

Abstract
TcdA and TcdB exotoxins are the main virulence factors of Clostridium difficile, one of the most deadly nosocomial pathogens. Recent data suggest that prophages can influence the regulation of toxin expression. Here we present the characterization of φCD38-2, a pac-type temperate Siphoviridae phage that stimulates toxin expression when introduced as a prophage into C. difficile. Host range analysis showed that φCD38-2 was able to infect 99/207 isolates of C. difficile representing 11 different PCR ribotypes. Of 89 isolates corresponding to the NAP1/027 hypervirulent strain, which recently caused several outbreaks in North America and Europe, 79 (89%) were sensitive to φCD38-2. The complete double-stranded DNA (dsDNA) genome was determined, and a putative function could be assigned to 24 of the 55 open reading frames. No toxins or virulence factors could be identified based on bioinformatics analyses. Our data also suggest that φCD38-2 replicates as a circular plasmid in C. difficile lysogens. Upon introduction of φCD38-2 into a NAP1/027 representative isolate, up to 1.6- and 2.1-fold more TcdA and TcdB, respectively, were detected by immunodot blotting in culture supernatants of the lysogen than in the wild-type strain. In addition, real-time quantitative reverse transcriptase PCR (qRT-PCR) analyses showed that the mRNA levels of all five pathogenicity locus (PaLoc) genes were higher in the CD274 lysogen. Our study provides the first genomic sequence of a new pac-type Siphoviridae phage family member infecting C. difficile and brings further evidence supporting the role of prophages in toxin production in this important nosocomial pathogen.
INTRODUCTION
Clostridium difficile is a Gram-positive, strictly anaerobic, spore-forming bacillus that causes infections with various symptoms ranging from asymptomatic carriage to fulminant diarrhea and pseudomembranous colitis (44). C. difficile infection is the most frequent cause of antibiotic-associated nosocomial diarrhea in industrialized countries (19). This opportunistic pathogen has caused severe outbreaks in North America and Europe over the last 8 years (22, 24, 38). TcdA and TcdB exotoxins are the main virulence factors of C. difficile and are encoded on a 19.6-kb chromosome region called the pathogenicity locus (PaLoc), which is found in all toxigenic isolates (44). A hypervirulent epidemic strain, called BI/NAP1/027, was shown to produce 16 times more TcdA and 23 times more TcdB in vitro than other isolates (45). The increased toxin production is thought to be responsible for the greater disease severity and higher mortality rates reported for patients infected with this particular strain (22, 24, 38).
The expression of C. difficile toxins is growth phase dependent. This regulation is achieved through the expression of TcdR, an alternative sigma factor that acts as a positive regulator of toxin expression, and TcdC, an early-expressed anti-sigma factor that prevents the TcdR-containing RNA polymerase from binding to toxin promoters (9, 29, 30, 32). A number of deletions were reported in the tcdC gene from various clinical isolates (6). A particular 1-bp deletion causing a −1 frameshift mutation and the expression of a truncated protein could possibly explain the increased toxin production observed in vitro for the NAP1/027 epidemic strain (6, 27, 32, 45).
Temperate bacteriophages (or simply phages) have played a determinant role in the virulence and evolution of major bacterial pathogens (5). Temperate phages can lead to lysogeny, which occurs when the phage integrates into the bacterial chromosome and remains as a “latent” prophage. During this lysogenic cycle, prophages sometimes modify the phenotype of their host, for example, by expressing highly potent toxins, like the Shiga toxins (Stx) in Escherichia coli, the cholera toxin (CT) in Vibrio cholerae, or the botulinum neurotoxins (BoNTs) in Clostridium botulinum (5).
A number of phages infecting C. difficile have been isolated and partially characterized so far (7, 12, 15–17, 20, 28, 34, 37, 40), and all of them are temperate. Besides the two prophages that were identified in the genome of C. difficile strain 630 (39), only four phages have been characterized at the molecular level, including complete genome sequencing, namely, φCD119 (17), φC2 (15), φCD27 (34), and φCD6356 (20). All of these phages are members of the Myoviridae family (phages with contractile tails), except φCD6356, which is the first and only Siphoviridae member (phage with a long noncontractile tail) from C. difficile for which a complete genome sequence is currently available (20). Hence, there is a clear lack of genomic data for this group of phages, especially those of the Siphoviridae family. So far, C. difficile phages have not been found to encode proven virulence factors or to convert nontoxigenic C. difficile isolates into toxin-producing lysogens (15, 17, 20, 34). Nevertheless, two recent studies suggest that phages may somehow contribute to the regulation of toxin production in C. difficile (14, 18), but the clear lack of data regarding phages of C. difficile makes it difficult to appreciate the real impact of prophages on C. difficile lifestyle and virulence.
In a previous study, we identified φCD38-2, a temperate Siphoviridae phage induced from a C. difficile clinical isolate (12). Here, we provide the full characterization of this phage, including whole genome sequencing and phenotypic characterization of lysogens. We also provide additional evidence supporting that prophages contribute to the virulence of this important nosocomial pathogen.
MATERIALS AND METHODS
Bacteria and growth conditions.
All strains used in this study were isolated from human fecal samples kindly provided by Louis Valiquette of the Université de Sherbrooke. Strain CD274, the host strain for φCD38-2, has all the characteristics of the BI/NAP1/027 hypervirulent strain (binary toxin positive, PCR ribotype 027, tcdC deletion at position 117). Bacteria were routinely grown inside a ThermoForma model 1025 anaerobic chamber (Fisher Scientific) under anaerobic atmosphere (10% H2, 5% CO2, and 85% N2) at 37°C in prereduced brain heart infusion (BHI) broth (Oxoid) or in TY broth (3% tryptose, 2% yeast extract, pH 7.4).
Bacterial DNA extraction and PCR ribotyping.
Three milliliters of an overnight C. difficile culture was centrifuged, and total genomic DNA was extracted using an Illustra bacterial genomic DNA extraction kit following the manufacturer's recommendations (GE Healthcare). PCR ribotyping was performed on an Eppendorf Mastercycler with 20 ng purified DNA and primers published by Bidet et al. (3), with modifications described previously (12). Band patterns were analyzed and compared using GelComparII (Applied Maths).
Prophage induction and phage propagation.
Phage φCD38-2 was isolated from a mitomycin C induction lysate (12). Three rounds of purification from single plaques were performed using the double agar overlay method (13) and 0.5 ml of a log-phase culture (optical density at 600 nm [OD600] of 0.4) of C. difficile strain CD274 as the sensitive host. The addition of 10 mM CaCl2 and 0.4 M MgCl2 into the soft agar was required to obtain plaques. For routine prophage induction, 10 μl of serial 10-fold dilutions of an overnight culture of C. difficile was spotted onto a BHI agar plate and incubated for 4 h at 37°C under anaerobic atmosphere to allow cells to reach the log phase. Plates were then irradiated under UV light (302 nm) for 10 s on a standard UV Transilluminator (GE Healthcare). A soft agar overlay was then poured on top of the plates as described above. Clear zones in the bacterial lawn were indicative of a successful prophage induction. Phages were then purified from an agar plug as described above. Standard procedures were used for amplification in BHI broth. CaCl2 and MgCl2 were added to a final concentration of 10 mM each, and phage titers were determined by the soft agar overlay method described above. Titers of ≥109 PFU/ml were easily obtained with this method.
Transmission electron microscopy (TEM).
Phage particles were washed with 0.1 M ammonium acetate, pH 7.5, deposited onto 400-mesh Formvar/carbon-coated copper grids (Cedarlane Laboratories), and negatively stained with 2% uranyl acetate (UA) as described before (12). The grids were observed at 60 kV with a Hitachi H-7500 transmission electron microscope equipped with a 1,000- by 1,000-pixel digital camera controlled with AMT software (Advanced Microscopy Techniques).
Host range determination and one-step growth curve assays.
A spot test on soft agar overlays prepared as described above was used to determine the host range of φCD38-2, with 10 μl of a 10-fold-diluted phage lysate and a collection of 207 clinical isolates representing 41 different PCR ribotypes. For one-step growth curve assays, cells were grown in prereduced BHI broth until the OD600 reached 0.8. Then, a phage aliquot was added to 2 ml of bacterial culture to obtain a multiplicity of infection (MOI) of 0.05. CaCl2 and MgCl2 were added to a final concentration of 10 mM each, followed by a 5-min incubation at 37°C to allow adsorption. One milliliter of the cell suspension was washed three times with prereduced BHI broth to remove nonadsorbed phages. Serial 10-fold dilutions were then made in 10 ml BHI broth containing 10 mM CaCl2 and MgCl2 and incubated at 37°C under anaerobic atmosphere. Aliquots were taken at fixed intervals over 180 min, and phage titers were determined as described above. The burst size was calculated as follows: (final phage titer − initial phage titer)/initial phage titer.
Analysis of structural proteins by SDS-PAGE and mass spectrometry.
Phage particles from a 1-liter cleared lysate (∼109 PFU/ml) were purified by two successive rounds of discontinuous cesium chloride gradient, as described previously (11). Twenty microliters of purified phage particles (5 × 1011 PFU/ml) was analyzed on a 12% denaturing SDS-polyacrylamide gel as described before (11). After Coomassie blue staining, protein bands were cut out of the gel, digested with trypsin, and analyzed by liquid chromatography-tandem mass spectrometry (LC–MS-MS) at the Proteomics Platform of the Génome Québec Innovative Center at McGill University (Montréal, Québec, Canada).
Phage DNA purification, restriction analysis, and Southern hybridization.
Small-scale preparations of whole phage DNA were obtained from cleared lysates by using a rapid miniprep protocol described elsewhere (35). For larger preparations, a maxi-Lambda DNA purification kit was used following the manufacturer's recommendations (Qiagen). Phage DNA was digested with various restriction enzymes (NEB, Roche), including EcoRV, HaeII, HindIII, and SwaI, and the digested products were heated at 75°C for 10 min and immediately run through a 0.8% agarose gel. Gels were stained with ethidium bromide, exposed to UV, and photographed using an ImageQuant 300 gel documentation system (GE Healthcare). Southern blot hybridizations were performed on restricted DNA as described before, with digoxigenin (DIG)-labeled probes consisting of PCR product A or B (primer sequences in Table S1 in the supplemental material) or whole phage genomic DNA (12).
Phage genome sequencing and bioinformatics analysis.
Whole phage genome sequencing and assembly were performed on a Roche 454 GS-FLX platform using the Titanium chemistry at the Génome Québec Innovation Center of McGill University (Montréal, Québec, Canada). Additional sequencing reactions were done directly on purified phage DNA with specific primers on an Applied Biosystems ABI 3730xl sequencer at the genomic platform of the CHUL research center (Québec, Canada). Additional sequence assembly was done using the Gap v4.10 application of the Staden package v1.6.0. Some editing was also done using BioEdit v7.0.5.3 and Artemis 11.22. Putative open reading frames (ORFs) were predicted using GeneMark.hmm for Prokaryotes v2.4 and Glimmer v3.02. The predicted proteins were compared with the BLASTp tools of the NCBI (2) and ACLAME (23) databases. Structural features and domains in predicted proteins were identified using InterProScan.
Isolation of lysogens.
Lysogens were created using a modified soft agar overlay method. Briefly, 0.1 ml of a φCD38-2 lysate (∼108 PFU/ml) was incorporated into BHI soft agar containing CaCl2 and MgCl2 that was then poured over BHI agar plates. Serial 10-fold dilutions of a log-phase (OD600 of 0.4) sensitive host were spread over this phage lawn and incubated overnight at 37°C under anaerobic atmosphere. Five phage-resistant colonies were picked and restreaked 3 times onto BHI agar plates without phages to purify the lysogens. The presence of the prophage in each lysogen was confirmed by PCR and Southern hybridization, and prophage functionality was assessed by UV induction followed by phage isolation, DNA extraction, and HindIII restriction profiling as described above.
Detection of toxins A and B.
An overnight preculture of C. difficile in TY broth was used to inoculate a fresh tube of the same broth (3% inoculum). Cells were grown as described above, and the OD600 was monitored over a 24-h period. Aliquots were taken at 2, 8, 12, 18, 24, and 48 h postinoculation. For extracellular toxin detection, cells were removed by centrifugation, and cleared supernatants were stored at −20°C until analysis. For intracellular toxin detection, bacteria from a 10-ml culture sample were collected by centrifugation and suspended in 0.5 ml phosphate-buffered saline (PBS). Cells were then broken with glass beads (≤106 μm; Sigma) using a FastPrep apparatus (MP Biomedicals). The lysate was cleared by centrifugation, and the supernatant was stored at −20°C until analysis. Detection of the toxins was done on appropriate dilutions using an enzyme-linked immunosorbent assay (ELISA) (Premier Toxins A and B kit; Meridian Biosciences), as recommended by the supplier. The ELISA unit definition corresponds to the absorbance at 450 nm of the ELISA reaction multiplied by the dilution factor and converted to a volume of 1 ml of cells (intracellular toxins), culture supernatant (extracellular toxins), or a combination of both (total toxins). An immunodot blot method was used to specifically detect TcdA and TcdB. For this, culture supernatants were serially diluted in TY broth and directly spotted (0.1 ml) onto nitrocellulose membranes using a 96-well dot blotter apparatus. All wells were washed twice with PBS, after which the membrane was allowed to air dry for 30 min. Toxins were detected with monoclonal anti-TcdA or anti-TcdB mouse antibody (Meridian Life Science) at a 1:3,000 or 1:1,000 dilution, respectively. A secondary anti-mouse IgG horseradish peroxidase (HRP)-linked antibody (Cell Signaling) was used at a 1:3,000 dilution, and the membranes were revealed with an ECL Plus Western blotting detection system (GE Healthcare) as recommended by the manufacturer, followed by exposition to Hyperfilm ECL autoradiography films (GE Healthcare) (33). Spot intensities were compared using ImageJ 1.42q software (http://rsbweb.nih.gov/ij/).
RNA extraction and gene expression analysis.
Total RNA was extracted from 10-ml culture samples after two successive treatments with TRIzol (Invitrogen). Cells were broken during the first treatment by adding glass beads (≤106 μm; Sigma) and using a FastPrep apparatus (MP Biomedicals). Total RNA was dissolved in RNase-free water, and 10 μg was treated with 6 units of RNase-free Turbo DNase I (Ambion) for 30 min at 37°C, as recommended by the manufacturer. The absence of contaminating genomic DNA was verified by performing a 40-cycle PCR with primers targeting the 16S rRNA gene in the presence of 200 ng total RNA. First-strand cDNA synthesis was performed on 3 μg total RNA using SuperScriptII RT (Invitrogen) with random primers (Promega) according to the manufacturer's specifications. Real-time quantitative reverse transcriptase PCRs (qRT-PCRs) were performed on a Mastercycler EP Realplex instrument (Eppendorf) in a total volume of 10 μl, with the following components: 1× PCR buffer (12 mM Tris-HCl, pH 8.3, 50 mM KCl, 8 mM MgCl2, 150 mM trehalose, 0.2% Tween 20, 0.2 mg/ml bovine serum albumin [BSA], 0.2× SYBR green [Roche]), 150 ng of template cDNA, and one of the primer sets specific for tcdA, tcdB, tcdC, tcdR, tcdE, or the 16S rRNA gene (see Table S1 in the supplemental material). The cycling conditions were as follows: 95°C for 2 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The ΔΔCT (threshold cycle) comparative method was used to calculate the relative expression of each gene, with the 16S rRNA gene as the reference gene.
Nucleotide sequence accession number.
The complete genome sequence of φCD38-2 has been submitted to GenBank under the accession number HM568888.
RESULTS
Phage isolation.
We have previously reported the induction of two temperate phages, φCD38-1 and φCD38-2, after mitomycin C treatment of CD38, a clinical isolate of C. difficile (12). Several C. difficile isolates were found to be sensitive to φCD38-2, including CD274, which has all the common characteristics of the hypervirulent NAP1/027 strain, which has caused several outbreaks in North America and Europe (ribotype 027, tcdC deletion, binary toxin positive). Phage φCD38-2 was purified from single plaques using CD274 as the host and further propagated in BHI broth to ≥109 PFU/ml. Transmission electron microscopy (Fig. 1) and DNA restriction profiling confirmed that the isolated phage, a member of the Siphoviridae family of the order Caudovirales (1), corresponded to the φCD38-2 phage that we described before (12).
Fig. 1.
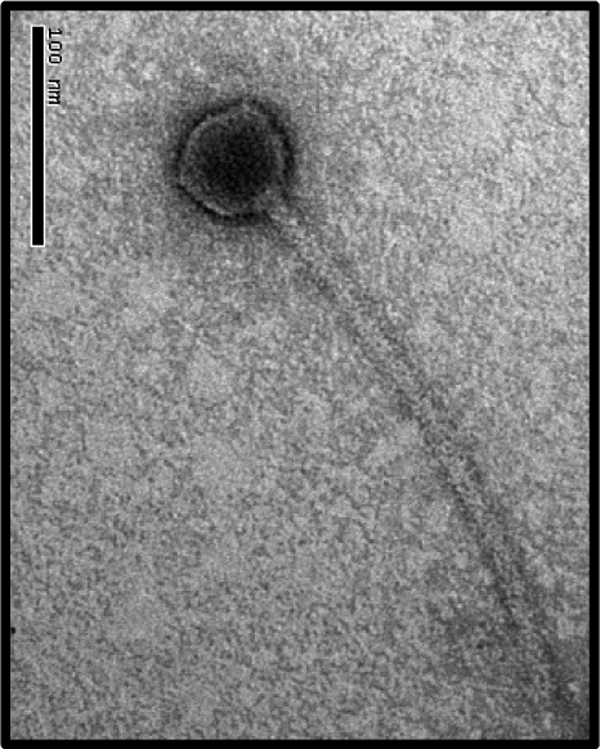
TEM picture of φCD38-2 negatively stained with 2% uranyl acetate. Bar, 100 nm.
Host range and lytic growth cycle.
The host range of φCD38-2 was determined using a collection of C. difficile clinical isolates and spot tests on soft agar overlays. φCD38-2 infected 99 of the 207 isolates tested (48%), among which 79 (80%) corresponded to the NAP1/027 epidemic strain (PCR ribotype 027). The other, non-NAP1/027 sensitive isolates represented 10 different PCR ribotypes (see Table S2 in the supplemental material). The lytic growth cycle of φCD38-2 was determined on strain CD274. The latent period was 95 min, and the burst size was 35 ± 11 PFU per infected cell, which is in the range reported for other C. difficile phages (16, 28).
Genome sequence.
Full genome sequencing was performed on a Roche 454 GS-FLX platform. A total of 17,659 sequence reads were obtained and assembled in two large contigs totaling 40,468 bp, with an average coverage of ∼90-fold. The two contigs were joined, and gaps were filled after sequencing reactions were performed directly on the phage DNA. The φCD38-2 genome is composed of a double-stranded DNA (dsDNA) molecule of 41,090 bp with a G+C content of 30.83%, which is a little above, but in the range reported for, that of other C. difficile phages (28.7 to 29.4%) (15, 17, 34) and of C. difficile strain 630 (29.06%) (39). Digestion of the purified phage DNA with various restriction enzymes gave profiles perfectly corresponding to a circular genomic map, except for a faint submolar fragment of ∼0.9 kb that was observed with EcoRV (see Fig. S1 in the supplemental material). Heating the digested DNAs at 75°C for 10 min prior to loading on the agarose gel did not reveal cohesive termini. Moreover, Southern hybridization of the restricted DNA with probe A, covering nucleotides 39450 to 40663, which we suspected to be the region containing the pac site, revealed the expected submolar fragments in all digestions (see Fig. S2 in the supplemental material). Thus, our data indicate that φCD38-2 is a pac-type phage that packages its DNA using a headful mechanism and that the pac site is located between orf53 and orf54. To our knowledge, this represents the first pac-type Siphoviridae phage to be described in C. difficile.
DNA homology and other similar prophages.
BLASTn analyses against the nonredundant nucleotide and whole genome shotgun databases at NCBI revealed the existence of unassembled genomic fragments nearly identical to φCD38-2 in two C. difficile isolates currently being sequenced at McGill University: strains QCD-37x79 (contig NZ_ABHG02000044) and QCD-63q42 (contigs NZ_ABHD02000046, NZ_ABHD02000048, NZ_ABHD02000049, and NZ_ABHD02000057).
Gene products and annotation.
Fifty-five putative orf genes encoding proteins of ≥30 amino acids were identified by GeneMark.hmm and Glimmer analyses using standard (ATG) and alternative (GTG, TTG, CTG) start codons. Manual validation of each orf was then performed, and the most probable start codon was selected based on the presence of a suitable ribosome-binding site complementary to the 3′ end of the 16S rRNA gene of C. difficile 630 (39). A genomic map of φCD38-2 is presented in Fig. 2.
Fig. 2.

Genetic organization of the complete φCD38-2 genome (41,090 bp). Predicted ORFs and their orientations are represented by arrows. Functional assignments are indicated above the ORFs, along with functional modules that were inferred based on gene annotation and whole genomic organization. Thick black arrows correspond to proteins identified by LC–MS-MS. The relative G+C content, calculated on a 10-base window along the whole φCD38-2 genome, is shown below the map to highlight a region of possible horizontal gene transfer (gray arrows). RBP, receptor-binding protein; SSB, single-strand binding protein.
All predicted ORFs were translated into proteins and compared against nonredundant protein sequences from GenBank and ACLAME databases using BLASTp. Putative functions were attributed to each ORF based on BLAST results, by comparison with homologous proteins found in the ACLAME database, and based on the presence of conserved domains found through searches in the conserved domain database (CDD) at NCBI and by InterProScan analyses. Overall, a putative function could be attributed to 23 of the 55 ORFs (42%), and the best BLAST hits corresponding to gene products identified in strain QCD-37x79 are listed in Table S3 in the supplemental material, along with a second relevant hit from another source, when available (excluding hits from strain QCD-63q42). The complete genome sequence of φCD6356, the first cos-type temperate Siphoviridae phage described in C. difficile, has recently been published (20). Protein similarity was found between the lysis, lysogeny control, and DNA replication, recombination, and modification modules of phages φCD38-2 and φCD6356, but their structural genes were unrelated. Also, only a few hits corresponded to gene products from φCD27 and φCD119, thus confirming that φCD38-2 is completely different from all Myoviridae phages of C. difficile described so far (15, 17, 34, 39). The gene organization and modular structure of the φCD38-2 genome are similar to those of other C. difficile phages and other temperate phages infecting low-G+C bacteria (25). Key features of the φCD38-2 annotation are described below.
Structural proteins.
Protein BLAST analyses showed that except for the two highly related prophages identified in C. difficile, strains QCD-37x79 and QCD-63q42, most structural proteins were related to other Clostridium genomic sequences, including C. perfringens, C. botulinum, and C. tetani (see Table S3 in the supplemental material). ORF18 corresponds to a tail protein with a putative endopeptidase activity and is probably the phage receptor-binding protein (RBP) responsible for host specificity. Structural proteins of CsCl-purified φCD38-2 particles were separated by SDS-PAGE, followed by LC–MS-MS analysis of the trypsin-digested protein bands (Fig. 3A). The experimental and calculated masses were in agreement, and peptide mapping did not reveal any evidence of posttranslational proteolytic processing (Fig. 3B). Based on local genomic organization and BLAST analyses, ORF7 was annotated as the major capsid protein (MCP) and ORF17 as the major tail protein (MTP). Because the boundary between the capsid and tail morphogenesis modules could not clearly be defined, ORF12 could be either a capsid or a tail protein, and as a consequence, it was annotated as a major structural protein (MSP), without reference to any particular virion structure (Fig. 3B).
Fig. 3.

Analysis of φCD38-2 structural proteins. (A) Coomassie brilliant blue staining of a 12% SDS-polyacrylamide gel, showing φCD38-2 structural proteins, along with a protein molecular mass marker (lane M). Arrows and letters on the right correspond to protein bands identified by LC–MS-MS analysis, which are further characterized in panel B.
Lysogeny control and putative lysogenic conversion genes.
In most phages infecting low-G+C Gram-positive bacteria, including C. difficile phages φC2, φCD27, and φCD119, the lysogeny module is located between the lysis cassette and the DNA replication and regulation module. This region generally encodes Cro and cI repressors, transcriptional regulators, and antirepressors, as well as the integrase (15, 17, 20, 26, 34). A lysogeny module could not clearly be defined in φCD38-2, and a different organization was observed. ORF39, a putative cI phage repressor based on BLAST results and on the presence of a helix-turn-helix (HTH) DNA-binding domain, was found approximately 10 kb downstream of the lysis cassette. ORF53, a phage integrase of the tyrosine recombinase/integrase family, was found 5 kb downstream of orf39 near ORF55, a protein of the SR serine recombinase family. In φCD6356, a site-specific recombinase (orf57) is also located apart from the lysogeny module (orf34 to orf40). However, in the latter case, 3 putative transcriptional regulators are clustered in the lysogeny module (orf35, orf37, and orf38), whereas in φCD38-2, we found only one.
A putative function could be assigned to only 8 (25%) of the 32 nonstructural genes (orf24 to orf55), of which 4 could be related to DNA replication, transcription, and gene regulation. InterProScan analyses predicted the presence of a signal peptide and/or transmembrane regions within ORF32, ORF33, and ORF34, suggesting that these proteins could potentially be targeted to the membrane or be secreted. Interestingly, the φCD38-2 genome showed a marked deviation in its G+C content from orf24 to orf34, where the average G+C content was 25.6% ± 1.2%, while it was 31.6% ± 3.1% in the rest of the genome (Fig. 2). Such deviations are often traces of past horizontal gene transfer (HGT) events. In line with this, a BLASTp analysis with ORF35 retrieved hits corresponding to DNase, including CDP07 (see Table S3 in the supplemental material), a putative DNase found on plasmid pCD630 (NCBI accession no. NC_008226.1) carried by C. difficile strain 630 (39). Moreover, a nucleotide BLAST analysis revealed a region extending from positions 31112 to 32991 in φCD38-2 sharing 65% identity with a region from plasmid pCD630 that corresponds to ∼2/3 of the gene coding for a DNase. Taken together, these data suggest that a portion of the φCD38-2 genome, located next to the lysis module, has been acquired through HGT and could possibly participate in lysogenic conversion of the host. Further experiments are needed to confirm this hypothesis. Note that we performed a similar analysis with φC2, φCD27, and φCD119, and only the last shared significant homology (68% nucleotide identity over 794 bp) with a region coding for a methyltransferase in plasmid pCKL555A (NCBI accession no. CP000674.1) of Clostridium kluyveri DSM 555 (17). Interestingly, a note in the ACLAME database mentions that pCKL555A is a prophage. These observations support the idea that other phages of C. difficile have probably recombined with plasmids as well.
Prophage maintenance as a circular plasmid.
The attachment site (attP) in most temperate phages is generally located near the integrase gene. Also, when a prophage integrates into the chromosome of its host, at least one band from the phage restriction profile shifts in the lysogen due to its fusion with bacterial DNA. In order to locate attP and to determine whether or not φCD38-2 integrates, we performed Southern blot hybridizations with DIG-labeled PCR probes covering the integrase region (probe A, orf53 to orf55) and tail (probe B, orf18 to orf20) genes (see Fig. S2 in the supplemental material). We also used the whole phage genome as a probe. As can be seen in Fig. S2, the whole restriction profiles and the sizes of specific fragments detected by the two PCR probes were identical in the purified phage and in the lysogen, except for the submolar fragment that was present only in the purified phage DNA. Because no visible shift in size could be observed with any bands and since the submolar fragment was absent from the lysogen, we concluded that φCD38-2 did not integrate and that its genome was circular in the lysogen (see Fig. S2). Interestingly, the presence of a 1.5-kb fragment sharing 65% identity at the DNA level with the pCD630 plasmid from C. difficile strain 630 further supports the evidence that the φCD38-2 prophage replicates as a circular plasmid. Also noteworthy to mention, the two contigs from strains QCD-37x79 and QCD-63q42 that are almost identical to φCD38-2 were found as unassembled fragments in public databases, suggesting that they could not be associated with bacterial DNA. Finally, a ParA homolog (ORF30) similar to a Spiroplasma citri Soj-like protein was found in φCD38-2 (ParA cd02042, 1e−3; Soj COG1192, 1e−18; CbiA pfam01656, 4e−9; SopA PHA02519, 3e−6) (see Table S3 in the supplemental material). Since ParA/Soj-like proteins are involved in chromosome segregation and plasmid maintenance (31), the presence of ORF30 in φCD38-2 supports a role in prophage maintenance as a plasmid. To our knowledge, this represents the first example of such a prophage in C. difficile.
Prophage-stimulated toxin production in φCD38-2 lysogens.
Previous reports have shown that toxin production in C. difficile can be affected by some prophages (14, 18). In order to test whether φCD38-2 could influence toxin production in C. difficile, we infected the host isolate CD274, which is a representative member of the hypervirulent strain BI/NAP1/027, to create lysogens. The growth profiles in TY broth and total biomass yields after 24 h were not significantly different between the CD274/φCD38-2 lysogen and the wild-type parental strain (Fig. 4A). Aliquots of cells and culture supernatants were collected at different time intervals, and intracellular and extracellular relative toxin levels were determined using a commercial ELISA. At 8 and 12 h, most of the toxins detected were intracellular, and the level increased 3-fold at 12 h, which is consistent with the entry into stationary phase. A slight and gradual decrease was observed afterward, but the levels did not differ significantly between the wild type and the lysogen (Fig. 4B). On the contrary, extracellular toxins accumulated faster and to a higher level in culture supernatants from the CD274/φCD38-2 lysogen, with 2.1-, 2.4-, and 2.0-fold more toxins than in wild-type CD274 after 12, 18, and 24 h of growth, respectively (P < 0.05 at 18 and 24 h). The proportion of extracellular toxins also represented ∼80 to 90% of the total toxins detected in culture samples. The amount of extracellular toxins reached a plateau at 24 h in supernatants from the wild-type strain but continued to accumulate gradually in the lysogen until 48 h (see Fig. S3 in the supplemental material). The total toxin production, expressed in ELISA units/ml of culture and obtained by combining the intracellular and extracellular toxin values, yielded 1.2-, 2.3-, and 1.8-fold higher toxin levels in the lysogen after 12, 18, and 24 h of growth, respectively (P < 0.05 at 18 and 24 h) (Fig. 4B). An immunodot blotting experiment using specific anti-TcdA and anti-TcdB antibodies revealed that both toxins, in particular, TcdB, accumulated to higher levels in culture supernatants of the lysogen (Fig. 5). Densitometry analysis of the dots showed that the amounts of TcdA and TcdB were ∼1.6- and 2.1-fold larger, respectively, in the CD274/φCD38-2 lysogen than in the wild-type CD274 strain. This was consistent with the results obtained with the ELISA (Fig. 4B; also see Fig. S3 in the supplemental material).
Fig. 4.
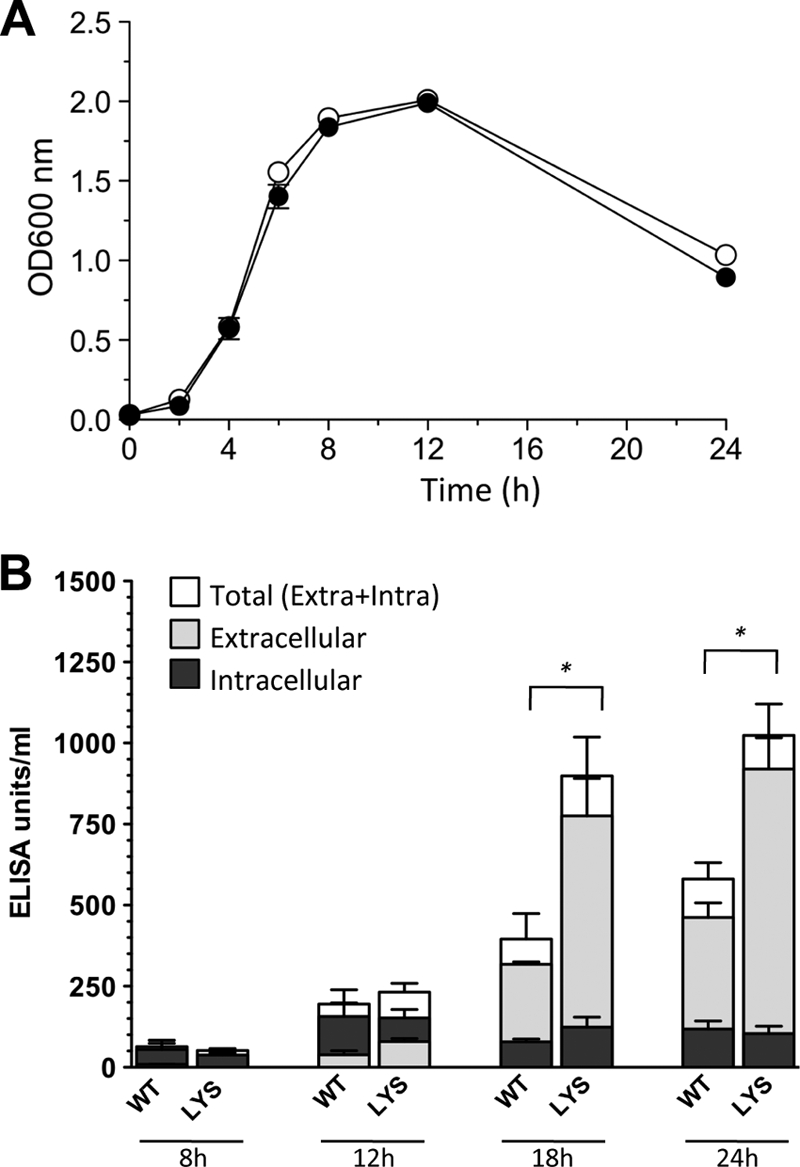
Growth and toxin production of wild-type CD274 and the CD274/φCD38-2 lysogen. (A) Growth of the wild type (white circles) and the lysogen (black circles) in TY broth was monitored by measuring the optical density at 600 nm over 24 h. (B) The relative amounts of TcdA and TcdB toxins were determined by a toxin A/B ELISA. Data represent the means ± standard deviations from three independent biological replicates. Extracellular, intracellular, and total (extracellular plus intracellular) toxin levels of the wild type (WT) and the lysogen (LYS) were compared by a Student t test. Significant differences (*, P < 0.05) were observed for total and extracellular, but not for intracellular, toxins.
Fig. 5.
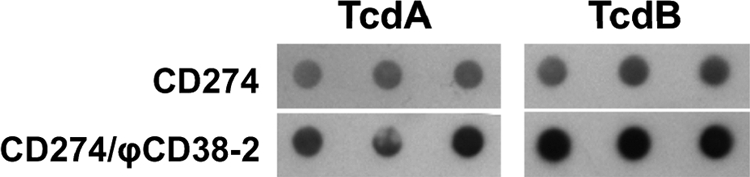
Immunodot blot detection of toxins in cleared supernatants from 24-h cultures of wild-type CD274 and a CD274/φCD38-2 lysogen. TcdA and TcdB toxins were detected using monoclonal anti-TcdA and anti-TcdB antibodies. Each spot represents an independent biological experiment.
We then performed real-time qRT-PCR assays to compare the relative expression levels of the five PaLoc genes in both strains. Total RNA was extracted from the CD274/φCD38-2 lysogen and the wild-type parental strain CD274 at 4, 12, 18, and 24 h postinoculation. Based on ΔCT values (compared to that for the 16S rRNA gene), the expression profiles were consistent with what we expected. For example, the expression of tcdR, tcdA, and tcdB increased sharply between 4 and 12 h and then decreased gradually afterwards. Expression of tcdC reached its maximum at 4 h and then gradually decreased. Finally, the expression of tcdE remained relatively constant over the first 18 h and then decreased at 24 h (data not shown). By use of the ΔΔCT relative comparison, the expression levels of all PaLoc genes were found to be similar in lysogenic and wild-type strains after 4 h of growth (ratio of ∼1), but after 24 h, the levels of tcdA, tcdB, tcdC, tcdE, and tcdR mRNA were, respectively, 2.7-, 2.9-, 2.7-, 5.7-, and 2.7-fold higher in the lysogen than in the wild-type strain (Fig. 6). Again, these data are in agreement with the toxin levels that were detected by ELISA and immunodot blotting. Our results show that the expression patterns are similar in both strains but that the presence of the prophage leads to higher expression of all PaLoc genes.
Fig. 6.

Relative expression of PaLoc genes in the CD274/φCD38-2 lysogen versus the wild-type CD274 strain at different time points. Data are presented as the fold change in gene expression in the lysogen relative to that for the wild-type strain and represent the means ± standard errors of the means from 4 independent biological replicates. A value of 1 means that there is no difference in mRNA levels between the two strains. For each gene, the fold change at 12, 18, and 24 h was compared to the value at 4 h by using the Student t test (*, P < 0.05; **, P < 0.01).
Four additional φCD38-2 lysogens were created in different genetic backgrounds to verify if the above-described observations made with the CD274 lysogen could be extended to other C. difficile isolates. Phage φCD38-2 was introduced into strains CD45 (PCR ribotype 035) and CD62 (PCR ribotype 014), as well as CD66 and CD111 (both PCR ribotype 027). Toxins A and B were detected in culture supernatants using the ELISA and were comparable to levels for strain CD274 (418 ELISA units/ml), with 379, 113, 780, and 724 ELISA units/ml for strains CD45, CD62, CD66, and CD111, respectively. When the amounts of toxins produced by the corresponding lysogens were compared, only the CD45 lysogen showed a 2-fold increase in extracellular toxins after 24 h compared to the level for the wild-type strain. Toxin levels in the supernatants of the CD62, CD66, and CD111 lysogens were, respectively, 1.3-, 0.6-, and 1.3-fold the level for the wild-type strain and were not significantly different. The amounts of tcdA and tcdB transcripts were determined by real-time qRT-PCR analysis of total RNA extracted from these lysogens and the corresponding parental strain after 24 h of growth in TY broth. The detected mRNA levels were higher in the CD45/φCD38-2 lysogen, with 29- and 22-fold increases in tcdA and tcdB mRNA, respectively (see Fig. S4 in the supplemental material). The CD62, CD66, and CD111 lysogens showed on average 5.76/5.57-, 0.75/0.44-, and 2.14/1.32-fold differences, respectively, in toxin A/B mRNA levels compared to levels for the parental strain. Five colonies were picked at the end of each culture experiment, and the presence of the prophage was confirmed by PCR in all cases, thus excluding prophage loss as a possible explanation for the observed variability in toxin production from one lysogen to another. Taken together, our results demonstrate that φCD38-2 can stimulate toxin production in some lysogens, including the NAP1/027 epidemic strain, by increasing mRNA transcription and/or stability and that this effect seems to be strain dependent.
DISCUSSION
The molecular basis for the hypervirulence and hyper-toxin-producing phenotype of the NAP1/027 epidemic strain is still unclear, and it is reasonable to presume that prophages might be involved. Here we report the microbiological and molecular characterization of φCD38-2, a temperate phage of the Siphoviridae family infecting Clostridium difficile, and our study provides further evidence that temperate phages can affect important virulence-associated phenotypes, like toxin production, in C. difficile.
The genomes of only a few phages of C. difficile have been fully sequenced to date. In addition, all currently available C. difficile phage sequences represent members of the Myoviridae family that are related genetically (15, 17, 34, 39). The only exception is phage φCD6356, a cos-type phage of the Siphoviridae family (20). The complete genome of φCD38-2 was sequenced, and protein comparisons revealed that the lysis and DNA replication/gene regulation modules of φCD38-2 and φCD6356 are related but that their structural genes are completely different. The fact that φCD38-2 is a pac-type Siphoviridae phage whereas φCD6356 is a cos-type phage confirms that they are part of two distinct phage families that package their DNA using two different mechanisms. The presence of a putative integrase gene (orf53) would have a priori suggested that φCD38-2 should integrate upon lysogenization, but our experimental data support the conclusion that φCD38-2 maintains itself as a circular plasmid and does not integrate into the chromosome of the lysogens that we tested. Of note, the genomic organization of φCD6356 is very similar to that of φCD38-2 regarding the nonstructural genes, and it would be interesting to know whether the φCD6356 prophage integrates or maintains itself as a circular plasmid, but this information was not provided by the authors (20). Also, the identification of a ParA homolog in φCD38-2, as in φCD6356 and φC2, was interesting because ParA/Soj-like proteins are involved in chromosome segregation and plasmid maintenance (31). ParA and ParB homologs were also shown to enable the temperate phage LE1 from Leptospira biflexa to replicate autonomously as a circular plasmid (4). Finally, we found an ∼1.9-kb fragment in the region encoding ParA in φCD38-2 that shares significant homology with the plasmid from C. difficile strain 630. This suggests that a past recombination event between a prophage and a plasmid occurred, leading to a chimeric phage that can autonomously replicate as a circular plasmid. To our knowledge, this represents the first example of such a prophage in C. difficile.
Prophage-stimulated toxin production in NAP1/027 lysogens.
TcdR and TcdC are positive and negative regulators of toxin production in C. difficile, respectively (44). A number of deletions were identified in tcdC, in particular, a 1-bp deletion at position 117 that leads to the synthesis of a severely truncated TcdC protein in the NAP1/027 epidemic strain (6, 27). This deletion is thought to be responsible for the increased toxin production reported in this strain (45). However, recent studies suggest that deletions in tcdC alone cannot explain hyper-toxin production and hypervirulence of NAP1/027 isolates (36, 43). The regulation of toxin production in C. difficile thus seems to be complex, and other mechanisms are likely involved in this process. A previous report by Goh et al. suggested that lysogens carrying the temperate phages φC2, φC6, and φC8 could modify toxin production in C. difficile (14). Interestingly, the PaLoc shares some sequence similarity with phage proteins, in particular, TcdE, suggesting that it is probably the remains of an ancient prophage. This also suggests that phage regulatory networks could be intertwined with those of the PaLoc (14, 42). Further evidence supporting a possible interconnection between prophages and the PaLoc was recently provided by Govind et al., who showed that during lysogeny, the RepR transcriptional regulator encoded by φCD119 was able to bind to a promoter region in the PaLoc upstream of tcdR, causing a downregulation of the expression of all PaLoc genes, including tcdA and tcdB (18).
Lysogenization of CD274 with φCD38-2 led to 1.6- and 2.1-fold increases in toxins A and B in culture supernatants, respectively (Fig. 4 and 5). Although not dramatic, the increase was significant. Also, the levels of intracellular toxins were not very different between the two strains, and most of the toxins (80 to 90%) were found in the culture supernatant. Using a real-time qRT-PCR approach, we demonstrated that the mRNA levels of all 5 PaLoc genes were higher in the lysogen carrying φCD38-2 than in the wild-type strain (Fig. 6). In addition, there seemed to be greater expression of tcdE than of the other PaLoc genes in the lysogen. Together, these results lead us to conclude that the lysogen carrying φCD38-2 synthesized and secreted more toxins, as a result of increased expression of PaLoc genes and especially tcdE. The net result is a higher extracellular toxin level in cultures of the lysogen and similar intracellular toxin levels in both strains. Our bioinformatics analyses identified only one HTH putative DNA-binding protein in the φCD38-2 genome (ORF39), and this protein is likely the cI phage repressor involved in lysogeny maintenance. The only other identifiable candidate that could possibly affect RNA transcription is a putative sigma factor (ORF52). This gene product could bind directly to promoter regions upstream of PaLoc genes and recruit the RNA polymerase to increase the rate of transcription initiation. Alternatively, it could interfere with TcdC. In the latter case, TcdC would be impaired in its ability to destabilize the TcdR-RNA polymerase holoenzyme, thus promoting transcription initiation through binding of TcdR. Because φCD38-2 does not seem to integrate into the chromosome of its host, disruption of a bacterial gene is unlikely to be the reason explaining the difference in PaLoc gene transcript levels. Further experiments are necessary to determine the exact mechanism leading to increased toxin expression.
Our study and those of Govind (18) and Goh (14) also showed that depending on the phage-host system and the bacterial genetic background, the impact of different prophages on toxin production varies. We found that transcription was significantly increased in some lysogens and was unaffected or slightly decreased in others (see Fig. S4 in the supplemental material). It is already known that toxin expression varies greatly from one strain to another and that several factors may participate in such regulation (10, 21). The strains we selected to create additional lysogens produced similar amounts of toxins. Hence, at least in our study, we can rule out the possibility that differences in basal toxin production explain the variable impact of φCD38-2 on these strains. The presence of a putative integrase gene in φCD38-2 suggests that the phage could potentially integrate into the chromosome of its host. However, consistent experimental evidence suggests that the φCD38-2 prophage replicates autonomously as a circular plasmid. Although we cannot completely exclude the possibility that this phage could integrate in some strains but not in others, the strain-dependent difference in toxin production that we observed in our study is unlikely due to gene disruption at different sites. We did not determine systematically the prophage content of the strains we used in our study, but we can confirm that strain CD111 contains at least one Siphoviridae phage different from φCD38-2 (unpublished data). We also found that strain CD274 contains a prophage of the Myoviridae family identical or highly similar to φCD5, which we previously characterized (12), and to φ027 from the epidemic strain R20291 (41) (NCBI accession no. accession number NCO13316). It is thus possible that multiple synergistic and/or antagonistic prophage interactions contribute to a complex network regulating toxin expression.
Conclusion.
In summary, we characterized and sequenced the first genome of a pac-type Siphoviridae phage infecting C. difficile. In addition, our data strongly suggest that φCD38-2 replicates as a circular plasmid and does not integrate into the chromosome of its host. Phage φCD38-2 is able to infect several isolates of the hypervirulent epidemic strain NAP1/027, which recently caused severe outbreaks in North America and Europe. Complete genome sequencing did not reveal the presence of identifiable virulence factors, but lysogenization of a NAP1/027 isolate with φCD38-2 led to increased in vitro toxin production through increased transcription of all PaLoc genes. Because φCD38-2 has the capacity to alter virulence-associated phenotypes through modulation of toxin expression, this phage represents a very interesting model to study phage-host interactions. The impacts of prophages in this clinically important pathogen remain relatively unexplored, and our study warrants further research in this area.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Louis Valiquette for providing clinical isolates of C. difficile. We also thank Pier-Luc Dudemaine for helping with the host range analysis.
This study was supported by the Centre de Recherche Clinique Étienne-Le Bel, by a discovery grant from the Natural Sciences and Engineering Council of Canada (NSERC), and by a seed grant from the Canadian Institutes of Health Research (CIHR).
L.-C.F. is a research scholar from the Fonds de la Recherche en Santé du Québec (FRSQ).
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 25 March 2011.
REFERENCES
- 1. Ackermann H. 2007. 5500 phages examined in the electron microscope. Arch. Virol. 152:227–243 [DOI] [PubMed] [Google Scholar]
- 2. Altschul S., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bidet P., Barbut F., Lalande V., Burghoffer B., Petit J. 1999. Development of a new PCR-ribotyping method for Clostridium difficile based on ribosomal RNA gene sequencing. FEMS Microbiol. Lett. 175:261–266 [DOI] [PubMed] [Google Scholar]
- 4. Bourhy P., et al. 2005. Complete nucleotide sequence of the LE1 prophage from the spirochete Leptospira biflexa and characterization of its replication and partition functions. J. Bacteriol. 187:3931–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brussow H., Canchaya C., Hardt W. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68:560–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Curry S., et al. 2007. tcdC genotypes associated with severe TcdC truncation in an epidemic clone and other strains of Clostridium difficile. J. Clin. Microbiol. 45:215–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dei R. 1989. Observations on phage-typing of Clostridium difficile: preliminary evaluation of a phage panel. Eur. J. Epidemiol. 5:351–354 [DOI] [PubMed] [Google Scholar]
- 8. Reference deleted.
- 9. Dupuy B., Govind R., Antunes A., Matamouros S. 2008. Clostridium difficile toxin synthesis is negatively regulated by TcdC. J. Med. Microbiol. 57:685–689 [DOI] [PubMed] [Google Scholar]
- 10. Dupuy B., Sonenshein A. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol. Microbiol. 27:107–120 [DOI] [PubMed] [Google Scholar]
- 11. Fortier L., Bransi A., Moineau S. 2006. Genome sequence and global gene expression of Q54, a new phage species linking the 936 and c2 phage species of Lactococcus lactis. J. Bacteriol. 188:6101–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fortier L., Moineau S. 2007. Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 73:7358–7366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fortier L., Moineau S. 2009. Phage production and maintenance of stocks, including expected stock lifetimes. Methods Mol. Biol. 501:203–219 [DOI] [PubMed] [Google Scholar]
- 14. Goh S., Chang B., Riley T. 2005. Effect of phage infection on toxin production by Clostridium difficile. J. Med. Microbiol. 54:129–135 [DOI] [PubMed] [Google Scholar]
- 15. Goh S., Ong P., Song K., Riley T., Chang B. 2007. The complete genome sequence of Clostridium difficile phage {phi}C2 and comparisons to {phi}CD119 and inducible prophages of CD630. Microbiology 153:676–685 [DOI] [PubMed] [Google Scholar]
- 16. Goh S., Riley T., Chang B. 2005. Isolation and characterization of temperate bacteriophages of Clostridium difficile. Appl. Environ. Microbiol. 71:1079–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Govind R., Fralick J., Rolfe R. 2006. Genomic organization and molecular characterization of Clostridium difficile bacteriophage {Phi}CD119. J. Bacteriol. 188:2568–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Govind R., Vediyappan G., Rolfe R., Dupuy B., Fralick J. 2009. Bacteriophage-mediated toxin gene regulation in Clostridium difficile. J. Virol. 83:12037–12045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gravel D., et al. 2009. Health care-associated Clostridium difficile infection in adults admitted to acute care hospitals in Canada: a Canadian Nosocomial Infection Surveillance Program study. Clin. Infect. Dis. 48:568–576 [DOI] [PubMed] [Google Scholar]
- 20. Horgan M., et al. 2010. Genome analysis of the Clostridium difficile phage PhiCD6356, a temperate phage of the Siphoviridae family. Gene 462:34–43 [DOI] [PubMed] [Google Scholar]
- 21. Hundsberger T., et al. 1997. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur. J. Biochem. 244:735–742 [DOI] [PubMed] [Google Scholar]
- 22. Kuijper E., Coignard B., Tull P. 2006. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin. Microbiol. Infect. 12(Suppl. 6):2–18 [DOI] [PubMed] [Google Scholar]
- 23. Leplae R., Hebrant A., Wodak S. J., Toussaint A. 2004. ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 32:D45–D49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loo V., et al. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N. Engl. J. Med. 353:2442–2449 [DOI] [PubMed] [Google Scholar]
- 25. Lucchini S., Desiere F., Brussow H. 1999. Comparative genomics of Streptococcus thermophilus phage species supports a modular evolution theory. J. Virol. 73:8647–8656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lucchini S., Desiere F., Brussow H. 1999. Similarly organized lysogeny modules in temperate Siphoviridae from low GC content gram-positive bacteria. Virology 263:427–435 [DOI] [PubMed] [Google Scholar]
- 27. MacCannell D. R., et al. 2006. Molecular analysis of Clostridium difficile PCR ribotype 027 isolates from eastern and western Canada. J. Clin. Microbiol. 44:2147–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mahony D., Bell P., Easterbrook K. 1985. Two bacteriophages of Clostridium difficile. J. Clin. Microbiol. 21:251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mani N., Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc. Natl. Acad. Sci. U. S. A. 98:5844–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mani N., et al. 2002. Environmental response and autoregulation of Clostridium difficile TxeR, a sigma factor for toxin gene expression. J. Bacteriol. 184:5971–5978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marston A., Errington J. 1999. Dynamic movement of the ParA-like Soj protein of B. subtilis and its dual role in nucleoid organization and developmental regulation. Mol. Cell 4:673–682 [DOI] [PubMed] [Google Scholar]
- 32. Matamouros S., England P., Dupuy B. 2007. Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol. Microbiol. 64:1274–1288 [DOI] [PubMed] [Google Scholar]
- 33. Matte I., et al. 2009. Anti-apoptotic proteins Bcl-2/Bcl-XL inhibit Clostridium difficile toxin A-induced cell death in human epithelial cells. Infect. Immun. 77:5400–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mayer M. J., Narbad A., Gasson M. J. 2008. Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J. Bacteriol. 190:6734–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moineau S., Pandian S., Klaenhammer T. R. 1994. Evolution of a lytic bacteriophage via DNA acquisition from the Lactococcus lactis chromosome. Appl. Environ. Microbiol. 60:1832–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murray R., Boyd D., Mulvey M., Levett P., Alfa M. 2009. Truncation in the tcdC region of the Clostridium difficile PathLoc of clinical isolates does not predict increased biological activity of toxin B or toxin A. BMC Infect. Dis. 9:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagy E., Foldes J. 1991. Electron microscopic investigation of lysogeny of Clostridium difficile strains isolated from antibiotic-associated diarrhea cases and from healthy carriers. APMIS 99:321–326 [DOI] [PubMed] [Google Scholar]
- 38. Pepin J., Valiquette L., Cossette B. 2005. Mortality attributable to nosocomial Clostridium difficile-associated disease during an epidemic caused by a hypervirulent strain in Quebec. CMAJ 173:1037–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sebaihia M., et al. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38:779–786 [DOI] [PubMed] [Google Scholar]
- 40. Sell T., Schaberg D., Fekety F. 1983. Bacteriophage and bacteriocin typing scheme for Clostridium difficile. J. Clin. Microbiol. 17:1148–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stabler R., et al. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tan K., Wee B., Song K. 2001. Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J. Med. Microbiol. 50:613–619 [DOI] [PubMed] [Google Scholar]
- 43. Verdoorn B., et al. 2010. High prevalence of tcdC deletion-carrying Clostridium difficile and lack of association with disease severity. Diagn. Microbiol. Infect. Dis. 66:24–28 [DOI] [PubMed] [Google Scholar]
- 44. Voth D. E., Ballard J. D. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Warny M., et al. 2005. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366:1079–1084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.