

Abstract
d-Amino acids are essential components for bacterial peptidoglycan, and these natural compounds are also involved in cell wall remodeling and biofilm disassembling. In Pseudomonas aeruginosa, the dadAX operon, encoding the d-amino acid dehydrogenase DadA and the amino acid racemase DadX, is essential for d- and l-Ala catabolism, and its expression requires a transcriptional regulator, DadR. In this study, purified recombinant DadA alone was sufficient to demonstrate the proposed enzymatic activity with very broad substrate specificity; it utilizes all d-amino acids tested as substrates except d-Glu and d-Gln. DadA also showed comparable kcat and Km values on d-Ala and several d-amino acids. dadRAX knockout mutants were constructed and subjected to analysis of their growth phenotypes on amino acids. The results revealed that utilization of l-Ala, l-Trp, d-Ala, and a specific set of d-amino acids as sole nitrogen sources was abolished in the dadA mutant and/or severely hampered in the dadR mutant while growth yield on d-amino acids was surprisingly improved in the dadX mutant. The dadA promoter was induced by several l-amino acids, most strongly by Ala, and only by d-Ala among all tested d-amino acids. Enhanced growth of the dadX mutant on d-amino acids is consistent with the finding that the dadA promoter was constitutively induced in the dadX mutant, where exogenous d-Ala but not l-Ala reduced the expression. Binding of DadR to the dadA regulatory region was demonstrated by electromobility shift assays, and the presence of l-Ala but not d-Ala increased affinity by 3-fold. The presence of multiple DadR-DNA complexes in the dadA regulatory region was demonstrated in vitro, and the formation of these nucleoprotein complexes exerted a complicated impact on promoter activation in vivo. In summary, the results from this study clearly demonstrate DadA to be the enzyme solely responsible for the proposed d-amino acid dehydrogenase activity of broad substrate specificity and the physiological functions of DadRAX in catabolism of several d-amino acids and support l-Ala as the signal molecule for induction of the dadAX genes through DadR binding to several putative operator sites.
INTRODUCTION
d-Amino acids are naturally synthesized in all living organisms and have a variety of specific functions. Examples include the well-known d-Ala and d-Glu, essential components of the bacterial cell wall, and d-Ser, serving as a neurotransmitter in humans. Recently, d-amino acids were reported to control growth phase-dependent cell wall remodeling (14) and to trigger biofilm disassembly (12). Therefore, it is important to understand how bacterial cells regulate d-amino acid homeostasis.
In living organisms, biosynthesis of free-form d-amino acids is catalyzed by racemases, with l-enantiomers as substrates. Peptidyl d-amino acids occur either by taking free d-amino acid as a substrate in the cell wall synthesis or by l-to-d epimerization, as in the nonribosomal peptide synthesis in microorganisms (4). In higher eukaryotic organisms, this process is infrequently catalyzed by enzyme-driven posttranslational isomerization (11). Amino acid racemization also occurs at an accelerated rate with physical and/or chemical treatments. For example, racemization of l-lysine at elevated temperatures has great potential as a commercial process of d-lysine production (20).
The biochemistry of d-amino acid catabolism has not been intensively studied in comparison to those of l-amino acids. In general, d-amino acids are metabolized either directly or after conversion into the l-enantiomers. Pseudomonas aeruginosa is able to utilize many d-amino acids as nutrients and hence serves as an excellent model organism to explore novel pathways and enzymes for d-amino acid metabolism. A new type of d-to-l arginine racemization by coupled catabolic and anabolic dehydrogenases encoded by the dauBA operon was recently reported by our group (15–16). Furthermore, the molecular structure of DauA, a flavin adenine dinucleotide (FAD)-dependent d-amino acid dehydrogenase, has been determined (8).
In contrast, l-alanine catabolism in Escherichia coli and other Gram-negative bacteria is mediated by DadX-dependent l-to-d racemization followed by DadA-dependent oxidative deamination of d-alanine (Fig. 1). An early report by Wasserman and coworkers established the presence of two alanine racemases, the importance of l-to-d racemization for l-Ala catabolism, and the physical proximity and coregulation of two genes encoding d-alanine dehydrogenase and catabolic alanine racemase in Salmonella enterica serovar Typhimurium (21). The same gene organization was later found in E. coli (17). The dadAX operon and its regulation by the leucine-responsive regulator Lrp and carbon catabolite repression in enteric bacteria have been characterized (23–24). DadA of E. coli has been reported as the “small subunit” of a membrane-associated FAD-dependent d-alanine dehydrogenase (18), and a similar description has been applied to its homologues in other bacteria. Whether this enzymatic activity requires the “large subunit” of unknown identity remained obscure. In Gram-positive bacteria, l-alanine is utilized directly by an l-alanine dehydrogenase; no l-to-d racemization is required.
Fig. 1.

(A) Gene organization of the dadRAX locus in Pseudomonas. Orthologues of dadRAX are shown in the same shade. PA, P. aeruginosa; PE, P. entomophila; PF, P. fluorescens; PM, P. mendocina; PP, P. putida; PSt, P. stutzeri; PSy, P. syringae. (B) The nucleotide sequence of the dadA regulatory region in P. aeruginosa PAO1. The −10 element and the transcriptional initiation site (+1) of the dadA promoter as described by Boulette et al. (1) are labeled, and the ribosome binding site in front of the ATG initiation codon of dadA is underlined. The 5′ ends of probes QA1 to -6 are labeled accordingly, the common 3′ end of these probes is highlighted and double underlined, and four putative DadR binding sites are numbered and underlined. (C) Proposed functions of the d-amino acid dehydrogenase DadA and the amino acid racemases DadX and Alr in alanine catabolism. PMS and INT are artificial electron acceptors in the assays for DadA as described in Materials and Methods.
The dadRAX locus (Fig. 1) of alanine catabolism and regulation in P. aeruginosa was initially identified in this laboratory during the study of putrescine and arginine utilization (6, 22), since l-alanine is generated from transamination reactions in the catabolic pathways of these compounds. Expression of dadAX in P. aeruginosa requires DadR (1), a transcriptional activator of the Lrp family (3, 7). The dadAX operon was also described by Boulette et al. for its importance in P. aeruginosa proliferation and infection in rat (1). The gene organization in the dadRAX locus is highly conserved among Pseudomonas bacteria (Fig. 1) except that there are three additional genes inserted into the dadA and dadR intergenic region in P. aeruginosa (Fig. 1) (www.pseudomonas.com).
In this study, we further characterized the biochemical properties of DadA and concluded that DadA is solely responsible for the dehydrogenase activity. The physiological functions of dadRAX in amino acid utilization and effects of various amino acids on the dadA promoter expression were analyzed in the respective mutants. The purified DadR protein was employed to demonstrate its DNA-binding activity. The results also support intracellular l-alanine instead of d-alanine as a signal for dadAX induction.
MATERIALS AND METHODS
Strains and growth conditions.
Bacterial strains used in the study include E. coli DH5α and Top10 (Invitrogen) and P. aeruginosa PAO1, PAO5710 (dadA), PAO5711 (dadX), and PAO5713 (dadR) as reported previously (6). Luria-Bertani (LB) medium was used for strain construction with the following supplements as required: ampicillin at 100 μg/ml (E. coli) and carbenicillin at 100 μg/ml (P. aeruginosa). Minimal medium P (MMP) (10) was used for the growth of P. aeruginosa supplemented with specific carbon (C) and nitrogen (N) sources as indicated.
Enzyme assays.
The substrate specificity of DadA was measured by a published method (15). A total of 18 d-amino acids were used, as needed. d-Cysteine (d-Cys) was not included because the SH group of d-Cys causes reduction of iodonitrotetrazolium chloride (INT). The reaction mixture contained 100 mM Tris-HCl (pH 8.7), 10 mM KCN, 0.2 mM FAD, 0.8 mM phenazine methosulfate (PMS), 0.8 mM INT, 6.7 μg/ml of DadA, and a 20 mM concentration of the tested d-amino acid in a total volume of 0.5 ml. The reaction was initiated by the addition of DadA and was continued for 4 min at 37°C before being stopped by 4 M HCl (50 μl). After addition of 5 μl Triton X-100 to dissolve the precipitates and appropriate dilution, A500 readings were taken. The background reading with double-distilled H2O substitution of d-amino acid in a parallel control reaction was subtracted from the readings with d-amino acids, and the specific activities were calculated thereafter. One unit of d-amino acid dehydrogenase activity was defined as the amount of enzyme that led to the reduction of one nanomole of INT per minute under the standard assay conditions.
After initial screening, several good substrates (d-Ala, d-His, d-Phe, d-Tyr, d-Pro, d-ser, d-Thr, d-Val, and d-Arg) were further analyzed to determine the catalytic parameters. The assay was carried out with the same procedure as described above, except that different concentrations of d-amino acids were used. The final concentration of purified protein used in the reaction was 7.22 μg/ml. The Km, kcat, and kcat/Km values were determined by nonlinear regression data analysis using the SigmaPlot 9.0 software program.
Construction of Φ(dadA′-lacZ+) transcriptional fusion plasmids.
dadA promoter fragments of different lengths were were PCR amplified from pZY6 (22) with specific pairs of oligonucleotide primers that carry either the BamHI or HindIII restriction site. After digestion with restriction enzymes, the purified PCR products were cloned into pQF50 to generate plasmids pQA1 to pQA6 (see Fig. 5). For the measurements of β-galactosidase activity, o-nitrophenyl-β-d-galactopyranoside (ONPG) was used as a substrate. The protein concentration was determined by the Bradford method with bovine serum albumin (BSA) as a standard (2).
Fig. 5.

Gel shift analysis of DadR and different dadA promoter interactions. (A) Schematic presentations of four putative DadR boxes (B1 to B4) relative to the −10 element of the dadA promoter. Also shown are probes QA1 to -6, which cover different locations in the dadA regulatory region, and sequence alignment of DadR Boxes 1 to 4. (B) Electromobility shift assays with probes QA1 to -6. Each reaction mixture contains specific probes, as indicated at the top, and a negative control DNA (N), except reactions with probe QA3, where the negative control DNA was omitted. Concentrations of the DadR protein in lanes 1 to 4 were 0, 20, 40, and 80 nM, respectively.
Expression of DadR and DadA in E. coli.
The structural genes dadR and dadA were amplified by PCR from the genomic DNA of PAO1 using the following primer pairs: 5′-TCC CCC GGG CGT ACC CAG CAT CAG AGC AAG-3′ and 5′-CCC AAG CTT CAA TCC GGA ACC GGT AGG TCG-3′ for dadR and 5′-CGA GTT CTG GTC CTT GGC AGC GGT GTC-3′ and 5′-CCC AAG CTT AGT GTG CTG GCG CTG GAT G-3′ for dadA. The resulting PCR product was digested with SmaI and HindIII, which are unique restriction sites introduced by the primers, and cloned into the expression vector pBAD-HisD (15) so that the N termini of DadA and DadR are fused in-frame with the six-histidine tag preceded by a ribosomal binding site and an arabinose-inducible promoter in the plasmid. The resulting plasmids, pCR6 and pCR7, were introduced into E. coli Top10. For overexpression of DadR, the recombinant strain of E. coli was grown in LB medium containing ampicillin (100 μg/ml) at 30°C until the optical density at 600 nm (OD600) reached 0.5, at which point 0.2% arabinose (wt/vol; final concentration) was added to the culture for induction. d-Ala was also added at a 5 mM concentration for overexpression of DadA. Culture growth was continued for another 2 h for DadA and 4 h for DadR expression under the same conditions, and cells were harvested by centrifugation.
Purification of hexahistidine-tagged DadR and DadA.
The cell pellets of dadR overexpression were suspended in phosphate buffer A (20 mM sodium phosphate, 1 M NaCl, 20 mM imidazole, pH 7.4) plus 1 mM phenylmethylsulfonyl fluoride (PMSF) as a protease inhibitor, and the cells were ruptured by an Aminco French press at 17,000 lb/in2. Cell debris was removed by centrifugation at 20,000 × g for 0.5 h, and the resulting cell extract was further precipitated by streptomycin sulfate and centrifuged again. The supernatant was applied to a HisTrap HP column (GE Healthcare) equilibrated with the same buffer. After the unbound proteins were washed off with equilibration buffer, His-tagged DadR was eluted at 50% of buffer B (20 mM sodium phosphate, 1 M NaCl, 1 M imidazole, pH 5.5). The target fractions were pooled together and concentrated using an Aminco Ultra-15 centrifugal filter unit (molecular mass cutoff, 10 kDa; Millipore) to change the buffer to 20 mM Tris-Cl (pH 7.6).
For DadA preparation, the same protocol as described above for DadR was followed except for the presence of 0.05% Triton X-100 in all buffers. The pooled DadA fractions from the affinity column were reloaded on MonoQ column (GE Healthcare) equilibrated with buffer C (20 mM Tris-Cl and 0.05% Triton X-100, pH 7.6). The target His-tagged DadA protein was eluted at 50% buffer D (20 mM Tris-Cl, 1 M KCl, and 0.05% Triton X-100, pH 7.6).
Chemical cross-linkage of DadR (40 to 60 μg) was conducted in 20 mM HEPES buffer (pH 7.5) in a total volume of 100 μl with 5 μl of 2.3% freshly prepared solution of glutaraldehyde for 2 to 5 min at 37°C. The reaction was terminated by addition of 10 μl of 1 M Tris-HCl (pH 8.0).
Electromobility shift assays.
DNA fragments covering the regulatory region of dadA were PCR amplified from pZY6 (22) with specific pairs of oligonucleotide primers. As a negative control, a DNA fragment of 600 bp covering the region downstream of the pmrA gene was amplified with the following primers: 5′-AAC GAA TTC GAG CAG AGC CTC TAC GGC TGG-3′ and 5′-TGG AAG CTT TCG ATG TTC AGC TGG AGC ACC-3′. For the binding reactions, the DNA probe at 0.2 nM was allowed to interact with different concentrations of DadR in a mixture of 20 μl containing 10 mM Tris-Cl (pH 7.5), 50 mM NaCl, 1 mM EDTA, 4 mM dithiothreitol (DTT), 5% (vol/vol) glycerol, 0.1 ng/μl negative-control DNA, and 300 μg/ml acetylated bovine serum albumin. When specified, l-alanine or d-alanine at 5 mM was added to the reaction mixtures. After incubation for 20 min at room temperature, 10 μl of each reaction mixture was loaded on a polyacrylamide gel (5%) in Tris-borate-EDTA buffer (pH 8.7). The gels were stained with SYBR green I solution (Invitrogen) for 20 min, washed twice with deionized H2O, and scanned with an imaging system (Omega UltraLum) with a setting for excitation at 473 nm and emission at 520 nm.
RESULTS
Substrate specificity and catalytic parameters of DadA.
As described in Materials and Methods, recombinant DadA with a hexahistidine tag at the N terminus was overexpressed in E. coli and purified by affinity chromatography (Fig. 2) in the presence of Triton X-100 due to its association with the cytoplasmic membrane (18). The substrate specificity of the purified recombinant DadA was determined as shown in Fig. 3. DadA has very broad substrate specificity; all the d-amino acids tested can be used as the substrate except d-Glu and d-Gln. Furthermore, the kcat and Km values of d-Ala and eight other substrates were determined as shown in Table 1. In contrast to DauA, which is subjected to substrate inhibition (15), no such property was observed for DadA with the tested amino acids. These results support DadA as an efficient d-amino acid dehydrogenase of broad substrate specificity.
Fig. 2.
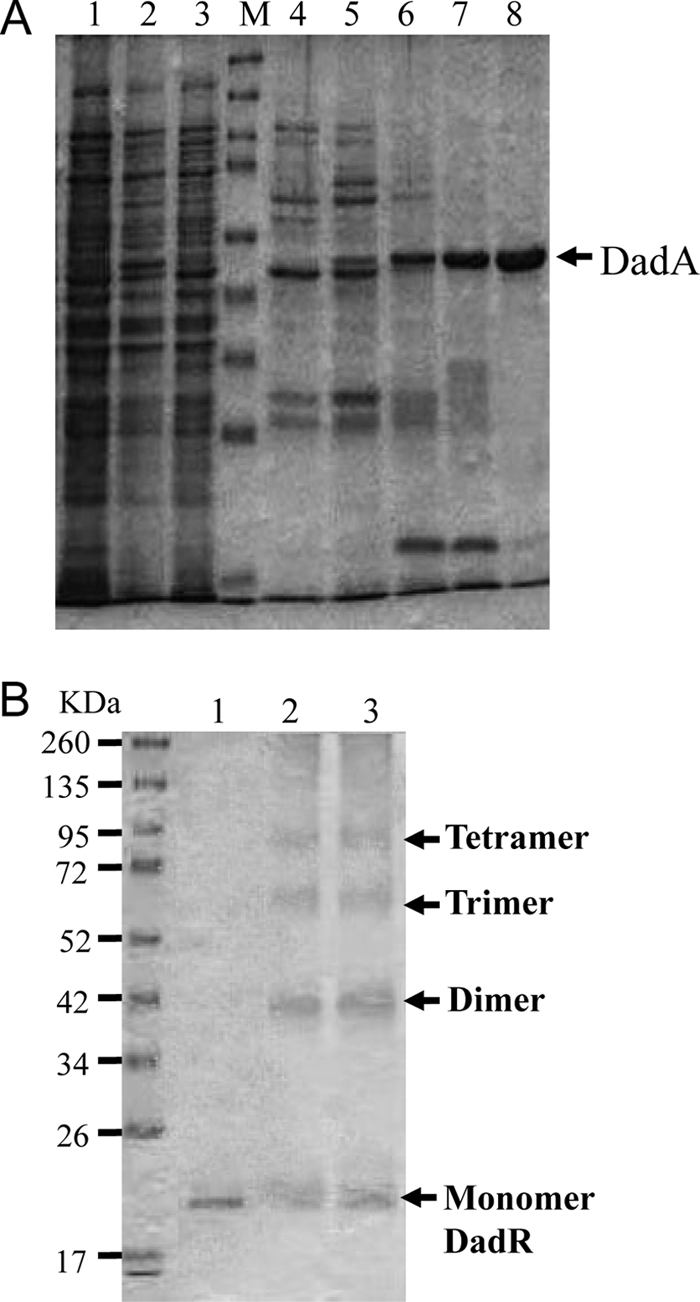
Purification of DadA and chemical cross-linkage analysis of DadR. (A) Protein samples during steps of DadA purification. Lane 1, no induction; lane 2, induction for 2 h; lane 3, induction for 3 h; lanes 4 to 7, fractions from HisTrap HP column; lane 8, purified DadA from the monoQ column. The sizes of protein markers (M) are the same as shown in the lower panel. DadA production repeatedly showed significantly higher yield in the culture induced for 2 h than in that induced for 3 h. (B) Cross-linkage of purified DadR with glutaraldehyde. Lane 1, purified DadR; lanes 2 and 3, 60 and 40 μg DadR treated by 2.3% glutaraldehyde.
Fig. 3.

Substrate specificity of DadA. The substrate specificity of DadA was measured with partially purified His-tagged DadA. Different substrates were added at a final concentration of 20 mM. Relative activity is in comparison to that of d-Ala. d-Tyr was not soluble in the assay mixture, but after DadA was added to initiate the reaction, this substrate was consumed gradually, and the mixture was as clear as other substrate reaction mixtures at the end.
Table 1.
Catalytic parameters of DadA
| Substrate | kcat (s−1)a | Km (mM)a | kcat/Km (M−1 s−1) |
|---|---|---|---|
| d-Alanine | 0.74 ± 0.02 | 0.46 ± 0.05 | 1.61 × 103 |
| d-Histidine | 0.84 ± 0.01 | 0.81 ± 0.07 | 1.04 × 103 |
| d-Phenylalanine | 1.10 ± 0.06 | 1.07 ± 0.22 | 1.03 × 103 |
| d-Tyrosine | 1.10 ± 0.09 | 1.78 ± 0.49 | 6.18 × 102 |
| d-Proline | 0.97 ± 0.03 | 1.82 ± 0.21 | 5.33 × 102 |
| d-Serine | 0.80 ± 0.02 | 1.65 ± 0.15 | 4.85 × 102 |
| d-Threonine | 0.68 ± 0.02 | 2.47 ± 0.18 | 2.75 × 102 |
| d-Valine | 0.78 ± 0.03 | 3.01 ± 0.29 | 2.59 × 102 |
| d-Arginine | 1.27 ± 0.07 | 5.37 ± 0.69 | 2.36 × 102 |
The values represent the averages of two measurements ± SD.
During overexpression of DadA, it was observed that the OD600 reading of the culture drops and cell debris appears after induction by l-arabinose. This was likely the result of d-Ala depletion and subsequent deterioration of cell wall synthesis. With this adverse effect of DadA overexpression considered, the induced culture was supplemented with d-Ala to improve growth. Even so, the cells could be induced only for 2 h for the maximal yield; a longer induction time resulted in a much lower level of DadA (Fig. 2, lanes 2 and 3).
Effects of dadA on d-amino acid utilization as a sole nitrogen source.
Among 19 d-amino acids tested, P. aeruginosa PAO1 was reported to utilize 12 of them as the sole source of nitrogen (16), and a mutation in dauA, encoding a d-arginine dehydrogenase (15), affects growth on d-Arg and d-Lys only. While the dadA gene is essential for d-alanine catabolism, its potential function in utilization of other d-amino acids was not clear. Therefore, we conducted a growth phenotype analysis of the dadA mutant in minimal medium supplemented with various d-amino acids. As shown in Table 2, utilization of five more d-amino acids other than d-alanine—His, Phe, Ser, Thr, and Val—was abolished in the dadA mutant.
Table 2.
Growth phenotypes of dadRAX mutants and effects of amino acids on dadA promoter activity
| Amino acid | Amino acid growth phenotype (d form)a |
dadA promoter activityb |
||||
|---|---|---|---|---|---|---|
| PAO1 | ΔdadR | ΔdadA | ΔdadX | d | l | |
| Ala | + | +/− | − | + | 12.1 | 28.9 |
| Val | + | +/− | − | + | 1.7 | 15.7 |
| Ser | + | +/− | − | + | 2 | 6.2 |
| Thr | + | +/− | − | + | 4.1 | 5.7 |
| Phe | + | +/− | − | + | 0.7 | 0.8 |
| His | + | +/− | − | + | 1 | 0.8 |
| Arg | + | + | + | + | 2 | 1.5 |
| Lys | + | + | + | + | 0.9 | 5.1 |
| Asn | + | + | + | + | 0.7 | 1.4 |
| Gln | + | + | + | + | 1 | 0.7 |
| Glu | + | + | + | + | 1 | 1 |
| Pro | + | + | + | + | 1.8 | 1.9 |
| Asp | − | − | − | − | 0.4 | 0.5 |
| Cys | − | − | − | − | 1.2 | 2.7 |
| Leu | − | − | − | − | 0.9 | 1.8 |
| Met | − | − | − | − | 1.4 | 1.4 |
| Trp | − | − | − | − | 1.4 | 8.4 |
| Tyr | − | − | − | − | 0.8 | 1.3 |
| Ile | ND | ND | ND | ND | ND | 1.2 |
| Gly | NA | NA | NA | NA | NA | 3 |
+, growth; +/−, poor growth,; −, no growth; NA, not available; ND, not determined. Growth was monitored at 37°C for 48 h in glucose minimal medium, with the indicated amino acid as the sole nitrogen source.
Cells were grown in minimal medium P with 20 mM glucose and 5 mM d-amino acids (d, d-amino acids; l, l-amino acids). Activity of the dadA promoter fused with lacZ in pZY6 (22) was measured. Values indicate fold changes in β-galactosidase activities in comparison to the level in Glu-grown cells. Each value represents the average from two measurements, with standard errors below 5%.
We also tested growth phenotypes of the dadR and dadX mutants with d-amino acids (Table 2). In the dadR mutant, devoid of a functional transcriptional activator for the dadA promoter, the level of DadA was expected to be constitutively low. Indeed, growth of the dadR mutant on the same set of d-amino acids identified from the analysis of the dadA mutant was found to be retarded in comparison to wild-type PAO1. Contrarily, the dadX knockout mutant exhibited 2- to 3-fold-enhanced growth on those d-amino acids as judged from the final cell density after growth for 48 h (data not shown). This unexpected finding of higher growth yields could be due to a very high basal level of dadA promoter activity in the dadX mutant, as described in Table 3.
Table 3.
Effects of dadRAX on regulation of dadA promoter activity
| Strain (genotype) | Growth conditiona | Sp act (nmol/min/mg)b | Fold change |
|---|---|---|---|
| PAO1 (wild type) | Glu | 100 | 1.0 |
| Glu + d-Ala | 780 | 7.8 | |
| Glu + l-Ala | 1,300 | 13.0 | |
| Glu + dl-Ala | 1,300 | 13.0 | |
| PAO5713 (dadR::Gm) | Glu | 4 | 1.0 |
| Glu + d-Ala | 4 | 1.0 | |
| Glu + l-Ala | 4 | 1.0 | |
| PAO5710 (dadA::Tc) | Glu | 620 | 1.0 |
| Glu + d-Ala | 3,300 | 5.3 | |
| Glu + l-Ala | 3,710 | 6.0 | |
| PAO5711 (dadX::Gm) | Glu | 2,890 | 1.0 |
| Glu + d-Ala | 800 | 0.3 | |
| Glu + l-Ala | 2,670 | 0.9 |
Cells were grown in minimal medium P with the indicated amino acid supplements.
Specific activities of β-galactosidase expressed from the ɸ(dadA-lacZ) translational fusion plasmid pZY6 (22). Values are the averages of two measurements for each growth condition, with standard errors of less than 5%.
We have reported that utilization of l-Ala as the sole source of carbon and nitrogen was abolished in mutants devoid of dadR, dadA, or dadX (6). The potential effects of dadRAX on the utilization of other l-amino acids were also tested by growth phenotype analysis. We found that utilization of l-Trp was abolished in the dadA mutant only. Since wild-type PAO1 does not grow on d-Trp (Table 2), this result suggests that l-Ala can be generated from and plays a pivotal role in l-Trp catabolism.
Effects of d- and l-amino acids on dadA expression.
Since DadA has broad substrate specificity and participates in the utilization of several d-amino acids, it was interesting to know how expression of dadA responds to the presence of different amino acids. PAO1 harboring pZY6 (22), a φ(dadA-lacZ) translational fusion, was grown in glutamate minimal medium in the presence or absence of d- and l-amino acids, and dadA promoter activities under these growth conditions were determined by measurements of β-galactosidase expression as shown in Table 2. Among l-amino acids, Ala exerted the strongest effect on dadA induction, followed by Val, Trp, Ser, Thr, Lys, Gly, and Cys. In comparison, this promoter can be induced by only two d-amino acids: Ala (significantly) and Thr (marginally). Since all these amino acids have very different chemical properties, it was unlikely that they all serve as signal molecules for dadAX induction. Instead, we hypothesized that l-Ala, which can be made from catabolism of other amino acids, serves as the authentic induction signal.
Regulation of the dadA promoter.
We have previously described DadR as a possible transcriptional regulator for dadAX. As shown in Table 3, activation of the dadA promoter by d- and l-alanine was completely abolished in the dadR mutant. Since the intracellular levels of d- and l-Ala could be modulated by DadA and DadX, we also measured the activity of the dadA promoter in the dadA and dadX mutants. When the dadA mutant was grown in glutamate minimal medium, the basal level of this promoter was more than 5-fold higher than that in wild-type PAO1, and exogenous d- and l-Ala were still able to induce this promoter further by 5- to 6-fold.
In the dadX mutant, the basal level of dadA promoter activity was more than 28-fold higher than that in PAO1. While no further induction by exogenous l-Ala can be detected, this promoter activity was surprisingly reduced 3-fold by d-Ala in the dadX mutant. Without a functional DadX racemase, l-to-d conversion of alanine might be greatly diminished, which could cause accumulation of l-Ala inside the cells and thus constitutive activation of the dadA promoter if l-Ala indeed serves as the signal molecule of dadAX expression. These results strongly suggest that a genetic lesion in the dadA or dadX gene can change the intracellular ratio of l-Ala and d-Ala, which subsequently affects promoter activation.
We also tested whether the stimulation effect of l-Ala on the dadA promoter can be inhibited by d-Ala in PAO1. As shown in Table 3, the level of dadA expression in the presence of l-Ala alone was comparable to that with a combination of d- and l-Ala of equal molar concentrations.
Subunit composition of His-tagged DadR.
DadR, a transcriptional regulator of the AsnC/Lrp family (PFAM accession no. PF01037), is essential for the expression of dadAX (Table 3). To test binding of DadR to the dadA regulatory region, we first constructed a plasmid for overexpression of His-tagged DadR in E. coli, and this recombinant DadR protein was purified by column chromatography (Fig. 2). To understand possible subunit configurations, DadR (0.5 mg/ml) was subjected to chemical cross-linkage with glutaraldehyde, followed by SDS-PAGE. Three additional polypeptides of higher molecular weights were detected (Fig. 2), corresponding to the estimated sizes of dimers, trimers, and tetramers of DadR. The subunit composition of DadR was also estimated by size exclusion column chromatography. With a relatively low concentration (<30 μg/ml), DadR under this condition was predominantly a dimer. These results indicated possible dimer-dimer interactions forming a tetrameric configuration of DadR at high concentrations.
Binding of DadR to the dadA promoter region.
The purified His-tagged DadR was applied in electromobility shift assays, using the same DNA fragment of 414 bp covering the entire intergenic region of dadA and its upstream gene as in the φ(dadA-lacZ) fusion plasmid pZY6. As shown in Fig. 4A, DadR reacts specifically with the dadA regulatory region, with an estimated apparent dissociation constant (Kd) of 28 nM. In the presence of l-alanine (5 mM), the Kd dropped to 9 nM (Fig. 4B) while d-alanine showed no apparent effect (Fig. 4C). These results support l-alanine as an internal signal compound for dadAX activation.
Fig. 4.

Electromobility shift analysis of DadR-dadA interaction. The assays were conducted in the absence of alanine (A), the presence of 5 mM l-alanine (B), or the presence of 5 mM d-alanine (C). Plots of free probe concentration as a function of DadR concentration are shown (D). F, free probe; N, negative control DNA; C1 and C2, DadR-DNA complexes 1 and 2.
During mobility shift analysis, we noticed that the free dadA probe of 414 bp migrated slower than the negative control DNA of 600 bp. Inspection of the nucleotide sequence of the dadA probe (Fig. 1) revealed multiple runs of 4 to 6 adenine base pairs (A tracts), which are known to have unique structures and cause intrinsic DNA bending and thus slower migration of DNA. Moreover, there are at least two apparent DadR-DNA nucleoprotein complexes (C1 and C2 in Fig. 4), suggesting the presence of multiple DadR binding sites in the dadA regulatory region.
To further analyze the locations of possible DadR binding sites, a series of deletion probes were generated for the mobility shift assays. As shown in Fig. 5, only one single nucleoprotein complex was observed with probes QA1 and QA2, and multiple nucleoprotein complexes were formed with probes QA3, QA4, QA5, and QA6. However, the affinity of DadR to the probe QA1 was significantly lower than that to the other five probes. These results suggested that the dadA promoter region may contain more than one DadR binding site.
Effects of multiple DadR binding sites on DadA promoter activity.
The observed formation of multiple DadR-DNA complexes in vitro may affect DadA promoter activity in vivo. To test this hypothesis, a set of Φ(dadA′-lacZ′) fusions was constructed by cloning the probes QA1 to QA6 into the vector pQF50, and the six resulting plasmids were introduced into PAO1 to check promoter activation by l-Ala. As shown in Table 4, pQA1, derived from the shortest probe, QA1, with low DadR affinity, was not induced by exogenous l-Ala, but alanine-dependent induction was recovered in pQA2. To our surprise, the induction effect disappeared in pQA3 and pQA4 but was restored again in pQA5 and pQA6. These results suggest that formation of different DadR-DNA complexes may have a complicated impact on promoter activation.
Table 4.
Effects of putative DadR boxes on dadA promoter activity
| Plasmid | DadR box(es) | Growth conditiona | Sp actb |
|---|---|---|---|
| pQA1 | 1 | Glu | 0.6 |
| Glu + Ala | 0.8 | ||
| pQA2 | 1, 2 | Glu | 1.0 |
| Glu + Ala | 5.0 | ||
| pQA3 | 1, 2, 3 | Glu | 0.5 |
| Glu + Ala | 1.0 | ||
| pQA4 | 1, 2, 3 | Glu | 0.4 |
| Glu + Ala | 1.2 | ||
| pQA5 | 1, 2, 3, 4 | Glu | 0.9 |
| Glu + Ala | 5.1 | ||
| pQA6 | 1, 2, 3, 4 | Glu | 1.0 |
| Glu + Ala | 7.3 |
The cells were grown in minimal medium in the presence of 10 mM l-glutamate (Glu) with or without 2 mM l-alanine (Ala). The host cell for these plasmids was P. aeruginosa PAO1.
Specific activities of β-galactosidase were determined using whole cells permeabilized with toluene and calculated as OD420/OD600/reaction time. Numbers shown here are relative activities, taking the specific activity of pQA6 in Glu as 1.0.
DISCUSSION
d-Amino acid utilization.
In P. aeruginosa, utilization of 18 proteinous d-amino acids (except d-Ile, due to the cost) as a sole nitrogen source was analyzed in glucose minimal medium with three strains: wild-type PAO1, the dauA mutant, devoid of catabolic d-Arg dehydrogenase, and the dadA mutant, devoid of d-Ala dehydrogenase. Based on the growth phenotypes, these d-amino acids were categorized into four groups: G1, nonutilization (Asp, Cys, Leu, Met, Trp, and Tyr); G2, DauA dependent (Arg and Lys); G3, DadA dependent (Ala, His, Phe, Ser, Thr, and Val); and G4, DauA/DadA independent (Pro, Glu, Gln, and Asn). These results revealed not only the importance of DadA and DauA but also the presence of novel enzymes yet to be identified in catabolism of d-Asn, Glu, Gln, and Pro.
d-Amino acid oxidase/dehydrogenase.
Like DauA and DadA, enzymes of this group contain FAD as a cofactor. Differentiation of oxidase and dehydrogenase depends on whether O2 (oxidase) or components of the electron transport chain (dehydrogenase) serve as a direct electron acceptor of FADH2 to recycle FAD and keep the enzyme active. Therefore, the oxidase activity can be measured by H2O2 production with a coupled peroxidase and its chromogenic substrate, while the dehydrogenase level can be determined by the presence of an artificial electron acceptor (e.g., phenazine methosulfate) and a coupled color reagent (e.g., iodonitrotetrazolium). DauA and DadA are true dehydrogenases.
There are 17 enzymes in this group (COG0665; cluster of orthologous groups [COG] classification) according to PAO1 genome annotations (www.pseudomonas.com), including DadA, DauA, and the sarcosine oxidase SoxB. PA1565 and PA5309 are likely to participate in polyamine catabolism (data not shown) and hence may not be related to d-amino acid catabolism. Based on its genetic proximity to PA1268, encoding the OH-l-Pro epimerase (9), PA1267 may encode oxidase/dehydrogenase for OH-d-Pro. Identification of enzymes in this group that may participate in d-Glu/Gln/Asn catabolism is currently in progress.
d-Alanine dehydrogenase is composed of DadA only.
DadA of E. coli was first characterized as the “small subunit” of a membrane-associated d-alanine dehydrogenase (18). This description has been applied to annotations of DadA homologues of many bacteria since then, but identity of the “large subunit” of this enzyme remains obscure. In this study we have successfully overexpressed DadA of P. aeruginosa as a soluble His-tagged recombinant protein. The results of enzyme assays have indicated that DadA is solely responsible for the observed dehydrogenase activity.
Alanine racemases.
Like E. coli, P. aeruginosa PAO1 possesses two alanine racemases, Alr and DadX (19). As shown in this study, the dadX mutant of PAO1 grew normally without the supplementing with d-Ala, suggesting that Alr is sufficient to fulfill the physiological demand of d-Ala biosynthesis in cell wall synthesis. However, the dadX mutant cannot grow on l-Ala as the sole source of carbon and nitrogen (6), suggesting that PAO1 needs to induce DadX synthesis to ensure sufficient l- to d-Ala conversion before serving as a substrate of DadA. While the dadRAX genes are highly conserved among pseudomonads, only P. aeruginosa and Pseudomonas mendocina contain the alr gene, and P. putida possesses a periplasmic basic amino acid-specific racemase encoded in an operon that is inducible by arginine (22) and lysine (unpublished data). The kinetic parameters of PAO1 Alr and DadX, which showed comparable values for l-Ala and d-Ala as substrates, have been reported (19). This therefore suggests that exogenous d-Ala might be converted into l isomers to trigger dadAX expression.
Regulatory mechanism of dadAX induction.
In this study we established DadR as a transcriptional regulator of dadAX by two lines of evidence: abolishment of dadAX induction by alanine in the dadR mutant (Table 3) and binding of purified DadR to the dadA regulatory region (Fig. 4 and 5). Although the dadA promoter can be induced by either l- or d-Ala, it is likely that intracellular l-Ala is the signal for induction. This is supported by the following observations. First, l-alanine exerts a stronger induction effect than d-Ala. Second, the dadA promoter is constitutively active in the dadX mutant, where intracellular l-Ala from intrinsic metabolism is expected to accumulate, and exogenous d-Ala reduces the promoter expression in this mutant. Finally, the DNA-binding activity of DadR in vitro is enhanced by the presence of l-Ala but not d-Ala. The recombinant His-tagged DadR protein used in this study has a major configuration as homodimers in low concentrations but is capable of forming homotetramers in higher concentrations. Neither l-Ala nor d-Ala exerted any significant effect on the subunit composition of DadR in vitro as analyzed by glutaraldehyde cross-linkage (data not shown).
The presence of multiple DadR-DNA complexes in the dadA regulatory region was also demonstrated in this study. Based on the results of electromobility shift experiments with different probes, four putative DadR boxes were proposed, as shown in Fig. 5. Among these four sites, the Box 1 sequence is more divergent from the sequences of Box 2, Box 3, and Box 4, which is consistent to the observation that DadR has much lower affinity to probe QA1, containing Box 1 only. A consensus sequence of 5′-KCGGWWTTTTWCCWG-3′ (K = C or G; W = A or T) can be derived from Boxes 2 to 4, which is somewhat similar to the consensus sequence of E. coli Lrp (8), YAGHAWATTWTDCTR (Y = C or T; H = not G; W = A or T; D = not C; R = A or G). The DadR protein was initially annotated as Lrp by the Pseudomonas genome project based on sequence comparison.
The DadR boxes may contain an intrinsic DNA bending site. Boxes 2 to 4 have a stretch of thymine (A/T tract), a sequence feature known to have a unique DNA structure and to cause DNA bending. Sequences other than the A/T tract may also cause intrinsic bending. Perhaps this bending structure plus conserved G/C bases on the flanking arms are essential for their recognition by DadR.
Although four DadR boxes were proposed in the entire dadA regulatory region, the results of fusion studies (Table 4) indicate that Box 1 alone cannot activate the dadA promoter by exogenous l-Ala in vivo. While a combination of Boxes 1 and 2 is as good as that of Boxes 1 to 4 for promoter activation, the efficiency is reduced significantly with the combination of Boxes 1 to 3. These results revealed that formation of different DadR-DNA complexes may have a complicated impact on activation of the dadA promoter. However, this layer of complexity could be species specific. Among all Pseudomonas species in the current genome website (www.pseudomonas.com), only in P. aeruginosa are the dadR and dadA genes separated by three additional genes of unknown function (Fig. 1), and the dadA regulatory region in PAO1 (357 bp) is much longer than the dadR-dadA divergent promoter region in other species (between 148 and 157 bp) to accommodate additional DadR boxes.
Why is dadAX induced by amino acids other than l-Ala?
We observed that several l-amino acids other than l-Ala are able to activate dadAX. However, utilization of these l-amino acids, except Ala and Trp, does not require dadAX. One possible explanation is that l-Ala is generated during metabolism of these amino acids through pyruvate-dependent transaminases. Specifically, a valine-pyruvate transaminase (e.g., orthologue of E. coli avtA) may convert l-Val to l-Ala. Catabolism of Gly, Thr, and Ser converges to make pyruvate and ammonium as final products, which could be used to generate l-Ala through ammonia assimilation to make glutamate and subsequent glutamate-pyruvate transamination. In the case of l-lysine, transamination of cadaverine, the product of lysine decarboxylase (5), could be catalyzed by SpuC, a pyruvate-dependent transaminase (6). Kynureninase (KynU) can hydrolyze kynurenine, an intermediate compound of l-Trp catabolism, and generate l-Ala and anthranilate (13). l-Trp utilization was completely blocked in the dadA mutant, strongly suggesting that transamination catalyzed by KyuU is the major or sole route of l-Trp deamination. l-Ala may serve as one of the important reservoirs for carbon and nitrogen storage. This hypothesis is supported by the observation that the dadA promoter was constitutively induced in the dadX mutant, where the intracellular l-Ala deriving from a variation of metabolic activities was expected to accumulate without conversion into d-Ala and therefore interacts with DadR to activate the dadA promoter.
In summary, from this study we clearly demonstrate that DadA per se is sufficient to catalyze the proposed d-amino acid dehydrogenase activity of broad substrate specificity. The DadR, DadA, and DadX proteins are essential for the maintenance of a balanced l-Ala/d-Ala ratio and play pivotal roles in the catabolism of alanine and several other d-amino acids. These results also support l-Ala as the signal molecule for induction of the dadAX genes through DadR binding to several putative operator sites.
ACKNOWLEDGMENT
This work was supported by National Science Foundation grant 0950217.
Footnotes
Published ahead of print on 4 March 2011.
REFERENCES
- 1. Boulette M. L., et al. 2009. Characterization of alanine catabolism in Pseudomonas aeruginosa and its importance for proliferation in vivo. J. Bacteriol. 191:6329–6334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bradford M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 3. Brinkman A. B., Ettema T. J., de Vos W. M., van der Oost J. 2003. The Lrp family of transcriptional regulators. Mol. Microbiol. 48:287–294 [DOI] [PubMed] [Google Scholar]
- 4. Challis G. L., Naismith J. H. 2004. Structural aspects of non-ribosomal peptide biosynthesis. Curr. Opin. Struct. Biol. 14:748–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chou H. T., Hegazy M., Lu C. D. 2010. L-Lysine catabolism is controlled by L-arginine and ArgR in Pseudomonas aeruginosa PAO1. J. Bacteriol. 192:5874–5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chou H. T., Kwon D. H., Hegazy M., Lu C. D. 2008. Transcriptome analysis of agmatine and putrescine catabolism in Pseudomonas aeruginosa PAO1. J. Bacteriol. 190:1966–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de los Rios S., Perona J. J. 2007. Structure of the Escherichia coli leucine-responsive regulatory protein Lrp reveals a novel octameric assembly. J. Mol. Biol. 366:1589–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu G., et al. 2010. Conformational changes and substrate recognition in Pseudomonas aeruginosa d-arginine dehydrogenase. Biochemistry 49:8535–8545 [DOI] [PubMed] [Google Scholar]
- 9. Goytia M., et al. 2007. Molecular and structural discrimination of proline racemase and hydroxyproline-2-epimerase from nosocomial and bacterial pathogens. PLoS One 2:e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haas D., Holloway B. W., Schambock A., Leisinger T. 1977. The genetic organization of arginine biosynthesis in Pseudomonas aeruginosa. Mol. Gen. Genet. 154:7–22 [DOI] [PubMed] [Google Scholar]
- 11. Jilek A., et al. 2005. Biosynthesis of a D-amino acid in peptide linkage by an enzyme from frog skin secretions. Proc. Natl. Acad. Sci. U. S. A. 102:4235–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kolodkin-Gal I., et al. 2010. D-Amino acids trigger biofilm disassembly. Science 328:627–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kurnasov O., et al. 2003. Aerobic tryptophan degradation pathway in bacteria: novel kynurenine formamidase. FEMS Microbiol. Lett. 227:219–227 [DOI] [PubMed] [Google Scholar]
- 14. Lam H., et al. 2009. D-amino acids govern stationary phase cell wall remodeling in bacteria. Science 325:1552–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li C., Lu C. D. 2009. Arginine racemization by coupled catabolic and anabolic dehydrogenases. Proc. Natl. Acad. Sci. U. S. A. 106:906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li C., Yao X., Lu C. D. 2010. Regulation of the dauBAR operon and characterization of D-amino acid dehydrogenase DauA in arginine and lysine catabolism of Pseudomonas aeruginosa PAO1. Microbiology 156:60–71 [DOI] [PubMed] [Google Scholar]
- 17. Lobocka M., Hennig J., Wild J., Klopotowski T. 1994. Organization and expression of the Escherichia coli K-12 dad operon encoding the smaller subunit of D-amino acid dehydrogenase and the catabolic alanine racemase. J. Bacteriol. 176:1500–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olsiewski P. J., Kaczorowski G. J., Walsh C. 1980. Purification and properties of D-amino acid dehydrogenase, an inducible membrane-bound iron-sulfur flavoenzyme from Escherichia coli B. J. Biol. Chem. 255:4487–4494 [PubMed] [Google Scholar]
- 19. Strych U., Huang H. C., Krause K. L., Benedik M. J. 2000. Characterization of the alanine racemases from Pseudomonas aeruginosa PAO1. Curr. Microbiol. 41:290–294 [DOI] [PubMed] [Google Scholar]
- 20. Takahashi E., Furui M., Seko H., Shibatani T. 1997. D-Lysine production from L-lysine by successive chemical racemization and microbial asymmetric degradation. Appl. Microbiol. Biotechnol. 47:347–351 [DOI] [PubMed] [Google Scholar]
- 21. Wasserman S. A., Walsh C. T., Botstein D. 1983. Two alanine racemase genes in Salmonella typhimurium that differ in structure and function. J. Bacteriol. 153:1439–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang Z., Lu C. D. 2007. Functional genomics enables identification of genes of the arginine transaminase pathway in Pseudomonas aeruginosa. J. Bacteriol. 189:3945–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhi J., Mathew E., Freundlich M. 1998. In vitro and in vivo characterization of three major dadAX promoters in Escherichia coli that are regulated by cyclic AMP-CRP and Lrp. Mol. Gen. Genet. 258:442–447 [DOI] [PubMed] [Google Scholar]
- 24. Zhi J., Mathew E., Freundlich M. 1999. Lrp binds to two regions in the dadAX promoter region of Escherichia coli to repress and activate transcription directly. Mol. Microbiol. 32:29–40 [DOI] [PubMed] [Google Scholar]