

Abstract
The Bacillus subtilis fadR regulon involved in fatty acid degradation comprises five operons, lcfA-fadR-fadB-etfB-etfA, lcfB, fadN-fadA-fadE, fadH-fadG, and fadF-acdA-rpoE. Since the lcfA-fadRB-etfBA, lcfB, and fadNAE operons, whose gene products directly participate in the β-oxidation cycle, had been found to be probably catabolite repressed upon genome-wide transcript analysis, we performed Northern blotting, which indicated that they are clearly under CcpA-dependent catabolite repression. So, we searched for catabolite-responsive elements (cre's) to which the complex of CcpA and P-Ser-HPr binds to exert catabolite repression by means of a web-based cis-element search in the B. subtilis genome using known cre sequences, which revealed the respective candidate cre sequences in the lcfA, lcfB, and fadN genes. DNA footprinting indicated that the complex actually interacted with these cre's in vitro. Deletion analysis of each cre using the lacZ fusions with the respective promoter regions of the three operons with and without it, indicated that these cre's are involved in the CcpA-dependent catabolite repression of the operons in vivo.
INTRODUCTION
Fatty acids are essential components of membranes and are important sources of metabolic energy in all organisms. Thus, the fatty acid degradation and biosynthesis pathways must be switched on and off according to the availability of fatty acids to maintain membrane lipid homeostasis. A soil bacterium of low GC content, Bacillus subtilis, possesses two transcriptional global regulators, FadR (formerly YsiA) (14) and FapR (22), which are involved in fatty acid degradation and biosynthesis, respectively (5). FadR is antagonized by long-chain acyl-coenzyme A (CoA) (14), whereas FapR is antagonized by malonyl-CoA, a building block for fatty acid biosynthesis (22). The FadR regulon comprises five fad operons, lcfA-fadR (ysiA)-fadB (ysiB)-etfB-etfA, lcfB (yhfL), fadN (yusL)-fadA (yusK)-fadE (yusJ), fadH (ykuF)-fadG (ykuG), and fadF (ywjF)-acdA-rpoE, the expression of which FadR represses (14) (Fig. 1). The first three operons are considered to be directly involved in the fatty acid β-oxidation cycle. LcfA and LcfB most likely encode long-chain fatty acid-CoA ligases, FadE likely encodes acyl-CoA dehydrogenase, FadB and FadN likely encode enoyl-CoA hydratase/3-hydroxylacyl-CoA dehydrogenase complexes, FadA likely encodes 3-ketoacyl-CoA thiolase, and EtfA and EtfB likely encode the electron transfer flavoprotein dehydrogenase α- and β-subunits.
Fig. 1.

The B. subtilis FadR regulon and its catabolite repression. The FadR regulon comprises the five operons (lcfA-fadRB-etfBA, lcfB, fadNAE, fadHG, and fadF-acdA-rpoE) that are involved in fatty acid β-oxidation and closely related functions (14). The lcfA-fadRB-etfBA operon possesses two promoters (PlcfA-fadRB-etfBA and PfadRB-etfBA). The FadR boxes located in the promoter regions (PfadRB-etfBA, PlcfB, PfadNAE, PfadHG, and PfadF-acdA-rpoE) are indicated by gray boxes, to which FadR binds to repress transcription from them. FadR is antagonized by long-chain acyl-CoA. Three of these five operons (lcfA-fadRB-etfBA, lcfB, and fadNAE) were found in this work to be under CcpA-dependent catabolite repression. The cre's (cre-lcfA, cre-lcfB, and cre-fadN) responsible for catabolite repression of the respective operons were located in the lcfA, lcfB, and fadN genes, to which the complex of CcpA and P-Ser-HPr binds. The formation of P-Ser-HPr is triggered by an increase in the concentration of fructose-1,6-bisphosphate (FBP). The gene products of the lcfA-fadRB-etfBA, lcfB, and fadNAE operons, which were under CcpA-dependent catabolite repression, are directly involved in fatty acid β-oxidative degradation, and their functions in β-oxidation are indicated on the right.
Growing B. subtilis is unable to degrade straight-chain fatty acids added to the culture medium (11). Since B. subtilis apparently possesses the genes involved in the β-oxidation cycle, this cycle appears to have a dispensable function under certain physiological conditions. In fact, the lcfA-fadRB-etfBA, lcfB, and fadNAE operons have been reported to be induced upon glucose starvation (13). Furthermore, the fadNAE operon is induced at the onset of sporulation (9). This induction requires the YvbA protein involved in cannibalism by sporulating cells. These findings imply that the three operons might be under catabolite repression, as the β-oxidation genes of Escherichia coli are positively regulated by the cyclic AMP receptor protein/cyclic AMP system for their catabolite repression (8, 18).
In B. subtilis, catabolite regulation occurs on the binding of the complex of the catabolite control protein (CcpA) and the seryl-phosphorylated form of a histidine-containing protein or its homologue (P-Ser-HPr or -Crh) to catabolite-responsive elements (cre's) of target operons (16); P-Ser-HPr or P-Ser-Crh was formed upon activation of HPr kinase/phosphorylase (HprK), which is triggered by an increase in the fructose-1,6-bisphosphate concentration in the presence of carbohydrates transported by the phosphoenolpyruvate-dependent phosphotransferase system, such as Glc (3, 7, 8, 10). The complex of CcpA and P-Ser-HPr (or P-Ser-Crh) triggers the expression of several genes, including those encoding the enzymes necessary for the synthesis of major extracellular products such as acetoin (3) and acetate (19). On the other hand, this complex represses many catabolic genes and operons, mainly those involved in carbon, nitrogen, and phosphate metabolism.
We investigated in this work whether or not the respective operons constituting the catabolic regulon of FadR involved in fatty acid β-oxidation are under catabolite repression. We found that out of the five fad operons, three (lcfA-fadRB-etfBA, lcfB, and fadNAE) encoding the constituents of the β-oxidation pathway are catabolite repressed. Their catabolite repression was dependent on CcpA and mediated by the respective cre located in the lcfA, lcfB, or fadN gene.
MATERIALS AND METHODS
Bacterial strains.
The B. subtilis strains used are listed in Table 1. Strain FU788 (the ΔfadR::cat mutant) was transformed with plasmid pCm::Tc (23) to change its chloramphenicol resistance to tetracycline resistance (10 μg/ml), which yielded strain FU884 (the ΔfadR::tet mutant). Strain FU1084 (the ΔfadR::tet ΔccpA::neo mutant) was constructed by the transformation of strain FU884 with DNA of strain FU402 (the ΔccpA::neo mutant) to neomycin resistant (15 μg/ml).
Table 1.
B. subtilis strains used in this work
| Strain | Genotype | Reference or source |
|---|---|---|
| 168 | trpC2 | 1 |
| FU788 | ΔfadR::cat trpC2 | 14 |
| FU884 | ΔfadR::tettrpC2 | This work |
| FU402 | ΔccpA::neotrpC2 | 24 |
| FU1084 | ΔfadR::tet ΔccpA::neo trpC2 | This work |
| FU989 | amyE::[cat PlcfA(−434/+467)(+cre)-lacZ] trpC2 | This work |
| FU987 | amyE::[cat PlcfA(−434/+437)(−cre)-lacZ] trpC2 | This work |
| FU1071 | amyE::[cat PlcfA(−434/+467)(+cre)-lacZ] ΔccpA::neotrpC2 | This work |
| FU990 | amyE::[cat PlcfA(−434/+467)(+cre)-lacZ] ΔfadR::tettrpC2 | This work |
| FU988 | amyE::[cat PlcfA(−434/+437)(−cre)-lacZ] ΔfadR::tet trpC2 | This work |
| FU1048 | amyE::[cat PlcfA(−434/+467)(+cre)-lacZ] ΔfadR::tetΔccpA::neotrpC2 | This work |
| FU961 | amyE::[cat PlcfB(−120/+514) (+cre)-lacZ] trpC2 | This work |
| FU960 | amyE::[cat PlcfB(−120/+484) (−cre)-lacZ] trpC2 | This work |
| FU1074 | amyE::[cat PlcfB(−120/+514) (+cre)-lacZ] ΔccpA::neotrpC2 | This work |
| FU964 | amyE::[cat PlcfB(−120/+514) (+cre)-lacZ] ΔfadR::tettrpC2 | This work |
| FU962 | amyE::[cat PlcfB(−120/+484) (−cre)-lacZ] ΔfadR::tet trpC2 | This work |
| FU965 | amyE::[cat PlcfB(−120/+514) (+cre)-lacZ] ΔfadR::tetΔccpA::neotrpC2 | This work |
| FU993 | amyE::[cat PfadN(−286/+1130)(+cre)-lacZ] trpC2 | This work |
| FU991 | amyE::[cat PfadN(−286/+1100)(−cre)-lacZ] trpC2 | This work |
| FU1051 | amyE::[cat PfadN(−286/+1130)(+cre)-lacZ] ΔccpA::neotrpC2 | This work |
| FU994 | amyE::[cat PfadN(−286/+1130)(+cre)-lacZ] ΔfadR::tettrpC2 | This work |
| FU992 | amyE::[cat PfadN(−286/+1100)(−cre)-lacZ] ΔfadR::tettrpC2 | This work |
| FU1053 | amyE::[cat PfadN(−286/+1130)(+cre)-lacZ] ΔfadR::tetΔccpA::neo trpC2 | This work |
To construct transcriptional fusion strains of the lcfA-fadRB-etfBA, lcfB, and fadNAE promoters with lacZ, their promoter regions (PlcfA, PlcfB, and PfadN) comprising nucleotides −434 to +467 and +437 (with and without the cre in lcfA), −120 to +514 and +484 (with and without the cre in lcfB), and −286 to +1130 and +1100 (with and without the cre in fadN) (+1 is the transcription initiation nucleotide; that of lcfA-fadRB-etfBA was determined in this work [Fig. 2 ]), were amplified by PCR using primer pairs (plcfA-F/plcfA-Rcre and plcfA-F/plcfA-Rdcre, plcfB-F/plcfB-Rcre and lcfB-F/plcfB-Rdcre, and pfadN-F/pfadN-Rcre and pfadN-F/pfadN-Rdcre, respectively) (Table 2), with the DNA of strain 168 as a template. Each of the PCR products, trimmed with XbaI and BamHI (or BglII for fadN) digestion, was cloned into the pCRE-test2 vector (15), which had been treated with the same restriction enzymes. Correct construction was confirmed by DNA sequencing. The resultant plasmids were linearized by PstI digestion and then integrated into the amyE locus of strain 168 through double-crossover transformation to obtain chloramphenicol resistance, which resulted in strains FU989 [PlcfA(+cre)-lacZ] and FU987 [PlcfA(−cre)-lacZ], FU961 [PlcfB(+cre)-lacZ] and FU960 [PlcfB(−cre)-lacZ], and FU993 [PfadN(+cre)-lacZ] and FU991 [PfadN(−cre)-lacZ]. The resultant strains were then transformed with the DNA of strain FU884 (the ΔfadR::tet mutant) to obtain tetracycline resistance, which resulted in strains FU990 and FU988, FU964 and FU962, and FU994 and FU992, whereas strains FU989, FU961, and FU993 were also transformed with the DNA of strain FU402 (the ΔccpA::neo mutant) to obtain neomycin resistance, which resulted in strains FU1071, FU1074, and FU1051, respectively. Doubly disrupted strains with respect to fadR and ccpA (strains FU1048, FU965, and FU1053) were obtained by transformation of strains FU988, FU964, and FU994 with DNA of strain FU402 to obtain neomycin resistance.
Fig. 2.
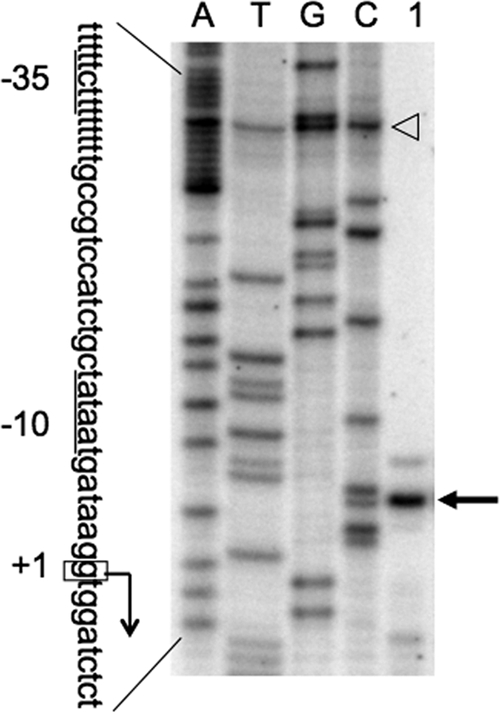
Determination of the transcription start site of the lcfA-fadRB-etfBA operon by primer extension analysis. Total RNA of strain FU988 [PlcfA(−cre)-lacZ; the ΔfadR mutant] (lane 1) was prepared and used for the reverse transcription reaction to generate runoff cDNA. Lanes A, T, G, and C contained the products of the dideoxy sequencing reactions obtained with the same primers as those used for reverse transcription; the open arrowhead indicates a false band observed in the A, T, G, and C lanes. The runoff cDNA is indicated by an arrow. The partial nucleotide sequence of the coding strand corresponding to the ladders is shown; the −35 and −10 sequences are underlined, and the transcription start site (+1) is enclosed in a box.
Table 2.
Oligonucleotide primers used in this work
| Oligonucleotide | Sequencea |
|---|---|
| ptsH-F | AGGAGAATGATACATATGGCACA |
| ptsH-R | TGGATCCTTGCATTACTCGCCGAGTCCTTC |
| hprK-F | ATCATATGGCAAAGGTTCGCACAAAAGACG |
| hprK-R | TAACGGATCCTCCTATTCTTCTTGTTCACC |
| lcfA-PE-F | GGCTTTCTTTTGTGGTGGCA |
| lcfA-PE-R | CGTTGGGATACTCGGCAAGC |
| lcfA-Nn-F | GTCGACCAGATTTTGATAACC |
| lcfA-Nn-R | CTGGATGGCTTCATGTTCATA |
| lcfB-Nn-F | GCGGCCGACATCTGCCGCTAAA |
| lcfB-Nn-R | GGCAAAATATCTCTCAGTGCCC |
| fadN-Nn-F | CCGTTTGCTGTGGAATATCAC |
| fadN-Nn-R | GTATCGTATAAAGATGCGGCC |
| fadH-Nn-F | GGCGGTAATTATTACCGGCGGG |
| fadH-Nn-R | CGGATAAGGATTCAGCCATTGCCC |
| fadF-Nn-F | CCGACGAAACGAAAGAAAGCTA |
| fadF-Nn-R | CGTCTCATGAAGCGGGTGCT |
| lcfA-FP-F | TTATGGCGTCTTGTTTGCCG |
| lcfA-FP-R | TTGTTTTTGCGTCAGCGGG |
| lcfB-FP-F | TGAGAGCATGCATGAATCGC |
| lcfB-FP-R | TTTTCCAGTCGTTCCTGACGT |
| fadN-FP-F | AAAAAGGGTGGATCGGAAGC |
| fadN-FP-R | TTCATGGCTTGGTCAATCGC |
| plcfA-F | TAGCTCTAGAAAAGGAAATCTTGCGGTGGC |
| plcfA-Rcre | GGAGGATCCGGTCGACTATTGATAACGTTTTC |
| plcfA-Rdcre | GGAGGATCCTTGCCTTCGGGAAAAGCAAATC |
| plcfB-F | TCTTCTAGAGGCCTTTGATCTGACAGAAG |
| plcfB-Rcre | GGAGGATCCGCAATATTTTTGCAAACGTTGTCAT |
| plcfB-Rdcre | GGAGGATCCTCCTGACCTCTGGGTCCGC |
| pfadN-F | TCGCTCTAGATGAAAAAGTGAAAGCAGCCG |
| pfadN-Rcre | AGAAGATCTCATCCGAATAAATAAGCGCTTTC |
| pfadN-Rdcre | AGAAGATCTCTTTTGTCCCTTTTGCCTGTTT |
Underlined sequences are XbaI and BamHI (or BglII) sites.
Preparation of the CcpA and P-Ser-HPr proteins.
The CcpA protein synthesized using the cells of E. coli strain JM109 bearing plasmid pCCPA19 (17) was purified to over 90% homogeneity by two steps of column chromatography (DEAE-Toyopearl 650 M [Tosoh Co., Tokyo, Japan] and heparin Sepharose [GE Healthcare]). The P-Ser-HPr protein was prepared as follows. The HPr protein was synthesized using cells of E. coli strain BL21(DE3) bearing plasmid pET-ptsH, which had been constructed by the cloning of the ptsH gene (PCR amplified with a primer pair [htsH-F/htsH-R] [Table 2]) into plasmid pET22b (+) (Novagen) using the NdeI and BamHI sites, and then purified to approximately 80% homogeneity by the column chromatography of DEAE-Toyopearl, 650 M. The His6-tagged HPr kinase was synthesized by the cells of E. coli strain BL21(DE3) bearing plasmid pET-hprK, which had been constructed by the cloning of the hprK gene (PCR amplified with a primer pair [hprK-F/hprK-R] [Table 2]) into plasmid pET16b using the NdeI and BamHI sites, and then purified to approximately 85% by column chromatography on Ni-nitrilotriacetic acid (NTA) agarose (Qiagen). Ser-46 of the HPr protein was phosphorylated in vitro to form P-Ser-HPr using the His6-tagged HPr kinase under the experimental conditions described (8). After the phosphorylation reaction, the kinase was removed from the reaction mixture by passage through an Ni-NTA agarose column.
Primer extension analysis.
B. subtilis strain FU988 cells were grown in S6 medium (4) containing 0.5% Casamino Acids (Difco) as a carbon source and 50 μg tryptophan per ml to an optimal density at 600 nm (OD600) of 1.0. Total RNA was extracted and purified from the cell pellet as described previously (26). To map the 5′ end of the lcfA-fadRB-etfBA transcript, total RNA (50 μg) was annealed to 1 pmol of primer lcfA-PE-R (Table 2), which had been 5′-end labeled with a Megalabel kit (Takara-Bio) and [γ-32P]ATP (MP Biomedicals), and then the primer extension reaction was conducted with ThermoScript reverse transcriptase (Invitrogen) as described previously (21). A template for the dideoxy sequencing reactions for ladder preparation was generated by PCR with the genomic DNA of strain 168 as a template and the primer pair of lcfA-PE-F/lcfA-PE-R (Table 2). An autoradiogram was obtained using a Typhoon 9400 variable image analyzer (GE Healthcare).
Northern analysis.
Total RNAs were extracted and purified as described above from cells of strains 168, FU884, FU402, and FU1084, which had been grown to an OD600 of 1 in S6 medium supplemented as described above, with and without 10 mM Glc, as described previously. RNA was electrophoresed in a glyoxal gel and then transferred to a Hybond-N membrane (GE Healthcare) (20). RNA transferred to the membrane was stained with 0.003% methylene blue to check its quality before hybridization. To prepare probes for the detection of the transcripts of lcfA-fadRB-etfBA, lcfB, fadNAE, fadHG, and fadF-acdA-rpoE, the respective lcfA, lcfB, fadN, fadH, and fadF fragments amplified by PCR using primer pairs (lcfA-Nn-F/lcfA-Nn-R, lcfB-Nn-F/lcfB-Nn-R, fadN-Nn-F/fadN-Nn-R, fadH-Nn-F/fadH-Nn-R, and fadF-Nn-F/fadF-Nn-R) (Table 2) and the chromosomal DNA of strain 168 as a template were labeled with a BcaBEST labeling kit (Takara-Bio, Kyoto, Japan) and [α-32P]-dCTP (MP Biomedicals). Hybridization and transcript detection were carried out as described previously (20).
DNase I footprinting.
DNase I footprinting experiments were performed essentially as described previously (6, 27). The lcfA, lcfB, and fadN probes for the footprinting of the CcpA and P-SerHPr proteins prepared as described above were synthesized by PCR with the genomic DNA of strain 168 as a template and primer pairs lcfA-FP-F/lcfA-FP-R, lcfB-FP-F/lcfB-FP-R, and fadN-FP-F/fadN-FP-R (Table 2), respectively. Prior to PCR amplification, the 5′ end of only one of each primer pair was labeled as mentioned above.
lacZ fusion analysis to monitor the promoters of lcfA-fadRB-etfBA, lcfB, and fadNAE.
B. subtilis cells were inoculated into 50 ml of S6 medium (4) supplemented as described above, with and without 10 mM Glc, to give an OD600 of 0.1. During cell growth at 37°C with shaking, 1-ml aliquots of the culture were withdrawn at 1-h intervals, and the β-galactosidase (β-Gal) activity was measured spectrophotometrically as described previously (25).
RESULTS AND DISCUSSION
Catabolite repression of the FadR regulon.
The B. subtilis fadR regulon involved in fatty acid degradation comprises five operons, lcfA-fadRB-etfBA, lcfB, fadNAE, fadHG, and fadF-acdA-rpoE (14) (Fig. 1). Although the transcription initiation nucleotides of lcfB, fadNAE, and fadF-acdA-rpoE were determined previously (14), that of lcfA-fadRB-etfBA was determined in this work to be a guanine at the 31st base upstream of the translation initiation base (Fig. 2). The presumed −10 sequence of the lcfA-fadRB-etfBA promoter (TATAAT) perfectly matched the consensus sequence of σA-recognized promoters, but the −35 sequence (TTCTTT) was very different from the consensus sequence.
Out of these operons, lcfA-fadRB-etfBA, lcfB, and fadNAE were found to be induced upon glucose starvation after glucose was exhausted from minimal medium by means of a DNA macroarray experiment (13), which was consistent with the DNA microarray results for glucose repression in the cells logarithmically grown in nutrient sporulation medium, except for in the case of lcfB, whose expression was not properly detected for an unknown reason (26) (http://www.genome.ad.jp/kegg/expression). In order to confirm these genome-wide profiling results, we performed Northern analysis of glucose repression of the above five operons belonging to the FadR regulon (Fig. 3). Total RNAs were prepared from cells of strains 168 (the wild type), FU884 (the ΔfadR mutant), FU402 (the ΔccpA mutant), and FU1084 (the ΔfadR ΔccpA mutant), which had been mid-logarithmically grown in the absence (−) and presence (+) of Glc. Figure 3A, B, C, D, and E show Northern blotting of the fad transcripts with the 32P-labeled probes of the PCR products of parts of the first genes of these operons (lcfA, lcfB, fadN, fadH, and fadF). The full-length 5.1-kb transcript of lcfA-fadRB-etfBA was too severely repressed by Glc to be visible in the wild-type fadR+ background (Fig. 3A). However, the 5.1-kb transcript was visible in the ΔccpA background even in the presence of Glc, so its synthesis was found to be repressed by Glc, with a repression ratio (band density for lane with Glc [+] divided by that without Glc [−]) of 0.29. This 5.1-kb transcript was truncated to 2.0 kb due to the presence of a transcription terminator in the 5′ region of the cat cassette that remained after the insertion of the tetracycline resistance gene (tet) into the cat gene through double-crossover transformation (23), which was observed in the ΔfadR background; the 2.0-kb transcript had also been observed with strain FU788 (the ΔfadR::cat mutant) previously (14). This 2.0-kb transcript was repressed by Glc with a repression ratio of 0.05 in the wild-type background. However, the 2.0-kb transcript was well derepressed, with a ratio of 0.28 in the ΔccpA background (this ratio is comparable to the 0.29 mentioned above for the 5.1-kb transcript in the ΔccpA background). This also indicated that the 2.0- and 5.1-kb transcripts were still significantly repressed by Glc even in the absence of CcpA. These results indicated that the transcription from the lcfA-fadRB-etfBA promoter is under CcpA-dependent catabolite repression as well as under significant but unknown CcpA-independent catabolite repression.
Fig. 3.

Northern analysis of catabolite repression of lcfA-fadRB-etfBA, lcfB, fadNAE, fadHG, and fadF-acdA-rpoE transcription. Total RNAs from strains 168 (wild type), FU884 (the ΔfadR mutant), FU402 (the ΔccpA mutant), and FU1084 (the ΔfadR ΔccpA mutant), which had been grown in the absence (−) and presence (+) of Glc, were subjected to Northern analysis using the lcfA, lcfB, fadN, fadH, and fadF probes (A, B, C, D, and E, respectively). The analysis was performed as described in the text. Each lane contained 10 μg of total RNA. The positions of size markers are indicated on the left. The open arrowheads indicate the positions of 23 and 16 rRNAs. Solid boxes beneath the genes (top) indicate the positions of the probes. Putative transcription terminators are given downstream of the last genes of the operons. The band densities were determined with ImageQuant TL (GE Healthcare). The repression ratios (band density determined with the cells grown with Glc divided by that without Glc) are given in parentheses.
The 2.0-transcript of lcfA was considered to be induced by FadR inactivation if the band densities of the 2.0- and 5.1-kb transcripts were compared (Fig. 3A), nevertheless there was found to be no FadR box regulating the lcfA-fadRB-etfBA promoter preceding lcfA (14). This might be consistent with the fact that the −35 sequence (TTCTTT) of this promoter is very different from the consensus sequence recognized by σA, implying the presence of an unknown transcriptional activator of this promoter that might be induced by FadR inactivation. This transcription activation system might possibly be under an unknown CcpA-independent catabolite repression.
The lcfB transcript of 1.7 kb was repressed by Glc in the wild-type and ΔfadR backgrounds (ratios of 0.20 and 0.19), respectively (Fig. 3B). These ratios were largely decreased in the ccpA background (ratios of 0.71 and 0.91), indicating that the lcfB expression is under CcpA-dependent catabolite repression. The full-length 5.5-kb transcript of the fadNAE operon was detected only in the ΔfadR background due to its strong repression by FadR (Fig. 3C). The 5.5-kb transcript was repressed by Glc (ratio = 0.20), whereas it was well derepressed in the ΔfadR ΔccpA background (ratio = 0.77). This finding indicates that the synthesis of the fadNAE transcript is under CcpA-dependent catabolite repression. The fadHG and fadF-acdA-rpoE operons were considered to be catabolite resistant, as judged from the above genome-wide mRNA profiling. Figure 3D shows that the fadHG operon is completely resistant to glucose repression not only in the wild type (ratio = 1.00) but also in the ΔfadR (ratio = 1.02) background. Figure 3E shows that the fadF-acdA-rpoE operon is slightly but significantly under catabolite repression in the ΔfadR background (ratio = 0.56). Unexpectedly, this operon was rather induced by Glc upon the introduction of ΔccpA (ratio = 2.60). This finding suggests that this operon might be very weakly subject to CcpA-dependent catabolite repression.
The above Northern blotting of catabolite repression of the FadR regulon clearly indicated in the ΔfadR genetic background that the synthesis of the transcripts of lcfA-fadRB-etfBA, lcfB, and fadNAE were repressed by Glc more than 5-fold, whereas that of fadHG and fadF-acdA-rpoE was hardly repressed (Fig. 3). The catabolite repression of lcfA-fadRB-etfBA, lcfB, and fadNAE was mediated by CcpA, whereas that of lcfA-fadRB-etfBA was only partially relieved by CcpA inactivation. As shown in Fig. 1, the gene products of the lcfA-fadRB-etfBA, lcfB, and fadNAE operons except for FadR (LcfA, LcfB, FadB, FadN, FadA, FadE, EtfA, and EtfB) are directly involved in fatty acid β-oxidation, the synthesis of which was clearly under CcpA-dependent catabolite repression. This implies that the production of acetyl-CoA as an energy supplier is not needed any more if Glc is present in the medium, even if the β-oxidation cycle functions only under certain specific physiological conditions, such as glucose starvation (13) and even if it does not degrade straight-chain fatty acids to support the growth of B. subtilis as a sole carbon source (11).
Analysis of the cre sequences involved in CcpA-dependent catabolite repression of the FadR regulon.
Since lcfA-fadRB-etfBA, lcfB, and fadNAE are under CcpA-dependent catabolite repression, we searched for cre sequences (3, 16) located in these operons, to which the complex of CcpA and P-Ser-HPr binds to exert catabolite repression (3, 7). A web-based cis-element search of the B. subtilis genome (http://dbtbs.hgc.jp/motiflocationsearch.html) using 50 known cre sequences (3) revealed the respective candidate cre sequences (ATGAAAACGTTATCA, ATGACAACGTTTGCA, and ATGAAAGCGCTTATT) in the lcfA, lcfB, and fadN genes (cre-lcfA, cre-lcfB, and cre-fadN), which comprise nucleotides +443 and +457, +490 and +504, and +1106 and +1020, respectively. We examined, by DNase I footprinting, whether or not the complex of CcpA and P-Ser-HPr is specifically bound. P-Ser-HPr was prepared by in vitro phosphorylation of HPr using His6-tagged HPr kinase as described above. A specific interaction of the complex of CcpA and P-Ser-HPr with each of the candidate lcfA, lcfB, and fadN cre's on the coding and noncoding strands was clearly observed, as shown in Fig. 4, in which the regions protected against DNase I are indicated by thick bars. As shown at the bottom of Fig. 4, each cre sequence is completely included in the respective protected regions of the coding and noncoding strands. Thus, the candidate cre's were highly likely involved in catabolite repression of the lcfA-fadRB-etfBA, lcfB, and fadNAE operons.
Fig. 4.

DNase I footprinting of the complex of CcpA and P-Ser-HPr with the cre-lcfA, cre-lcfB, and cre-fadN sequences. Analysis using the 238-bp lcfA, 211-bp lcfB, and 225-bp fadN probes was performed as described in the text. The left and right ladders are DNA footprints of the 5′-end-labeled coding and noncoding strands, respectively, of the lcfA, lcfB, and fadN probes. Lanes 1 to 4 contained 0.05 pmol of the 32P-labeled probe DNA in the reaction mixture (50 μl). Lanes 1 and 4 contained no protein. Lane 2 contained 6 μM CcpA and 8.6 μM P-Ser-HPr, and lane 3 contained 12 μM CcpA and 16.6 μM P-Ser-HPr. The assay mixture containing CcpA and P-Ser-HPr was first incubated for 10 min at 30°C before addition of the DNA probe. Lanes A, T, G, and C contained the products of the corresponding dideoxy sequencing reactions performed with the same primers as those used for probe preparation. The protected regions in ladders are indicated by black vertical bars. The nucleotide sequences of the protected regions of the coding and noncoding strands of each probe (thick horizontal lines) are shown beneath the footprints of the respective probes; the cre sequences are boxed.
There exists another promoter for the transcription of fadRB-etfBA (Fig. 1) (14). However, we found no candidate cre in the promoter and transcribed region of fadRB-etfBA by a web-based cis-element search of the B. subtilis genome mentioned above. Actually, the 2.0-kb transcript of lcfA was derepressed with a glucose repression ratio of 0.28 in the ΔccpA background, which is comparable to 0.29 for a 5.1-kb transcript in theΔccpA background (Fig. 3A). This coincides with the lcfA-fadRB-etfBA region containing only one cre in lcfA. Hence, it was considered that the transcription of fadBR-etfBA could not be under the CcpA-dependent catabolite repression.
Deletion analysis of each of the cre's in the lcfA, lcfB and fadN genes.
To examine whether or not cre-lcfA, cre-lcfB, and cre-fadN are each actually involved in catabolite repression in vivo, the promoter regions of lcfA-fadRB-etfBA, lcfB, and fadNAE with and without them [PlcfA(+cre) and PlcfA(−cre); PlcfB(+cre) and PlcfB(−cre); and PfadN(+cre) and PfadN(−cre)] were fused with lacZ, and the fusions were integrated into the amyE loci of the wild-type, ΔccpA, ΔfadR, and ΔfadR ΔccpA strains, respectively. First, cells of the PlcfA(+cre)-lacZ strains, i.e., FU989 (the wild type), FU1071 (the ΔccpA mutant), FU990 (the ΔfadR mutant), and FU1048 (the ΔccpA ΔfadR mutant) (Fig. 5 A, columns 1, 2, 5 to 8, 11, and 12, respectively), and PlcfA(−cre)-lacZ strains, FU987 (the wild type) and FU988 (the ΔfadR mutant) (Fig. 5A, columns 3, 4, 9, and 10), were grown in the absence (−) and presence (+) of Glc, and then the β-Gal activities in the cells were determined at an OD600 of 1. The activities in the wild-type and ΔfadR mutant cells grown without Glc (Fig. 5A, columns 1 and 7) are higher than those with Glc (Fig. 5A, columns 2 and 8). β-Gal activity ratios (+Glc/−Glc) of columns 2/1 and 8/7 in Fig. 5A are 0.40 and 0.40, respectively; the values (Fig. 5A, columns 1 and 2) are quite low due to repression by FadR, exhibiting relatively high standard deviations. However, the activities in the PlcfA(−cre)-lacZ and PlcfA(+cre)-lacZ ΔccpA mutant cells, which were grown with Glc, were very close to those without Glc (Fig. 5A, columns 4/3 and 10/9, 0.75 and 0.84; and columns 6/5 and 12/11, 0.57 and 0.85, respectively); the cre or CcpA dependency of the catabolite repression of PlcfA-fadRB-etfBA is clearly observed in its well-derepressed state of the ΔfadR mutant (Fig. 5A, columns 9 to 12). These results indicate clearly that cre-lcfA is actually involved in CcpA-dependent catabolite repression of lcfA-fadRB-etfBA in vivo. The CcpA-independent catabolite repression of PlcfA-fadRB-etfBA detected in Northern blotting (Fig. 3A) was observed somewhat in the lacZ fusion experiments (Fig. 5A).
Fig. 5.

Examination of lacZ expression under the control of each of the lcfA-fadRB-etfBA, lcfB, and fadNAE promoters (PlcfA, PlcfB, and PfadN) in the ΔfadR and (or) ΔccpA backgrounds, and deletion analysis of the cre-lcfA, cre-lcfB, and cre-fadN sequences. The lcfA, lcfB, and fadN promoter regions with and without the cre were placed upstream of lacZ, and each of the resultant lacZ fusions (PlcfA-lacZ, PlcfB-lacZ, and PfadN-lacZ with and without the cre) was integrated into the amyE loci of the wild-type, ΔfadR, ΔccpA, and ΔfadR ΔccpA strains. (A) lacZ expression under the control of each of the promoters was monitored during growth with and without Glc, as described in the text, using PlcfA-lacZ strains FU989 (wild type, +cre) (columns 1 and 2, −/+ Glc), FU987 (wild type, −cre) (3 and 4, −/+ Glc), FU1071 (the ΔccpA mutant, +cre) (5 and 6, −/+ Glc), FU990 (the ΔfadR mutant, +cre) (7 and 8, −/+ Glc), FU988 (the ΔfadR mutant, −cre) (9 and 10, −/+Glc), and FU1048 (the ΔfadR ΔccpA mutant) (11 and 12, −/+ Glc). PlcfB-lacZ strains (B), FU961 (wild type, +cre), FU960 (wild type, −cre), FU1074 (the ΔccpA mutant, +cre), FU964 (the ΔfadR mutant, +cre), FU962 (the ΔfadR mutant, −cre), and FU965 (the ΔfadR ΔccpA mutant), and PfadN-lacZ strains (C), FU993 (wild type, +cre), FU991 (wild type, −cre), FU1051 (the ΔccpA mutant, +cre), FU994 (the ΔfadR mutant, +cre), FU992 (the ΔfadR mutant, −cre), and FU1053 (the ΔfadR ΔccpA mutant), were also used; the column assignments for PlcfB-lacZ and PfadN-lacZ are the same as those for the PlcfA-lacZ strains. The average β-Gal activities at an OD600 of 1, with standard deviations, which were obtained in two independent lacZ monitoring experiments, are shown. In the case that the LacZ activity was less than 2 nmol/min per OD, the maximal activity in the y axis is also set to 2 nmol/min per OD. The glucose-repression ratios of each strain are shown on bold gray bars. Significant differences between the average β-Gal activities are shown by ** (P < 0.05) and * (0.05 < P < 0.15) (Student's t test).
Similarly, the β-Gal activities in the PlcfB(+cre)-lacZ and PlcfB(−cre)-lacZ cells were determined (Fig. 5B, columns 1 to 12). cre-lcfB in the wild-type and ΔfadR mutant cells caused catabolite repression of PlcfB (Fig. 5B, columns 2/1 and 8/7, 0.31 and 0.29), respectively; this repression was not observed in the PlcfB(−cre)-lacZ strains (Fig. 5B, columns 4/3 and 10/9, 0.69 and 0.97, respectively) or in the PlcfB(+cre)-lacZ ΔccpA strains (Fig. 5B, columns 6/5 and 12/11, 0.86 and 0.93, respectively). The results clearly indicate that the cre-lcfB is actually involved in CcpA-dependent catabolite repression of lcfB in vivo.
The β-Gal activities in the wild-type and ΔccpA, ΔfadR, and ΔccpA ΔfadR mutant cells carrying PfadN(+cre)- and PfadN(−cre)-lacZ, which were grown in the absence and presence of Glc, were also determined (Fig. 5C, columns 1 to 12). The cre-fadN in the wild-type and ΔfadR mutant cells caused catabolite repression of PfadN (Fig. 5C, columns 2/1 and 8/7, 0.36 and 0.19, respectively). The activities in the PfadN(−cre)-lacZ and PfadN(+cre)-lacZ ΔccpA mutant cells were less repressed by Glc (Fig. 5C, columns 4/3 and 10/9, and 6/5 and 12/11, 0.44 and 0.59, and 0.75 and 0.55, respectively), but the ratios were significantly lower than the corresponding ratios for the PlcfA- and PlcfB-lacZ strains described above. These results suggest that CcpA-dependent catabolite repression involving the cre-fadN is not so strict, with the result that residual catabolite repression was observed even in the ccpA background. This might be possibly due to the relatively low affinity of the cre-fadN toward the complex of CcpA and P-Ser-HPr in vivo and (or) the presence of another cre upstream of it for more strict catabolite repression, as in the case of the iol operon (15). Anyway, it was obvious that the cre-fadN also functions in the catabolite repression of the fadNAE operon in vivo.
As reviewed previously (3), the complex of CcpA and P-Ser-HPr triggers the expression of several genes involved in the formation of acetate and acetoin, major extracellular products of B. subtilis grown on Glc. It also triggers the expression of an anabolic operon (ilv-leu) involved in the biosynthesis of branched-chain amino acids (24). On the other hand, this complex represses many genes and operons, including an entrance gene for the tricarboxylic acid (TCA) cycle (citZ) (12), several transporter genes for TCA cycle intermediates, some respiration genes, and many catabolic and anabolic genes involved in carbon, nitrogen, and phosphate metabolism, as well as ones for certain extracellular enzymes and secondary metabolites. It was demonstrated in this work that the complex also repressed the catabolic operons involved in the fatty acid β-oxidation cycle (lcfA-fadRB-etfBA, lcfB, and fadNAE).
ACKNOWLEDGMENTS
We are grateful to M. Kashimura and N. Tsuchiya for their technical help.
This work was supported by the Strategic Support Project of Research Infrastructure Formation for Private Universities, the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Y. Fujita).
Footnotes
Published ahead of print on 11 March 2011.
REFERENCES
- 1. Anagnostopoulos C., Spizizen J. 1961. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81:741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clark D. P., Cronan J. E., Jr 1996. Two-carbon compounds and fatty acids as carbon sources, p. 343–357 In Neidhardt F. C., et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol. 1. ASM Press, Washington, DC [Google Scholar]
- 3. Fujita Y. 2009. Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci. Biotechnol. Biochem. 73:245–259 [DOI] [PubMed] [Google Scholar]
- 4. Fujita Y., Freese E. 1981. Isolation and properties of a Bacillus subtilis mutant unable to produce fructose-bisphosphatase. J. Bacteriol. 145:760–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fujita Y., Matsuoka H., Hirooka K. 2007. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 66:829–839 [DOI] [PubMed] [Google Scholar]
- 6. Fujita Y., Miwa Y. 1989. Identification of an operator sequence for the Bacillus subtilis gnt operon. J. Biol. Chem. 264:4201–4206 [PubMed] [Google Scholar]
- 7. Fujita Y., Miwa Y., Galinier A., Deutscher J. 1995. Specific recognition of the Bacillus subtilis gnt cis-acting catabolite-responsive element by a protein complex formed between CcpA and seryl-phosphorylated HPr. Mol. Microbiol. 17:953–960 [DOI] [PubMed] [Google Scholar]
- 8. Galinier A., et al. 1998. New protein kinase and protein phosphatase families mediate signal transduction in bacterial catabolite repression. Proc. Natl. Acad. Sci. U. S. A. 95:1823–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. González-Pastor J. E., Hobbs E. C., Losick R. 2003. Cannibalism by sporulating bacteria. Science 301:510–513 [DOI] [PubMed] [Google Scholar]
- 10. Henkin T. M., Grundy F. J., Nicholson W. L., Chambliss G. H. 1991. Catabolite repression of α-amylase gene expression in Bacillus subtilis involves a trans-acting gene product homologous to the Escherichia coli lacI and galR repressors. Mol. Microbiol. 5:575–584 [DOI] [PubMed] [Google Scholar]
- 11. Kaneda T. 1971. Incorporation of branched chain C6-fatty acid isomers into the related long-chain fatty acids by growing cells of Bacillus subtilis. Biochemistry 10:340–347 [DOI] [PubMed] [Google Scholar]
- 12. Kim H. J., Roux A., Sonenshein A. L. 2002. Direct and indirect roles of CcpA in regulation of Bacillus subtilis Krebs cycle genes. Mol. Microbiol. 45:179–190 [DOI] [PubMed] [Google Scholar]
- 13. Koburger T., Weibezahn J., Bernhardt J., Homuth G., Hecker M. 2005. Genome-wide mRNA profiling in glucose starved Bacillus subtilis cells. Mol. Genet. Genomics 274:1–12 [DOI] [PubMed] [Google Scholar]
- 14. Matsuoka H., Hirooka K., Fujita Y. 2007. Organization and function of the YsiA regulon of Bacillus subtilis involved in fatty acid degradation. J. Biol. Chem. 282:5180–5194 [DOI] [PubMed] [Google Scholar]
- 15. Miwa Y., Fujita Y. 2001. Involvement of two distinct catabolite-responsive elements in catabolite repression of the Bacillus subtilis myo-inositol (iol) operon. J. Bacteriol. 183:5877–5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miwa Y., Nakata A., Ogiwara A., Yamamoto M., Fujita Y. 2000. Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res. 28:1206–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miwa Y., Saikawa M., Fujita Y. 1994. Possible function and some properties of the CcpA protein of Bacillus subtilis. Microbiology 140:2567–2575 [DOI] [PubMed] [Google Scholar]
- 18. Pauli G., Ehring R., Overath P. 1974. Fatty acid degradation in Escherichia coli: requirement of cyclic adenosine monophosphate and cyclic adenosine monophosphate receptor protein for enzyme synthesis. J. Bacteriol. 117:1178–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Presecan-Siedel E., et al. 1999. Catabolite regulation of the pta gene as part of carbon flow pathways in Bacillus subtilis. J. Bacteriol. 181:6889–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sambrook J., Russell D. W. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21. Satomura T., et al. 2005. Enhancement of glutamine utilization in Bacillus subtilis through the GlnK-GlnL two-component regulatory system. J. Bacteriol. 187:4813–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schujman G. E., Paoletti L., Grossman A. D., de Mendoza D. 2003. FapR, a bacterial transcription factor involved in global regulation of membrane lipid biosynthesis. Dev. Cell 4:663–672 [DOI] [PubMed] [Google Scholar]
- 23. Steinmetz M., Richter R. 1994. Plasmids designed to alter the antibiotic resistance expressed by insertion mutations in Bacillus subtilis, through in vivo recombination. Gene 142:79–83 [DOI] [PubMed] [Google Scholar]
- 24. Tojo S., et al. 2005. Elaborate transcription regulation of the Bacillus subtilis ilv-leu operon involved in the biosynthesis of branched-chain amino acids through global regulators of CcpA, CodY and TnrA. Mol. Microbiol. 56:1560–1573 [DOI] [PubMed] [Google Scholar]
- 25. Yoshida K., et al. 2000. Systematic study of gene expression and transcription organization in the gntZ-ywaA region of the Bacillus subtilis genome. Microbiology 146:573–579 [DOI] [PubMed] [Google Scholar]
- 26. Yoshida K., et al. 2001. Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Res. 29:683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida K., et al. 2004. Bacillus subtilis LmrA is a repressor of the lmrAB and yxaGH operons: identification of its binding site and functional analysis of lmrB and yxaGH. J. Bacteriol. 186:5640–5648 [DOI] [PMC free article] [PubMed] [Google Scholar]