

Abstract
Agrobacterium VirB7, VirB9, and VirB10 form a “core complex” during biogenesis of the VirB/VirD4 type IV secretion system (T4SS). VirB10 spans the cell envelope and, in response to sensing of ATP energy consumption by the VirB/D4 ATPases, undergoes a conformational change required for DNA transfer across the outer membrane (OM). Here, we tested a model in which VirB10 regulates substrate passage by screening for mutations that allow for unregulated release of the VirE2 secretion substrate to the cell surface independently of target cell contact. One mutation, G272R, conferred VirE2 release and also rendered VirB10 conformationally insensitive to cellular ATP depletion. Strikingly, G272R did not affect substrate transfer to target cells (Tra+) but did block pilus production (Pil−). The G272R mutant strain displayed enhanced sensitivity to vancomycin and SDS but did not nonspecifically release periplasmic proteins or VirE2 truncated of its secretion signal. G272 is highly conserved among VirB10 homologs, including pKM101 TraF, and in the TraF X-ray structure the corresponding Gly residue is positioned near an α-helical domain termed the antenna projection (AP), which is implicated in formation of the OM pore. A partial AP deletion mutation (ΔAP) also confers a Tra+ Pil− phenotype; however, this mutation did not allow VirE2 surface exposure but instead allowed the release of pilin monomers or short oligomers to the milieu. We propose that (i) G272R disrupts a gating mechanism in the core chamber that regulates substrate passage across the OM and (ii) the G272R and ΔAP mutations block pilus production at distinct steps of the pilus biogenesis pathway.
INTRODUCTION
The Agrobacterium VirB/VirD4 type IV secretion system (T4SS) mediates the transfer of DNA and protein substrates to plant and other cells by a cell contact-dependent mechanism (1). This T4SS, assembled from the 11 VirB proteins and the VirD4 substrate receptor, is composed of two terminal structures, a cell envelope-spanning translocation channel and an extracellular fiber termed the T pilus (16). Early biochemical studies showed that the lipoprotein VirB7, outer membrane (OM)-associated VirB9, and bitopic VirB10 form a machine subassembly that is both intrinsically stable and stabilizing for other VirB subunits (15, 21). This subassembly, termed the core complex, is phylogenetically and functionally conserved among T4SSs carried by Gram-negative bacteria (22).
A core complex from the Escherichia coli pKM101 conjugation system was purified and structurally resolved by cryo-electron microscopy (14). The complex, composed of 14 copies each of the VirB7, VirB9, and VirB10 homologs, is a large ring-shaped structure of 185 Å in height and width, composed of two layers surrounding a central chamber of ∼80 Å at its widest point. More recently, a crystal structure of the OM-spanning part of this complex was solved (23), identifying an α-helical structure in the VirB10-like TraF protein (termed the antenna projection [AP]) that forms the OM pore. While in the cryo-electron microscopy structure, this region is constricted to an inner diameter of about 10 Å, in the crystal structure, it forms a pore of ∼30 Å in diameter. These structures revealed that VirB10-like TraF spans the entire cell envelope to form channels in both the inner membrane (IM) and the OM (14, 23). There are many examples of bitopic proteins that function in a complex with other IM or OM subunits to regulate OM translocation or integrity, e.g., E. coli TonB (34), TolA (10), bacteriophage spanins (8, 45), and OM bitopic subunits of dedicated secretion systems (29). However, VirB10-like TraF is unique among bacterial proteins in possessing both IM- and OM-spanning, pore-forming regions (14, 23, 27). Such a transenvelope topology is ideally suited for VirB10-like proteins to serve as structural scaffolds for the assembly of multicomponent T4SSs (16, 22, 27).
Besides its role in machine morphogenesis, VirB10 undergoes a structural transition upon sensing of ATP binding or hydrolysis activities by the IM VirD4 and VirB11 ATPases (11). This conformational switch was observed as a difference in protease susceptibility of VirB10 in wild-type (WT) cells versus cells lacking one or both of these ATPases, cells producing Walker A mutant forms of either ATPase, or cells depleted of ATP energy (11). Previously, it was also shown with a formaldehyde (FA) cross-linking assay that translocating DNA substrates form close contacts with VirD4, VirB11, VirB6, and VirB8, which are likely important for substrate docking and transfer across the IM, and with VirB2 pilin and the OM-associated VirB9, which are postulated to form the portion of the channel extending through the periplasm and OM (12). VirB10 does not form an FA-cross-linkable contact with the translocating DNA substrate; however, a virB10 null mutation blocks FA cross-linking of the DNA with VirB2 and VirB9. This suggests that ATP energy at the IM might activate a structural transition in VirB10 required for OM channel formation or gating (12).
The biochemical findings, together with the recently determined pKM101 core structure, thus support a role for the C-terminal region of VirB10 in regulating passage of secretion substrates across the OM. To test this model, here we screened for VirB10 mutations that confer a gating defect in the T4SS channel. We report the isolation of a mutation that confers unregulated release of the VirE2 secretion substrate to the cell surface, as well as enhanced uptake of detergent and a large antibiotic. This mutation is located near the AP pore and renders VirB10 conformationally insensitive to cellular ATP levels. Our findings support a proposal that G272R mediates substrate release by “locking” VirB10 in an energy-activated, open conformation.
MATERIALS AND METHODS
Bacterial strains and induction conditions.
Agrobacterium tumefaciens and Escherichia coli strains and plasmids used in this study are listed in Table 1. A Spcr derivative of strain PC1010 was obtained by serial passage on increasing concentrations of medium containing spectinomycin to 500 μg/ml. Conditions for growth of A. tumefaciens and E. coli and for vir gene induction with 100 μM acetosyringone (AS) in induction medium (ABIM) have been described previously (7). Plasmids were maintained in E. coli and A. tumefaciens by addition of carbenicillin (100 μg/ml), kanamycin (100 μg/ml), tetracycline (5 μg/ml), and gentamicin sulfate (20 μg/ml for E. coli and 100 μg/ml for A. tumefaciens).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| A. tumefaciens strains | ||
| A348 | C58 chromosome, pTiA6NC (octopine-type) Ti plasmid | 42 |
| A348Spcr | Spcr, A348 derivative | 38 |
| Ax56 | A348 virB10::Tn5virB; nonpolar on virB11 | 19 |
| PC1010 | A348 virB10 deletion, nonpolar on virB11 | 7 |
| PC1010Spcr | Spcr, PC1010 derivative | This study |
| E. coli strains | ||
| DH5α | F− φ80dlacZΔM15 endA1 recA1hsdR17(rK− mK−) supE44 thi-1 gyrA96 Δ(lacZYA-argF)U169 | 39 |
| XL1-Red | endA1gyrA96thi-1hsdR17supE44relA1lacmutD5mutSmutT Tn10 Tetr | Stratagene |
| Plasmids | ||
| pBSIISK+ | Ampr, ColE1 cloning vector | Stratagene |
| pTJS140 | Carr, IncP broad-host-range vector with RK2 and ColE1 replicons | 40 |
| pUCD2 | Tetr Spcr Kanr Crbr; IncW broad-host-range vector | 18 |
| pED1 | Ampr, pUC119 with PvirB | 47 |
| pED10 | Tetr, pTJS75-Tet with PvirB-virB10,virB11 | 47 |
| pKT230 | Kanr, plasmid RSF1010 (IncQ) derivative | 5 |
| pJB31 | Spcr, pKT230 with Spcr and deleted of Kanr | 6 |
| pML122Genr | Genr, plasmid RSF1010 (IncQ) derivative | 23 |
| pLB1300 | Carr, pTJS140 (IncP) with PvirB at BamHI site | This study |
| pLB1310 | Carr, pLB1300 (IncP) with PvirB-virB10 | This study |
| pLB1311 | Carr, pLB1310 (IncP) with PvirB-virB10.G272R | This study |
| pLS23 | Spcr, PvirE-virE1,FL-virE2 in an IncP vector | 43 |
| pLS27 | Tetr Spcr Kanr, pUCD2 with PvirE-virE1,FL-virE2 from pLS23 | L. Stahl and A. Binns |
| pPC203 | Tetr Spcr Kanr, pUCD2 with PvirE-virE1,FL-virE2Δ525-533 | This study |
| pKVDB129 | Carr, Genr pBBR1MCSGen producing VirB10ΔAP | 26 |
Plasmid constructions.
Plasmid pLB1300 was constructed by cloning a 530-bp fragment containing the virB promoter from pED1 (47), which was modified to include BamHI ends, into the unique BamHI site of the broad-host-range vector pTJS140 (40). The 1,240-bp EcoRI fragment from pED10 (47), containing the entire virB10 coding sequence, was cloned into the adjacent unique EcoRI site in pLB1300 to yield pLB1310. Plasmid pLS23 carries a 3.0-kb BamHI fragment comprising the virE promoter, virE1, and virE2 with a FLAG epitope tag inserted after the start codon of virE2 (43). This BamHI fragment was inserted into the BamHI site of pBSIISK+ such that it was flanked by XbaI and PstI restriction endonuclease recognition sites. The XbaI site was made blunt ended, and the blunt-ended XbaI-PstI fragment was introduced into EcoRV/PstI-digested vector pUCD2 (18). For construction of FL-VirE2Δ525-533, pBSIISK+ carrying the BamHI fragment was digested with XbaI, made blunt ended, and then digested with SalI (a unique SalI site is located at codon 525 of virE2), and the resulting blunt-ended XbaI-SalI fragment was introduced into EcoRV/SalI-digested pUCD2. The resulting construct encodes VirE2 missing a portion of it C-terminal secretion signal (48, 49). Insertions of the Xba-SalI fragments carrying full-length FL-virE2 and FL-virE2Δ525-533 into pUCD2 resulted in plasmids pLS27 and pPC203, respectively.
Mutagenesis and FLAG-VirE2 release screen.
Plasmid pLB1310 was subjected to random mutagenesis in vivo using the XL1-Red E. coli mutator strain (Stratagene, La Jolla, CA). Mutated plasmids isolated from pools of XL1-Red E. coli were introduced into Ax56(pLS27) (19). Colonies were scraped off the plate, resuspended in ABIM, and diluted so that they could be plated at ∼100 CFU per plate on ABIM agar. The resulting colonies were screened for FL-VirE2 release by colony immunoblotting with M2 anti-FLAG monoclonal antibody (Sigma) (26). Four primers (5′AAGCGCGGACGAACTC3′, 5′ACTCAAGAGCATGGCT3′, 5′GGTGAATGAACAACGCA3′, and 5′ACCTCAATCGGAGCTG3′) were used to sequence both strands of the virB10 genes of mutated pLB1310 derivatives.
Virulence assays.
A. tumefaciens strains were tested for virulence by inoculating wound sites of Kalanchoe daigremontiana leaves. As controls, all leaves were coinoculated with WT A348 and isogenic null mutants as positive and negative controls (7). Relative virulence was assessed by inoculation of serially diluted cultures adjusted to the same optical density at 600 nm (OD600) on plant wound sites. Tumor formation was monitored over a period of 6 weeks and scored on a scale of − (avirulent) to +++ (WT virulence). Tumorigenesis was further assessed by inoculation on Nicotiana tabacum cv. Havana 425 leaf explants (43). The number of tumors per leaf explant was determined after 14 to 21 days.
Conjugation assays.
Conjugative transfer of mobilizable RSF1010 (IncQ) plasmids was carried out at 18°C as described previously (25) (see Table 2 for results of mating assays monitoring transfer of pML122Genr [23]; similar results were obtained in replicates of these assays using two other RSF1010 derivatives, pKT230 [4] and pJB31 [5]). Strain A348Spcr or PC1010Spcr served as the recipient for tests of pML122Genr or pKT230 transfer; strain A348 or PC1010 carrying pLB1310 or pLB1311 (Crbr) served as the recipient for tests of pJB31 transfer. Assays were performed in triplicate at least three times, and transfer frequencies are presented as the number of transconjugants per donor for a representative experiment.
Table 2.
Effects of G272R mutation on DNA transfer
| Virulence on Kalanchoea | Donor strainb (+ pML122Genr) | Recipient strain | Transfer frequency (no. of transconjugants/donor)c |
|---|---|---|---|
| +++ | A348 | A348Spcr | 2.2 (±0.6) × 10−5 |
| − | PC1010 | A348Spcr | <10−8 |
| ++ | PC1010(pLB1310) | A348Spcr | 9.3 (±0.8) × 10−6 |
| ++ | PC1010(pLB1311) | A348Spcr | 9.7 (±0.4) × 10−6 |
| A348 | PC1010Spcr(pLB1311) | 2.3 (±1.3) × 10−5 | |
| PC1010 | PC1010Spcr(pLB1311) | <10−8 | |
| PC1010(pLB1310) | PC1010Spcr(pLB1311) | 1.1 (±1.5) × 10−5 | |
| A348 | PC1010Spcr | 4.8 (±0.6) × 10−8 | |
| A348 | PC1010Spcr(pLB1310) | 3.5 (±0.4) × 10−5 |
Inoculated K. daigremontiana leaves were scored for tumor formation on a scale of no tumors (−) to tumors incited by WT A348 (+++). Results are for strains without the IncQ plasmid.
A348, WT; PC1010, ΔB10. pLB1310 produces native VirB10, and pLB1311 produces VirB10.G272R.
Data are the means of triplicate determinations (± standard deviation) from a single experiment and are representative of three or more independent assays.
Protein analysis and immunoblotting.
Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) or with a Tricine-SDS-PAGE system as previously described (35). Proteins were transferred to nitrocellulose membranes, and immunoblots were developed with goat anti-rabbit antibodies conjugated to alkaline phosphatase and histochemical substrates. Alternatively, blots were developed with anti-rabbit antibodies conjugated to horseradish peroxidase, and antibody-antigen interactions were visualized by chemiluminescence (Pierce, Thermo Scientific). Molecular size markers were prestained SDS-PAGE broad-range markers from Bio-Rad.
T pilus isolation and extracellular blot assay.
A. tumefaciens strains were grown to an OD600 of 0.5 in MG/L medium at 28°C and then induced with acetosyringone (AS) for expression of the vir genes for 6 h at 22°C, as described previously (20). The induced culture (500 μl) was spread on ABIM agar plates, and the plates were incubated for 4 days at 18°C. Cells were then gently scraped off the plates in 1 ml 50 mM KPO4 buffer (pH 5.5) or buffer A (100 mM HEPES [pH 7.5], 250 mM sucrose, 25 mM MgCl2, 0.1 mM KCl) and passed through a 25-gauge needle to collect flagella, pili, and surface proteins. The sheared material was centrifuged at 14,000 × g for 30 min at 4°C, and the supernatant was filtered through a 0.22-μm-pore-size cellulose acetate membrane to remove intact cells and cell debris. The filtered fraction was centrifuged at 100,000 × g for 1 h at 4°C to recover T pili. T pili were suspended in 50 μl of Laemmli's buffer, boiled, and subjected to Tricine-SDS-PAGE as described above. Surface-exposed VirB2 and VirB5 were detected by colony immunoblotting as described previously (26).
Protease accessibility assay.
The protease susceptibility of VirB10 in energized cells and cells depleted of ATP energy by treatment with arsenate was determined as described previously (11).
Growth curves.
A. tumefaciens strains were grown to an OD600 of 0.5 in MG/L medium at 28°C and then induced in ABIM for 4 h at room temperature (RT). Cells were normalized and diluted in ABIM (OD600 = 0.1). Greinier Bio-One flat-bottom, 24-well PS microplates (Sigma-Aldrich, Prague, Czech Republic) were filled with the cell suspension (1 ml in each well). The plates were covered with a sealing membrane (Breathe-Easy; Sigma-Aldrich). Growth over time (18 h) was monitored using a Synergy Mx Multi-Mode microplate reader (BioTek Instruments). The absorbance in each well was measured at 600 nm at given intervals (30 min) with continuous shaking of the microplate. The absorbance data, with background absorbance subtracted, were exported to MS Excel for further processing. Experiments were repeated three times, and data for a representative experiment are shown.
Assays for vancomycin and SDS sensitivity.
The growth of each strain in ABIM supplemented with 0.5% SDS or vancomycin (50 μg/ml and 100 μg/ml) was monitored over 18 h using a Synergy Mx Multi-Mode microplate reader (BioTek Instruments) as described above.
Assays for outer membrane integrity.
RNase release from intact cells was assayed as described by Lazzaroni and Portalier (28). Briefly, ABIM-induced cells were normalized (OD 600 = 0.6), and 20 μl was spotted on ABIM RNase agar (standard ABIM agar supplemented with 0.1% yeast extract and 1% RNA type VI from Torula yeast [Sigma]). After overnight growth at 28°C, RNA was precipitated by flooding the plate with cold 10% trichloroacetic acid (TCA), and RNase leakage was detected by identification of clear zones surrounding colonies. For detection of periplasmic RNase activity, plate-induced cultures were scraped from the plates, resuspended in 1 ml ABIM, and subjected to osmotic shock. The shockate fractions were spotted on fresh ABIM RNase agar for 1 h prior to TCA precipitation and monitored for clearing. ChvE release was monitored by colony immunoblotting with anti-ChvE antibody (26). For detection of total periplasmic ChvE, induced cultures were treated as described above, and the osmotic shockates were spotted on a 0.45-mm nitrocellulose membrane, allowed to sit for 5 min, and developed by immunostaining as described above (26). Digital images were obtained using a Canon Scan LiDE 25 scanner.
RESULTS
Isolation of a virB10 mutant conferring surface exposure of VirE2.
A. tumefaciens translocates substrates through the VirB/VirD4 T4SS to target cells without releasing detectable amounts to the milieu. To identify mutations conferring a T4SS gating defect, we used a nondenaturing colony dot blot assay to screen strains expressing virB10 mutant alleles for host cell contact-independent surface exposure of functional, FLAG-tagged VirE2 (FL-VirE2). VirE2 is a single-stranded DNA-binding protein that is translocated to plant cells independently of the T-DNA but upon translocation binds the T-DNA to protect it from nucleases and promote further delivery to the nucleus (13, 48, 49). ΔvirB10 mutant strains lacking or trans-expressing wild-type virB10 do not release detectable amounts of secretion substrates to the cell surface (Fig. 1A). Among the ∼12,000 strains screened, we identified one mutant that accumulated appreciable amounts of surface-exposed FL-VirE2 (Fig. 1A). The virB10 allele carried a single mutation that resulted in substitution of Arg for Gly272. We reintroduced the G272R codon substitution into wild-type virB10 by site-directed mutagenesis and determined that the corresponding mutant strain phenocopied the original mutation, confirming that the G272R mutation was responsible for surface exposure of FL-VirE2. The G272R mutant protein accumulated at abundant levels and assembled into cross-linkable complexes indistinguishable from those in wild-type cells (Fig. 1A and data not shown). Furthermore, its production did not affect steady-state accumulation of FL-VirE2 or other core components (VirB7 or VirB9) (Fig. 1A). The G272R mutant strain also appeared identical to the isogenic VirB10-producing strain in broth culture and growth as colonies on plates, and it displayed a WT growth rate in ABIM inducing medium (Fig. 1B and data not shown).
Fig. 1.

Effects of the G272R mutation on VirE2 release, VirB protein accumulation, and cell growth. (A) Surface exposure of FLAG-tagged VirE2 (FL-VirE2). Upper panel, FL-VirE2 surface display as monitored by immunostaining of colony blots with anti-FLAG antibodies. Lower panels, total cellular levels of FL-VirE2, VirB10, VirB9, and VirB7 as monitored by immunostaining of Western blots with anti-FLAG and anti-VirB antibodies. Strains: nonpolar ΔvirB10 mutant (ΔB10; PC1010) without (−) or with native VirB10 (B10) produced from pLB1310 or the G272R mutant (G272R) from pLB1311; PC1010 also produced FL-VirE2 from pLS27 or FL-VirE2Δ525-533 (FL-E2ΔC9) from pPC203. (B) Growth of PC1010 producing native VirB10 or the G272R mutant in vir induction medium.
Despite the absence of a detectable effect on cell growth, the G272R mutation could nonspecifically disrupt OM integrity. We assayed strains producing native VirB10 or the G272R mutant protein for growth in the presence of detergents or antibiotics that ordinarily do not cross the OM and for release of periplasmic proteins. The G272R mutant strain did display enhanced sensitivity to both vancomycin and SDS compared to the VirB10-producing strain (Fig. 2A and B). However, the mutant strain did not release detectable amounts of periplasmic proteins, including RNase or the sugar-binding protein ChvE (Fig. 2C). These findings suggest that the G272R mutation confers leakiness to the T4SS channel without disrupting the overall integrity of the OM barrier. The leaky channel allows for substrate release to the cell surface and uptake of detergent and vancomycin across the OM, presumably into the periplasm.
Fig. 2.

Effects of G272R on membrane integrity, as monitored by sensitivity to vancomycin and SDS and release of periplasmic proteins. (A and B) Strain PC1010 (ΔB10) producing native VirB10 (B10) or the G272R mutant was suspended in vir induction medium containing vancomycin (50 or 100 μg/ml) or SDS (0.5%), and growth or cell lysis, respectively, was monitored for 18 h. (C) Strain PC1010 (ΔB10) cells lacking or producing native VirB10 or the G272R mutant were assayed for RNase release from intact cells on RNA-containing plates. The presence of cellular RNase was confirmed by addition of osmotic shockates to RNA-containing plates. Strains were assayed for release of periplasmic ChvE as monitored by development of colony blots with anti-ChvE antibodies. Production of cellular ChvE was assessed by spotting osmotic shockates on nitrocellulose and immunostaining with anti-ChvE antibodies.
To confirm that the mutation mediated release only of a secretion-competent substrate, we also assayed for leakage of VirE2Δ525-533, a mutant lacking a portion of the C-terminal signal sequence; this truncated protein is defective in binding the VirD4 substrate receptor and translocation to target cells (43, 48, 49). The G272R-producing strain did not accumulate detectable levels of VirE2Δ525-533 on the cell surface, even though the truncated protein was abundantly produced (Fig. 1A). This indicates that the G272R mutation confers translocation only of bone fide secretion substrates and also acts distal in the translocation pathway to VirD4 and other possible T4SS substrate specificity filters.
G272R is locked in an energy-activated conformation.
In response to sensing of ATP binding or hydrolysis by the VirD4 and VirB11 ATPases, VirB10 undergoes a structural transition, detectable as a change in susceptibility to Streptomyces griseus protease. Specifically, protease treatment of spheroplasts from vir-induced wild-type (WT) cells results in degradation of the 48-kDa native VirB10 to a 40-kDa species (Fig. 3) (11). In contrast, no degradation is observed upon protease treatment of spheroplasts from ATP-depleted WT cells or cells lacking the VirD4 or VirB11 ATPases. If our model that a VirB10 structural transition couples ATP energy to substrate transfer across the OM is correct, the G272R mutation might affect energy sensing or the ATP-mediated structural transition at the distal portion of the translocation channel. To test for a possible structural effect of the G272R mutation, we assayed for protease susceptibility of the G272R mutant from cells that were or were not depleted of cellular ATP by arsenate treatment (11). Like for native VirB10, the G272R mutant protein was degraded to the 40-kDa species in energized cells (Fig. 3). However, in contrast to the native protein, the G272R mutant also was degraded to the 40-kDa species with protease treatment of ATP-depleted cells (Fig. 3). These findings suggest that the G272R might “lock” VirB10 in the energized, open-channel conformation, resulting in the unregulated release of VirE2 to the cell surface.
Fig. 3.

Effects of G272R on the energy-dependent VirB10 conformational switch. A348 (WT) or PC1010 (ΔB10) cells lacking or producing native VirB10 (B10) or the G272R mutant were treated with the energy poison arsenate or H2O as a control. Spheroplasts from these cells were incubated in the presence (+) or absence (−) of the S. griseus protease, and VirB10 degradation was monitored by development of immunoblots with anti-VirB10 antibodies. The positions of full-length VirB10 (B10; ∼48 kDa) and the proteolytic cleavage product (B10*; ∼40 kDa) are indicated at right.
G272R is permissive for intercellular substrate transfer but blocks T pilus biogenesis.
Next, we sought to determine whether the G272R mutation disrupts substrate transfer to target cells or assembly of the T pilus. Translocation was monitored with a plant infection assay and conjugative transfer of an IncQ plasmid to agrobacterial recipients. The G272R mutant incited formation of plant tumors with kinetics and morphologies similar to those for the wild-type strain, and it also transferred the IncQ plasmid at levels comparable to those for the wild-type strain (Table 2). These findings suggest that the G272R mutation exerts its effects specifically on channel gating and does not disrupt establishment of the donor-target cell junction or substrate delivery across the target cell envelope.
In previous studies, it was shown that agrobacterial donor cells conjugatively transfer IncQ plasmids to agrobacterial recipients engineered to synthesize the VirB proteins at appreciably higher frequencies than recipients lacking the VirB proteins (9, 30). In the case of VirB10, a ΔvirB10 null mutant acquires the IncQ plasmid at a frequency of ∼2 to 3 orders of magnitude lower than that for the isogenic strain producing native VirB10 in trans (Table 2). We asked whether the G272R mutation affected VirB10-mediated plasmid acquisition with matings between wild-type A348 donor cells and ΔvirB10 cells producing either the native VirB10 or the G272R mutant as recipients. As shown in Table 2, the G272R mutant strain acquired the IncQ plasmid at efficiencies comparable to those for the isogenic strain producing native VirB10 (9). The G272R mutation therefore does not alter the capacity of VirB10 to mediate productive cell-cell contacts, regardless of whether the mutant protein is produced in agrobacterial donor or recipient strains.
Cells producing native VirB10 possess abundant amounts of extracellular VirB2 pilin and VirB5, a protein shown to bind the tip of the T pilus (2), as monitored by a colony immunoblot assay or immunostaining of the shear fraction (Fig. 4). Intriguingly, however, the G272R mutant strain did not accumulate detectable levels of surface-exposed VirB2 pilin or VirB5 with either assay, although in these cells both pilin proteins accumulated to high levels (Fig. 4). The G272R mutation therefore falls in a class of previously identified Tra+ Pil− “uncoupling” mutations (25, 26) but with the additional novel feature that it also confers substrate exposure at the cell surface.
Fig. 4.
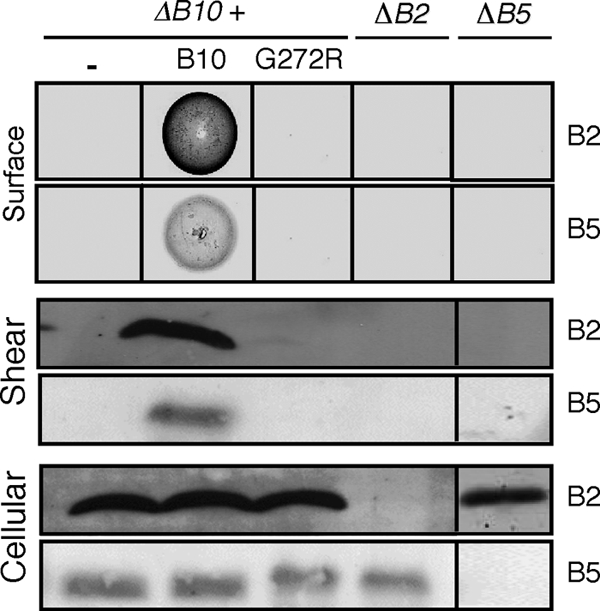
Effects of the G272R mutation on T pilus production, as monitored by assaying for the presence of VirB2 pilin and pilus-associated VirB5 on the cell surface by Western blotting and immunostaining of colonies and shear fractions with anti-VirB2 and anti-VirB5 antibodies, respectively. Corresponding analyses of the nonpolar ΔvirB2 and ΔvirB5 mutants show absence of antibody cross-reactivity and codependence of VirB2 and VirB5 for surface display. Bottom, steady-state cellular levels of VirB2 and VirB5.
Distinct effects of G272R and AP deletion mutations on T4SS function.
VirB10 shares extensive sequence relatedness with TraFpKM101 and other homologs throughout the region surrounding Gly272 (14, 17). Modeling of Gly272 on the recently derived TraFpKM101 X-ray structure showed that this residue sits at the distal edge of the β-barrel domain, near the putative AP pore domain (Fig. 5A). Correspondingly, in the space-filling model of the pKM101 core complex, Gly272 is exposed in the chamber of the core complex (Fig. 5B). A positively charged Arg residue at this position might disrupt gating of the channel, possibly by altering a core complex interaction with a channel-gating regulatory domain. Previously, we reported that partial deletions of the AP resemble the G272R mutation in conferring a Tra+ Pil− phenotype (26). According to the TraF structure, the VirB10 AP is predicted to span residues 285 to 334, whereas the “uncoupling” deletion mutations leave the transmembrane helix, α2, intact, but delete either the loop between α2 and α3 (α2-α3 loop; Δ308-330) or the α2-α3 loop and the lumen-facing α3 helix (Δ308-337). The partial AP deletion mutant strains do not elaborate T pili but rather shed VirB2 into the milieu as monomers, short oligomers, or aggregates (26).
Fig. 5.

Modeling of G272R in the pKM101 crystal structures. (A) Ribbon model of VirB10 (TraF) showing the β-barrel (in gray) and antenna projection (AP) (green). The G272R mutation sits in an unstructured domain between the β-barrel and AP. (B) Space-filling model of the pKM101 core complex comprised of homologs of VirB7 (TraN) (pink), VirB9 (TraO) (blue), TraF (VirB10) β-barrel/linker domains (gray), and AP domain (α2, α3 helices, and intervening loop) (green). VirB10 G272R, corresponding to the TraF G294R mutation, is indicated in red. The AP domain spans residues 285 to 334 and the Δ308-337 mutation encompassing the AP loop and α3 helix confers a Tra+ Pil− phenotype (26).
In view of these findings, we sought to determine whether a partial AP deletion mutation (Δ308-337) also confers increased surface exposure of the VirE2 secretion substrate. However, we found that the partial ΔAP mutant did not possess VirE2 on the cell surface at levels greater than observed with the isogenic strain producing native VirB10 (Fig. 6A), nor did it exhibit enhanced sensitivity to vancomycin or SDS (Fig. 6C and D). Like the G272R mutant, the AP deletion mutant did not show altered growth rates (Fig. 6B) or release of periplasmic RNase or ChvE to the cell surface (Fig. 6E). The partial ΔAP mutation thus appears to specifically block T pilus polymerization at a step distinct from that affected by the G272R mutation.
Fig. 6.

Effects of the ΔAP (Δ308-337) mutation on VirE2 release, DNA transfer, cell growth, and outer membrane integrity. (A) Effect of the ΔAP mutation on release of Flag-VirE2. Samples were prepared and analyzed as described for Fig. 1A. The ΔAP mutant strain displayed only slightly reduced levels of substrate transfer to plants, as monitored by virulence assays, and IncQ plasmid transfer to agrobacterial recipients. Strains: B10, PC1010 producing native VirB10; ΔAP, PC1010 producing VirB10Δ308-337. (B to E) As described in the Fig. 2 legend, showing that this partial ΔAP mutation did not confer diminished cell growth, enhanced sensitivity to vancomycin or SDS, or release of periplasmic RNase or ChvE. The partial ΔAP mutation disrupts polymerization of VirB2 into the T pilus (26).
DISCUSSION
Conjugation systems and most other T4SSs translocate DNA or protein substrates by a process requiring direct contact with target cells (1). Although we have identified a number of altered-function mutations through random mutagenesis of virB10, the G272R substitution was unique in conferring host cell contact-independent surface exposure of VirE2. The mutant strain displayed WT colony morphology and growth in rich and vir-inducing media but did not nonspecifically release the periplasmic RNase or ChvE, or VirE2 deleted of its C-terminal secretion signal, consistent with a model in which the G272R mutation specifically disrupts VirB/D4 channel gating. The G272R mutant did exhibit enhanced sensitivity to molecules that normally do not pass through OM porins, e.g., SDS and vancomycin, further suggesting that the gating defect allows for bidirectional movement of molecules too large to diffuse through porins across the OM. Gating mutations of other OM channels display similar phenotypes; recently, such mutations were isolated in the filamentous bacteriophage f1 OM secretin, and most of these mutations similarly conferred enhanced sensitivity to detergents and large antibiotics (43).
The region surrounding G272 bears a sequence (S262xGxDxLG269xxG272xxG275xVDxH280) that is highly conserved among VirB10 homologs associated with Gram-negative bacterial T4SSs (see a Pro Dom analysis of VirB10 homologs at http//:mmg.uth.tmc.edu/webpages/faculty/pchristie.html). Most notable are invariant Gly residues corresponding to VirB10 Gly269 and Gly272 and a strongly conserved residue, Gly275. In the first X-ray structure of a VirB10 homolog, Helicobacter pylori ComB10, a domain spanning residues 268 to 287 (A. tumefaciens VirB10 numbering) formed an α helix projecting from the side of a large β-barrel domain; this projection, originally termed the α1 helix, was implicated in stabilizing VirB10 homodimers or multimers (46). In the context of this structure, the mechanistic consequences of the G272R mutation were not immediately evident. However, in the more recent pKM101 X-ray structure, the corresponding Gly residues of the 14 TraF subunits encircle the interior of the core chamber near the OM (Fig. 5) (14); this region of the core complex is ideally positioned to participate in regulation of substrate transfer across the OM.
Defining the nature of gating mechanisms for various OM channels of Gram-negative bacteria has received considerable attention in recent years. A general mechanism, exemplified with TonB-activated transporters of small molecules (31), the P pilus biogenesis system (37), and larger OM channels formed by secretins (24, 36, 40), involves the formation of a plug domain or a constriction in the β-barrel channel subunit. In the closed configuration, the plug or constriction prevents the passage of substrate and other molecules, e.g., detergents and large antibiotics. The conformational switch to the open state might be induced by substrate binding to regulatory domains and perhaps other activating signals, e.g., IM energy from ATP hydrolysis or the electrochemical gradient (36, 43). Recent structural studies have identified plug or constriction domains in OM secretins (36, 37, 41) and have also shed light on conformational switches accompanying the closed-to-open channel transitions (36, 37). Corresponding mutational studies have also identified motifs in the primary sequences of OM channel complexes that participate in channel gating (3, 44). In the case of the filamentous bacteriophage f1, for example, the pIV secretin assembles as a dodecameric barrel structure in which the C-terminal ring forms the OM pore (32), and “leaky” mutations were clustered in two sequence motifs, GATE1 and GATE2, in the adjacent middle ring (44). Interestingly, despite structural differences in the pIV and T4SS core complexes (44), the gating mutations in the pIV secretin and the G272R mutation in the T4SS core complex are in the interiors of the respective chambers near the OM channels. However, in contrast to the GATE motifs, which are not well conserved among members of the secretin superfamily (44), the GxxGxxG motif is strongly conserved among VirB10 homologs, which is suggestive of a gating mechanism that is fundamental to all T4SS machines of Gram-negative bacteria.
The T4SS core complex does not possess an obvious plug, in contrast to the TolC, PapC usher, and secretin complexes (14, 23). However, the full-length T4SS core structure clearly resolves a constriction of about 5 to 10 Å, which is too small for passage of substrates. In contrast, the crystal structure of the outer membrane part of the core complex (the O layer) obtained by proteolysis of the full-length core complex displays a larger opening of about 30 Å. Conceivably, the proteolytic removal of the IM part of the core complex (the I layer) resulted in conformational changes in the 14-helix OM channel so that the crystal structure reflects the open conformation. In the context of the intact VirB/VirD4 channel, other VirB subunits would regulate the gating of the OM pore. A likely mechanistic consequence of the G272R mutation is that the long Arg side chain or the introduction of a positive charge at this site provides a steric block that disrupts core-VirB channel contacts. Two VirB subunits, VirB2 pilin and VirB9, have been proposed to regulate substrate translocation across the OM (12). VirB2 pilin is required for pilus production and for substrate transfer; however, the isolation of Tra+ Pil− “uncoupling” mutations establishes that an intact pilus is not required for substrate transfer (12, 16, 25, 26). The T4SSs thus might exist in or transition between two states: a secretion-competent state and a pilus biogenesis-competent state. In a secretion-competent state, the pilus would be short but still sufficiently long to exert mechanical forces on the constricted OM channel, thereby inducing its opening. In the pilus biogenesis-competent state, the pilus would grow out through the OM channel. The G272R steric block might restrict pilus outgrowth, resulting in a pilus polymer sufficiently long to trigger OM channel opening and consequently VirE2 release but not long enough to produce a visible pilus, hence showing the Pil− phenotype.
An effect of the G272R mutation on VirB9 activity also cannot be excluded at this time. VirB9-like subunits possess two highly conserved domains separated by a nonconserved linker region (25). In the pKM101 X-ray structure, the C-terminal domain (CTD) of VirB9-like TraO lines the outside, not the inside, of the core chamber; therefore, this domain probably does not interact directly with the interior of the core near Gly272 (14). The N-terminal domain (NTD) likely extends along the portion of the core complex projecting through the periplasm toward the cytoplasmic membrane. Besides detection of an FA-cross-linkable contact between VirB9 and DNA substrates (12), two-residue insertion mutations were isolated in the VirB9 NTD that selectively permit translocation of two different DNA substrates, T-DNA or an IncQ plasmid, to target cells. These were termed substrate discrimination mutations, and it was postulated that the VirB9 NTD modulates substrate trafficking through differential recognition/binding of relaxases covalently bound at the 5′ ends of DNA substrates (25). Conceivably, the G272R mutation acts indirectly by imposing a structural change on the VirB9 NTD that affects its ability to regulate substrate trafficking. At this point, however, we have determined that the G272R mutant forms precipitable complexes with partner subunits VirB7 and VirB9 in energized and ATP-depleted cells and also forms chemical cross-linked species indistinguishable from native VirB10, arguing against a gross perturbation by the G272R mutation of the overall core complex structure.
While further studies are needed to define the molecular basis for gating of the VirB/D4 OM channel, here we also showed that the G272R mutation rendered VirB10 sensitive to protease even in ATP energy-depleted cells (Fig. 3). These findings suggest that the G272R mutation induces a structural state resembling the open-channel conformation of WT cells, in effect bypassing the requirement for the coupling of ATP hydrolysis by the VirB/D4 ATPases to OM translocation. It is interesting to note, however, that in energized WT cells, the energy-activated (protease-susceptible) form of native VirB10 is also detectable (11), yet WT cells do not release detectable amounts of VirE2 to the cell surface (Fig. 1). This indicates that energization of VirB10 is necessary, but not sufficient, for gating of the OM channel. Another activating signal must be required for gating the OM channel, and the G272R mutation apparently also bypasses this requirement. One such activating signal could be the “mating signal” identified in early studies of the F plasmid transfer system (33). This signal is propagated from recipient cells to donor cells upon contact with the F pilus or donor cell envelope to stimulate plasmid transfer. Although the nature of the mating signal remains unknown, the G272R mutation might bypass a requirement for signal activation through direct contact with recipient cells.
Of further interest, the G272R mutation did not disrupt intercellular translocation, suggesting that it does not perturb establishment of the donor-recipient cell mating junction. At this time, the architecture of the T4SS channel at the cell surface is poorly defined, but the fact that the G272R mutation permits substrate transfer while blocking surface accumulation of pilins and assembly of T pili adds to the argument that pilins/pili are not required for formation of the VirB/VirD4 mating junction, in apparent contrast to the situation for the F plasmid transfer system (4).
In sum, we have presented evidence that Gly272 within a conserved GxxGxxG sequence motif and the AP domain of VirB10 contribute in distinct ways to regulate substrate transfer across the OM and biogenesis of the T pilus. The G272R mutation selectively disrupts gating of the secretion channel at the OM, allowing for release of substrates to the cell surface and small-molecule uptake. This mutation also blocks T pilus (or pilin) extrusion but not establishment of critical donor-target cell contacts required for intercellular substrate translocation. In contrast, the AP partial deletions do not disrupt gating of the secretion channel or formation of the mating junction but instead impose a block in a terminal stage of T pilus assembly involving the polymerization process. Ongoing structure-function studies are focused on defining the nature of contacts between these domains or other channel constituents in regulating substrate transfer, the role(s) of ATP energy and other possible contact-dependent signals in activating OM translocation, and the mechanistic contributions of the GxxGxxG motif and the AP on pilus biogenesis.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM48746 to P.J.C. and NSF grants MCB9506144, MCB9905126, and MCB0416471 to L.M.B. J. E. Kerr was supported in part by Molecular Basis of Infectious Diseases (MBID) training grant 1 T32 AI55449.
We thank Andrew Binns and Lisa Stahl for construction of plasmid pLS27 and Liane D'Allesandro and Gape Machao for technical assistance.
Footnotes
Published ahead of print on 18 March 2011.
REFERENCES
- 1. Alvarez-Martinez C. E., Christie P. J. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 73:775–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aly K. A., Baron C. 2007. The VirB5 protein localizes to the T-pilus tips in Agrobacterium tumefaciens. Microbiology 153:3766–3775 [DOI] [PubMed] [Google Scholar]
- 3. Augustus A. M., Celaya T., Husain F., Humbard M., Misra R. 2004. Antibiotic-sensitive TolC mutants and their suppressors. J. Bacteriol. 186:1851–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Babic A., Lindner A. B., Vulic M., Stewart E. J., Radman M. 2008. Direct visualization of horizontal gene transfer. Science 319:1533–1536 [DOI] [PubMed] [Google Scholar]
- 5. Bagdasarian M., et al. 1981. Specific-purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 16:237–247 [DOI] [PubMed] [Google Scholar]
- 6. Beaupre C. E., Bohne J., Dale E. M., Binns A. N. 1997. Interactions between VirB9 and VirB10 membrane proteins involved in movement of DNA from Agrobacterium tumefaciens into plant cells. J. Bacteriol. 179:78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berger B. R., Christie P. J. 1994. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J. Bacteriol. 176:3646–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berry J., Savva C., Holzenburg A., Young R. 2010. The lambda spanin components Rz and Rz1 undergo tertiary and quaternary rearrangements upon complex formation. Protein Sci. 19:1967–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bohne J., Yim A., Binns A. N. 1998. The Ti plasmid increases the efficiency of Agrobacterium tumefaciens as a recipient in virB-mediated conjugal transfer of an IncQ plasmid. Proc. Natl. Acad. Sci. U. S. A. 95:7057–7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cascales E., et al. 2007. Colicin biology. Microbiol. Mol. Biol. Rev. 71:158–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cascales E., Christie P. J. 2004. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc. Natl. Acad. Sci. U. S. A. 101:17228–17233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cascales E., Christie P. J. 2004. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science 304:1170–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cascales E., Christie P. J. 2003. The versatile bacterial type IV secretion systems. Nat. Rev. Microbiol. 1:137–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chandran V., et al. 2009. Structure of the outer membrane complex of a type IV secretion system. Nature 462:1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christie P. J. 1997. Agrobacterium tumefaciens T-complex transport apparatus: a paradigm for a new family of multifunctional transporters in eubacteria. J. Bacteriol. 179:3085–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Christie P. J. 2009. Structural biology: translocation chamber's secrets. Nature 462:992–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christie P. J., Atmakuri K., Krishnamoorthy V., Jakubowski S., Cascales E. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Close T. J., Zaitlin D., Kado C. I. 1984. Design and development of amplifiable broad-host-range cloning vectors: analysis of the vir region of Agrobacterium tumefaciens plasmid pTiC58. Plasmid 12:111–118 [DOI] [PubMed] [Google Scholar]
- 19. Dale E. M., Binns A. N., Ward J. J. 1993. Construction and characterization of Tn5virB, a transposon that generates nonpolar mutations, and its use to define virB8 as an essential virulence gene in Agrobacterium tumefaciens. J. Bacteriol. 175:887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fernandez D., et al. 1996. The Agrobacterium tumefaciens virB7 gene product, a proposed component of the T-complex transport apparatus, is a membrane-associated lipoprotein exposed at the periplasmic surface. J. Bacteriol. 178:3156–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernandez D., Spudich G. M., Zhou X. R., Christie P. J. 1996. The Agrobacterium tumefaciens VirB7 lipoprotein is required for stabilization of VirB proteins during assembly of the T-complex transport apparatus. J. Bacteriol. 178:3168–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fronzes R., Christie P. J., Waksman G. 2009. The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7:703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fronzes R., et al. 2009. Structure of a type IV secretion system core complex. Science 323:266–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hodgkinson J. L., et al. 2009. Three-dimensional reconstruction of the Shigella T3SS transmembrane regions reveals 12-fold symmetry and novel features throughout. Nat. Struct. Mol. Biol. 16:477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jakubowski S. J., Cascales E., Krishnamoorthy V., Christie P. J. 2005. Agrobacterium tumefaciens VirB9, an outer-membrane-associated component of a type IV secretion system, regulates substrate selection and T-pilus biogenesis. J. Bacteriol. 187:3486–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jakubowski S. J., et al. 2009. Agrobacterium VirB10 domain requirements for type IV secretion and T pilus biogenesis. Mol. Microbiol. 71:779–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karuppiah M., Berry J.-L., Derrick J. P. 2011. Outer membrane translocons: structural insights into channel formation. Trends Microbiol. 19:40–48 [DOI] [PubMed] [Google Scholar]
- 28. Lazzaroni J. C., Portalier R. C. 1981. Genetic and biochemical characterization of periplasmic-leaky mutants of Escherichia coli K-12. J. Bacteriol. 145:1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li G., Howard S. P. 2010. ExeA binds to peptidoglycan and forms a multimer for assembly of the type II secretion apparatus in Aeromonas hydrophila. Mol. Microbiol. 76:772–781 [DOI] [PubMed] [Google Scholar]
- 30. Liu Z., Binns A. N. 2003. Functional subsets of the VirB type IV transport complex proteins involved in the capacity of Agrobacterium tumefaciens to serve as a recipient in virB-mediated conjugal transfer of plasmid RSF1010. J. Bacteriol. 185:3259–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oke M., et al. 2004. The plug domain of a neisserial TonB-dependent transporter retains structural integrity in the absence of its transmembrane beta-barrel. FEBS Lett. 564:294–300 [DOI] [PubMed] [Google Scholar]
- 32. Opalka N., et al. 2003. Structure of the filamentous phage pIV multimer by cryo-electron microscopy. J. Mol. Biol. 325:461–470 [DOI] [PubMed] [Google Scholar]
- 33. Ou J. T. 1975. Mating signal and DNA penetration deficiency in conjugation between male Escherichia coli and minicells. Proc. Natl. Acad. Sci. U. S. A. 72:3721–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Postle K., Larsen R. A. 2007. TonB-dependent energy transduction between outer and cytoplasmic membranes. Biometals 20:453–465 [DOI] [PubMed] [Google Scholar]
- 35. Rashkova S., Zhou X.-R., Christie P. J. 2000. Self-assembly of the Agrobacterium tumefaciens VirB11 traffic ATPase. J. Bacteriol. 182:4137–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reichow S. L., Korotkov K. V., Hol W. G., Gonen T. 2010. Structure of the cholera toxin secretion channel in its closed state. Nat. Struct. Mol. Biol. 17:1226–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Remaut H., et al. 2008. Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell 133:640–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sagulenko Y., Sagulenko V., Chen J., Christie P. J. 2001. Role of Agrobacterium VirB11 ATPase in T-pilus assembly and substrate selection. J. Bacteriol. 183:5813–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J. E., Fritsch F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 40. Schmidhauser T. J., Ditta G., Helinski D. R. 1987. Broad host range plasmid cloning vectors in gram-negative bacteria, p. 287–332 In Rodriquez R. L., Denhardt D. T. (ed.), Vectors: a survey of molecular cloning vectors and their uses. Buttersworth Publishers, Stoneham, MA [Google Scholar]
- 41. Schraidt O., et al. 2010. Topology and organization of the Salmonella typhimurium type III secretion needle complex components. PLoS Pathog. 6:e1000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sciaky D., Montoya A. L., Chilton M. D. 1978. Fingerprints of Agrobacterium Ti plasmids. Plasmid 1:238–253 [DOI] [PubMed] [Google Scholar]
- 43. Simone M., McCullen C. A., Stahl L. E., Binns A. N. 2001. The carboxy-terminus of VirE2 from Agrobacterium tumefaciens is required for its transport to host cells by the virB-encoded type IV transport system. Mol, Microbiol. 41:1283–1293 [DOI] [PubMed] [Google Scholar]
- 44. Spagnuolo J., et al. 2010. Identification of the gate regions in the primary structure of the secretin pIV. Mol. Microbiol. 76:133–150 [DOI] [PubMed] [Google Scholar]
- 45. Summer E. J., et al. 2007. Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J. Mol. Biol. 373:1098–1112 [DOI] [PubMed] [Google Scholar]
- 46. Terradot L., Bayliss R., Oomen C., Leonard, Baron C., Waksman G. 2005. Structures of two core subunits of the bacterial type IV secretion system, VirB8 from Brucella suis and ComB10 from Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 102:4956–4961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ward J. E. J., Dale E. M., Christie P. J., Nester E. W., Binns A. N. 1990. Complementation analysis of Agrobacterium tumefaciens Ti plasmid virB genes by use of a vir promoter expression vector: virB9, virB10, and virB11 are essential virulence genes. J. Bacteriol. 172:5187–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao Z., Sagulenko E., Ding Z., Christie P. J. 2001. Activities of virE1 and the VirE1 secretion chaperone in export of the multifunctional VirE2 effector via an Agrobacterium type IV secretion pathway. J. Bacteriol. 183:3855–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou X.-R., Christie P. J. 1999. Mutagenesis of Agrobacterium VirE2 single-stranded DNA-binding protein identifies regions required for self-association and interaction with VirE1 and a permissive site for hybrid protein construction. J. Bacteriol. 181:4342–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]