F., Tamames J., Fernandes P. A., Ramos M. J. 2007. Analysis of zinc-ligand bond lengths in metalloproteins: trends and patterns. Proteins 69:466–475 [DOI] [PubMed] [Google Scholar]
- 57. Terwilliger T. C. 2003. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D Biol. Crystallogr. 59:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Terwilliger T. C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56:965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Terwilliger T. C., Berendzen J. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55:849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Theobald D. L., Wuttke D. S. 2006. THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics 22:2171–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tibazarwa C., Wuertz S., Mergeay M., Wyns L., van Der Lelie D. 2000. Regulation of the cnr cobalt and nickel resistance determinant of Ralstonia eutropha (Alcaligenes eutrophus) CH34. J. Bacteriol. 182:1399–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. UniProt Consortium 2010. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38:D142–D148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van Stelten J., Silva F., Belin D., Silhavy T. J. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vogt S. L., et al. 2010. The Cpx envelope stress response both facilitates and inhibits elaboration of the enteropathogenic Escherichia coli bundle-forming pilus. Mol. Microbiol. 76:1095–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Winn M. D., Isupov M. N., Murshudov G. N. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57:122–133 [DOI] [PubMed] [Google Scholar]
- 66. Wriggers W. 2010. Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2:21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yamamoto K., Ishihama A. 2006. Characterization of copper-inducible promoters regulated by CpxA/CpxR in Escherichia coli. Biosci. Biotechnol. Biochem. 70:1688–1695 [DOI] [PubMed] [Google Scholar]
- 68. Yamamoto K., Ishihama A. 2005. Transcriptional response of Escherichia coli to external copper. Mol. Microbiol. 56:215–227 [DOI] [PubMed] [Google Scholar]
- 69. Yamamoto K., Ogasawara H., Ishihama A. 2008. Involvement of multiple transcription factors for metal-induced spy gene expression in Escherichia coli. J. Biotechnol. 133:196–200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.