

Abstract
An alcohol dehydrogenase (ADH) from hyperthermophilic archaeon Thermococcus guaymasensis was purified to homogeneity and was found to be a homotetramer with a subunit size of 40 ± 1 kDa. The gene encoding the enzyme was cloned and sequenced; this gene had 1,095 bp, corresponding to 365 amino acids, and showed high sequence homology to zinc-containing ADHs and l-threonine dehydrogenases with binding motifs of catalytic zinc and NADP+. Metal analyses revealed that this NADP+-dependent enzyme contained 0.9 ± 0.03 g-atoms of zinc per subunit. It was a primary-secondary ADH and exhibited a substrate preference for secondary alcohols and corresponding ketones. Particularly, the enzyme with unusual stereoselectivity catalyzed an anti-Prelog reduction of racemic (R/S)-acetoin to (2R,3R)-2,3-butanediol and meso-2,3-butanediol. The optimal pH values for the oxidation and formation of alcohols were 10.5 and 7.5, respectively. Besides being hyperthermostable, the enzyme activity increased as the temperature was elevated up to 95°C. The enzyme was active in the presence of methanol up to 40% (vol/vol) in the assay mixture. The reduction of ketones underwent high efficiency by coupling with excess isopropanol to regenerate NADPH. The kinetic parameters of the enzyme showed that the apparent Km values and catalytic efficiency for NADPH were 40 times lower and 5 times higher than those for NADP+, respectively. The physiological roles of the enzyme were proposed to be in the formation of alcohols such as ethanol or acetoin concomitant to the NADPH oxidation.
INTRODUCTION
Alcohol dehydrogenases (ADHs) are ubiquitous in three life domains and a family of oxidoreductases that catalyze the NAD(P)H-dependent interconversion between alcohols and the corresponding aldehydes or ketones (53). Among them, the medium-chain ADHs have been studied extensively, and these ADHs usually contain zinc. Zinc-containing ADHs constitute a big protein family with various enzyme activities, including alcohol dehydrogenase, polyol dehydrogenase, and cinnamyl alcohol dehydrogenase activities (37). A large number of zinc-containing ADHs, including those from hyperthermophiles Thermococcus kodakaraensis, Pyrococcus horikoshii, Aeropyrum pernix, and Sulfolobus solfataricus, contain catalytic and structural zincs (5, 11, 20, 25, 30). The zinc-containing ADHs from the mesophile Clostridium beijerinckii and thermophiles Thermoanaerobacter brockii and Thermoanaerobacter ethanolicus contain only catalytic zinc (34).
Hyperthermophiles are a group of microorganisms growing optimally at ≥80°C, of which anaerobic heterotrophs have increasingly attracted attention for the use of fermentation at the elevated temperatures (31). All members of the genus Thermococcus are chemoorganotrophs which can grow on peptide-containing substrates (6, 50), and some of them are able to utilize carbohydrates, including starch and chitin as a carbon source (6), though some are capable of producing H2 from CO (38, 61). Glycolysis from glucose to pyruvate in Thermococcus celer and Thermococcus litoralis appears to occur via a modified Embden-Meyerhof (EM) pathway containing ADP-dependent hexose kinase and phosphofructokinase and a tungsten-containing glyceraldehyde-3-phosphate, ferredoxin oxidoreductase (59). Among strains of the genus Thermococcus, Thermococcus strain ES1 was first reported to produce ethanol under S0-limiting conditions (42), one of the attractive renewable energy resources. Subsequently, native ADHs have been purified and characterized, and all of these ADHs are iron containing and proposed to be responsible for ethanol formation (3, 39, 42, 43).
In addition to interests in their physiological roles in production of alcohols, ADHs from hyperthermophiles, particularly zinc-containing ADHs and short-chain ADHs, are highly desired as promising catalysts in chiral synthesis because of the features such as solvent tolerance, stereoselectivity, and thermostability (5, 44, 62, 70). A zinc-containing ADH from the anaerobic archaeon Pyrococcus furiosus underwent asymmetric ketone reduction to the corresponding chiral alcohols (44, 70). The presumably zinc-containing l-threonine dehydrogenase from T. kodakaraensis was highly stable, and this dehydrogenase had a half-life at 85°C of more than 2 h (5). In the family of zinc-containing ADHs, those from the hyperthermophilic archaea T. kodakaraensis, P. horikoshii, S. solfataricus, and A. pernix contain both catalytic and structural zincs and have been extensively studied in terms of structure, catalysis, function, or regulation (2, 16, 19, 25–28, 52). However, no ADHs containing only catalytic zinc have been characterized for Thermococcus species yet.
Thermococcus guaymasensis is a hyperthermophilic starch-utilizing archaeon, producing acetate, propionate, isobutyrate, isovalerate, CO2, and H2S if sulfur is present in growth medium (15). Here, we report the production of ethanol and acetoin in glucose fermentation of T. guaymasensis and the properties of an ADH from this hyperthermophile. Moreover, the purified ADH from T. guaymasensis is the most thermostable zinc-containing ADH characterized from Thermococcus species, which has high solvent tolerance, unusual stereoselectivity, and broad substrate specificity.
MATERIALS AND METHODS
Chemicals and organisms.
All chemicals were commercially available. (R)-(−)-2-Butanol, (S)-(+)-2-butanol, (2R,3R)-(−)-2,3-butanediol, (2S,3S)-(+)-2,3-butanediol, and meso-2,3-butanediol were purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). T. guaymasensis DSM 11113T was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), Braunschweig, Germany. The pGEM-T easy vector (Promega, Madison, WI) was used for the cloning of PCR products.
Growth conditions.
T. guaymasensis was cultured in the medium as described previously (15), with modifications. The medium at pH 7.0 contained the following chemicals and other components (in g/liter unless otherwise specified): KCl, 0.33; MgCl2·2H2O, 2.7; MgSO4·7H2O, 3.4; NH4Cl, 0.25; CaCl2·2H2O, 0.14; K2HPO4, 0.14; Na2SeO3, 0.01 mg; NiCl2·6H2O, 0.01 mg; NaHCO3, 1.0; NaCl, 18; resazurin, 0.001; cysteine·HCl·H2O, 0.5; Na2S·9H2O, 0.5; Bacto-yeast extract, 10; Trypticase soy broth, 10; elemental sulfur, 10; dextrose, 5; HEPES, 5.2; trace mineral solution, 10 ml; and vitamin solution, 10 ml. The preparation of trace mineral and vitamin solutions was done as described previously (4). In a large scale of cultivation, it was routinely cultured in a 20-liter glass carboy at 88°C in which elemental sulfur and HEPES were omitted. The resulting cell pellet after centrifugation was frozen in liquid nitrogen immediately and stored at −80°C until use.
Preparation of cell extract.
The frozen cells (50 g) of T. guaymasensis were resuspended in 450 ml of 10 mM Tris-HCl anaerobic buffer (pH 7.8) containing 2 mM dithiothreitol (DTT), 2 mM sodium dithionite (SDT), and 5% (vol/vol) glycerol. The suspension was incubated at 37°C for 2 h under stirring. The supernatant was collected as the cell extract after 30 min of centrifugation at 10,000 × g.
Purification of T. guaymasensis ADH.
All the purification steps were carried out anaerobically at room temperature. The cell extract of T. guaymasensis was loaded onto a DEAE-Sepharose column (5 by 10 cm) that was equilibrated with buffer A (50 mM Tris-HCl [pH 7.8] containing 5% [vol/vol] glycerol, 2 mM DTT, 2 mM SDT). T. guaymasensis ADH that bound weakly to the column was eluted out while buffer A was applied at a flow rate of 3 ml min−1. A linear gradient (0 to 0.5 M NaCl) was further applied onto the column. Fractions containing ADH activity were then pooled and loaded onto a hydroxyapatite column (2.6 by 15 cm) at a flow rate of 2 ml min−1. The column was applied with a gradient (0 to 0.5 M potassium phosphate in buffer A), and ADH started to elute from the column at a concentration of 0.25 M potassium phosphate. Fractions containing enzyme activity were pooled and applied to a phenyl-Sepharose column (2.6 by 10 cm) equilibrated with 0.8 M ammonia sulfate in buffer A. A linear gradient (0.82 to 0 M ammonia sulfate in buffer A) was applied at a flow rate of 2 ml min−1, and the ADH started to elute at a concentration of 0.4 M ammonia sulfate. Fractions containing ADH activity were desalted and concentrated by ultrafiltration using 44.5 mm YM-10 membranes (Millipore Corporation, Bedford, MA). The concentrated samples were applied to a Superdex-200 gel filtration column (2.6 by 60 cm) equilibrated with buffer A containing 100 mM KCl at a flow rate of 2.5 ml min−1. The purity of the fractions containing ADH activity was verified using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (36).
Enzyme assay and protein determination.
The catalytic activity of T. guaymasensis ADH was measured at 80°C by monitoring the substrate-dependent absorbance change of NADP(H) at 340 nm (ε340 = 6.3 mM−1 cm−1). Unless otherwise specified, the enzyme assay was carried out in duplicate using the assay mixture (2 ml) for alcohol oxidation containing 50 mM 2-butanol and 0.4 mM NADP+ in 100 mM CAPS [3-(cyclohexylamino)-1-propanesulfonic acid] buffer (pH 10.5). The assay mixture (2 ml) for the reduction of ketone/aldehyde contained 6 mM 2-butanone and 0.2 mM NADPH in 100 mM HEPES (pH 7.5). The addition of the purified enzyme (0.25 μg) initiated the enzyme assay. One unit of the activity is defined as 1 μmol NADPH formation or oxidation per min. The protein concentrations of all samples were determined using the Bradford method, and bovine serum albumin served as the standard protein (12).
Determination of catalytic properties.
The effect of pH on the enzyme activities was determined over a range of 5.5 to 11.4. The buffers (100 mM) used were phosphate (pH 5.5 to 8.0), EPPS [4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid; 8.0 to 9.0], glycylglycine (9.0 to 9.7), and CAPS (9.7 to 11.4). The effect of the temperature on the enzyme activity was examined at temperatures from 30 to 95°C. Enzyme thermostability was determined by incubating the enzyme in sealed serum bottles at 80°C and 95°C. Residual activity was assayed at various time intervals under the standard assay conditions. Substrate specificity was determined using primary and secondary alcohols (50 mM), diols and polyols (50 mM), or aldehydes and ketones (6 mM) under standard assay conditions. The effect of cations, EDTA, or DTT on enzyme activities was carried out by measuring the reduction of 2-butanone in 100 mM HEPES buffer (pH 7.0), considering the low solubility of cations at alkaline pHs.
Enzyme kinetic parameters were determined using different substrates and coenzymes (NADP+ or NADPH). Concentrations of substrates were ≥10× the apparent Km unless specified for NADPH, sec-butanol, NADP+, and 2-butanone, while concentrations of the corresponding cosubstrates were kept constant and higher than 10× the apparent Km. Apparent values of Km and Vmax were calculated using the curve fittings of SigmaPlot (Systat Software, Inc., San Jose, CA).
Metal analyses.
The metal contents of T. guaymasensis ADH were determined by using inductively coupled plasma mass spectrometry (ICP-MS; VG Elemental PlasmQuad 3 ICP-MS at the Chemical Analysis Laboratory, University of Georgia). The purified enzyme was pretreated to wash off nonbinding metals in the anaerobic chamber, where the oxygen level was kept below 1 ppm. The washing buffer used was 10 mM Tris-HCl containing 2 mM DTT (pH 7.8). The washing procedure was carried out using YM-10 Amicon centrifuge tubes, including 7 repeats of centrifugation (concentration and refilling of buffers). The pass-through solution was collected as its control.
Ketone reduction coupled with NADPH regeneration.
The reaction mixture (2 ml) contained 100 mM HEPES buffer (anaerobic, pH 7.5), 50 mM 2-butanone or 2-pentanone, 25 μg T. guaymasensis ADH, 500 mM isopropanol, and 1 mM NADPH. The reaction was carried out at 30°C for 24 h unless otherwise specified. The reactants (butanone/2-butanol and 2-pentanone/2-pentanol) were determined with a Shimadzu GC-14A gas chromatograph (GC) equipped with a flame ionization detector (FID; 250°C). The GC analyses were performed under the following conditions: column, MXT-624 (0.53-mm inside diameter [ID] by 30-m length; Restek, Bellefonte, PA); FID sensitivity range, 102; carrier gas, helium at a linear velocity of 80 cm s−1. For the 2-pentenone/2-pentanol determination, the temperature program included: isotherm at 60°C for 3 min, 30°C/min ramp to 110°C, and isotherm at 110°C for 2 min. For the 2-butanone/2-butanol determination, the temperature program included isotherm at 40°C for 3 min, a 30°C/min ramp to 100°C, and isotherm at 100°C for 2 min. The reaction mixture (1 μl) was directly applied onto the injector (200°C) for GC analyses. The peak areas were quantitated using specific external standards.
Analyses of fermentation products.
The possible fermentation products, such as ethanol, acetoin, and 2,3-butanediol, were measured using the above-mentioned GC systems, with modifications. The temperature program was modified at the FID sensitivity range of 10: isotherm at 80°C for 3 min, a 10°C/min ramp to 150°C, and isotherm at 150°C. Prior to the analyses, the culture medium was centrifuged at 10,000 × g for 5 min and the supernatant was filtered to remove the residual cells.
Stereoselective conversion between acetoin and 2,3-butanediol.
The reaction mixture (2 ml) of 2,3-butanediol oxidation contained 100 mM CAPS buffer (anaerobic, pH 10.5), 50 mM (2R,3R)-(−)-2,3-butanediol or meso-2,3-butanediol, 25 μg T. guaymasensis ADH, 500 mM acetone, and 1 mM NADP+. The reaction mixture (2 ml) of acetoin reduction contained 100 mM HEPES buffer (anaerobic, pH 7.5), 50 mM racemic R/S-acetoin, 25 μg T. guaymasensis ADH, 500 mM isopropanol, and 1 mM NADPH. All the reactions were carried out at 30°C for 24 h unless specified. After that, the reaction mixture (1 ml) was extracted with 1 ml ethyl acetate or dichloride methane with shaking at 350 rpm for 30 min (room temperature). The stereoselectivity of the purified enzyme was determined using a Shimadzu GC-14A gas chromatograph equipped with a CP-Chirasil-Dex CB column (0.25-mm ID by 25-m length; Varian, Inc., Palo Alto, CA). The GC operating conditions described below included the FID detector (250°C) at a sensitivity range of 10 and helium as the carrier gas at a linear velocity of 40 cm s−1. The temperature program for R/S-acetoin, (2R,3R)-(−)-2,3-butanediol, (2S,3S)-(+)-2,3-butanediol, and meso-2,3-butanediol was listed as the following: isotherm at 60°C for 5 min, a 30°C/min ramp to 90°C, and isotherm at 90°C for 6 min. The reaction mixture after extraction (0.5 μl) was directly applied onto the injector (200°C) for each assay. The peak areas were quantitated using specific external standards. To identify whether T. guaymasensis ADH catalyzed asymmetric reduction of 2-butanone to chiral 2-butanols, the formation of R-2-butanol or S-2-butanol was verified by using the same GC operating conditions described in this section except that the temperature program was set isothermally at 45°C for 10 min.
Determination of amino-terminal and internal sequences.
To determine internal sequences, the purified enzyme was run on 12.5% SDS-PAGE gel and subjected to in-gel trypsin digestion. The resulting peptides were extracted and cleaned with the procedures described previously (60). The resulting samples were applied for mass spectrometry analyses on a Waters Micromass Q-TOF Ultima using nanospray injection as the sample delivery method (Mass Spectrometry Facility, University of Waterloo, Waterloo, ON, Canada). The PEAKS software program (BSI, Waterloo, ON, Canada) was used for tandem mass spectrometry (MS-MS) profiling. The amino-terminal sequence of T. guaymasensis ADH was determined by Edman degradation (Molecular Biology Core Facility, Dana Farber Cancer Institute, Boston, MA).
Cloning of the T. guaymasensis ADH gene.
The genomic DNA used as the template of PCR was extracted using the solution of phenol, chloroform, and isoamyl alcohol (25:24:1). Based on amino-terminal and internal sequences, two oligonucleotides, TGADHNF (5′-AARATGMGNGGTTTTGCAATG-3′) and TGADHIR (5′-GGAGTGCTGGTGATATCC-3′), were synthesized and used as forward and reverse PCR primers, respectively. A PCR was performed using Hot Start KOD polymerase with 15 pmol of each primer against 100 ng of genomic DNA isolated from T. guaymasensis cells. The thermal program consisted of 40 cycles of denaturation at 95°C for 20 s, annealing at 56°C for 20 s, and extension at 70°C for 30 s. The generated DNA fragment was modified using the procedure of addition of 3′-A overhangs (A-tailing) and ligated into the pGEM-T easy vector. Escherichia coli DH5α was transformed with this construct. After the blue/white screening, the recombinant plasmid was extracted, purified (57), and sequenced (Core Molecular Biology and DNA Sequencing Facility, York University, ON, Canada). The inverse PCR was designed for obtaining the flanking sequences by using two primers, TGMAYN01 (5′-TCTCCTTCTCAATCCACTCG-3′) and TGMAY28C02 (5′-GCAATAACTCCCGACTGG-3′). The genomic DNA was digested by the restriction enzyme HindIII for 6 h at 37°C and then incubated at 65°C for half an hour to denature the enzyme. The digested product was ligated to circle DNA by using T4 DNA ligase overnight at room temperature, which was used as the template in inverse PCRs (65). After preheating to 94°C for 5 min, 36 cycles were performed, consisting of denaturation at 95°C for 20 s, primer annealing at 58°C for 20 s, and elongation at 70°C for 1 min. The resulting 1.4-kb product of inverse PCRs was sequenced by the dye termination method using several primers designed on raw sequence information (Molecular Biology Core Facility, University of Waterloo, Waterloo, ON, Canada).
Data mining.
The homologues of T. guaymasensis ADH were identified by performing BLASTP searches (1). Phylogenetic analyses were performed by aligning T. guaymasensis ADH and close homologues of zinc-containing families using the program Clustal W (64). The primary structure analysis was estimated using the program ProtParam on the ExPASy server (22). The secondary and tertiary structure prediction was performed using the SWISS-MODEL server (33, 58). The software program PyMOL was used to analyze and visualize the tertiary structure of the T. guaymasensis ADH monomer (17).
Nucleotide sequence accession number.
The nucleotide sequence of T. guaymasensis ADH has been submitted to the GenBank database under accession number HQ415820.
RESULTS
Growth and alcohol formation of T. guaymasensis.
T. guaymasensis is a heterotrophic archaeon and could grow in the absence of sulfur during glucose fermentation. Ethanol was found to be one of the end products in the tested culture, and its production was in small amounts (<1 mM) and appeared to be correlated with the growth (Fig. 1). Hydrogen was a major end product when sulfur was omitted in the culture medium, and the highest level of hydrogen production (28.7 mmol per liter culture medium) was observed when the culture reached the maximal cell density (X. Ying and K. Ma, unpublished data). Whether T. guaymasensis was cultured in the presence of 20 mM HEPES buffer and 0.5% sulfur or not, the final pH of the medium after 48 h growth was around 6.0. Acetoin was detected as a metabolite in the spent medium, with a relatively higher level (2 to 3 mM). The acetoin formation of T. guaymasensis was detectable after 2 h in incubation and accumulated as the cell density increased under all tested conditions (Fig. 2), implying that its production might not be as a response to the pH change. 2,3-Butanediol could not be detected in the fermentation culture.
Fig. 1.
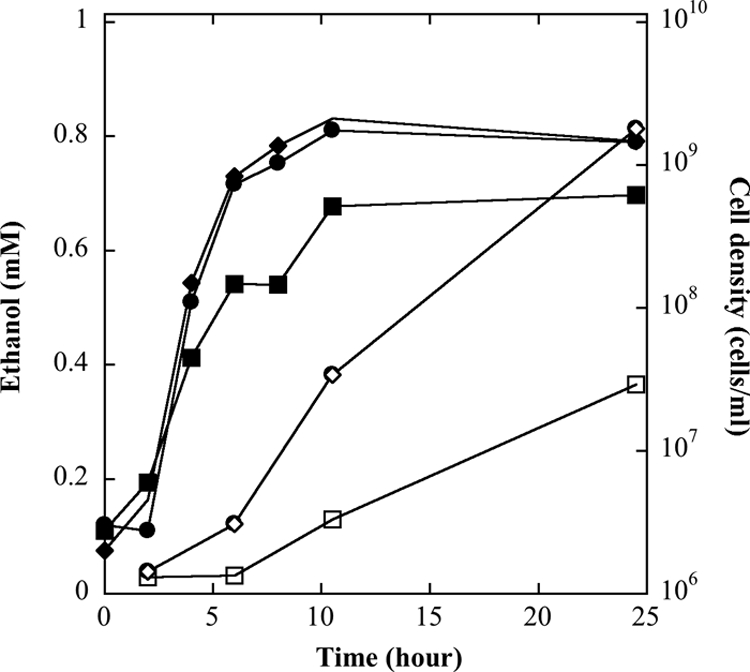
Growth and ethanol production of T. guaymasensis. T. guaymasensis can grow on glucose and was cultured under different sulfur concentrations: 0% (squares), 0.5% (circles), and 2% sulfur (diamonds). The cell density (filled symbols) and ethanol production (open symbols) were determined using direct cell counting and gas chromatography, respectively.
Fig. 2.
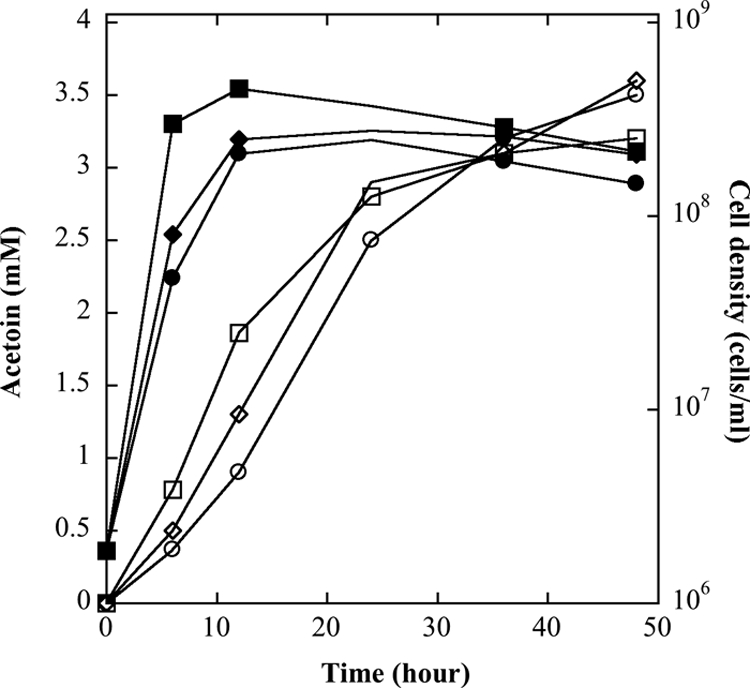
Growth and acetoin production of T. guaymasensis. T. guaymasensis was cultured under different conditions: 0.5% (wt/vol) elemental sulfur with 20 mM HEPES buffer (squares), no sulfur and no buffer (circles), and 20 mM HEPES buffer but no sulfur (diamonds). The cell density (filled symbols) and acetoin production (open symbols) were determined using direct cell counting and gas chromatography, respectively, as described in Materials and Methods.
ADH activities in the cell extract of T. guaymasensis.
The production of ethanol and acetoin indicated that T. guaymasensis might harbor multiple ADHs or a dominant ADH with multifunctions. The cell extract was prepared to investigate the presence of ADH activities. Alcohols such as glycerol, 1-butanol, 2-butanol, 2,3-butanediol, and 1,4-butanediol were used as assay substrates to differentiate the possible types of ADH activities, such as polyol, primary-alcohol, secondary-alcohol, and diol dehydrogenase activities. The results showed that all ADH activities in the cell extract of T. guaymasensis were NADP+ dependent. The types of ADH activities were diverse, and the activities were detectable with the use of all the alcohol substrates mentioned above. The highest ADH activity (18.9 U mg−1) showed when 2-butanol was used as the assay substrate.
Purification of T. guaymasensis ADH.
Considering the possibility of multiple ADHs in the cell extract, the ADH activities were traced using 2-butanol, 1-butanol, and glycerol as assay substrates during the purification procedure. ADH activities appeared in a single peak from all liquid chromatography columns, and the ratios of ADH activities among 2-butanol, 1-butanol, and glycerol (200:10:0.5 to 1) were almost constant until the enzyme was purified to homogeneity by a four-step procedure using fast protein liquid chromatography (FPLC). The ADH in T. guaymasensis was partially eluted during the loading of the sample onto the DEAE-Sepharose column, suggesting that the enzyme might have higher isoelectric point (pI) value, which was confirmed later by theoretical pI calculated from the deduced amino acid sequence. The enzyme could be completely eluted by using buffer A, and such property significantly facilitated the separation of the ADH from other proteins in the cell extract. Subsequently, the ADH activity was eluted out as a predominant single peak in the following chromatography. The purified ADH after the gel-filtration chromatography had a specific activity of 1,149 U mg−1, with a yield of 17% (Table 1). The native molecular mass was determined using gel filtration to be 135 ± 5 kDa. The SDS-PAGE analyses of the purified enzyme yielded a single band with a molecular mass of 40 ± 1 kDa (Fig. 3). Thus, the purified ADH appeared to be a homotetramer.
Table 1.
Purification of ADH from T. guaymasensis
| Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| Cell extract | 3718.5 | 1.2 × 105 | 33.7 | 1 | 100 |
| DEAE-sepharose | 446.8 | 6.3 × 104 | 142 | 4.2 | 52 |
| Hydroxyapatite | 58.7 | 5.7 × 104 | 970 | 28.8 | 47 |
| Phenyl-sepharose | 30.7 | 3.4 × 104 | 1,099 | 32.6 | 28 |
| Gel filtration | 17.4 | 2.0 × 104 | 1,149 | 34.1 | 17 |
Fig. 3.

SDS-PAGE (12.5%) of the purified ADH from T. guaymasensis. Lane 1, molecular markers; lane 2, 1.5 μg purified T. guaymasensis ADH.
Catalytical and physical properties of the purified T. guaymasensis ADH.
T. guaymasensis ADH was NADP+ dependent. The optimal pHs of the enzyme were found to be 10.5 for the 2-butanol oxidation and 7.5 for the 2-butanone reduction. The purified enzyme from T. guaymasensis was thermophilic, and its activity increased along with the elevated temperatures up to 95°C. The oxygen sensitivity of the enzyme was monitored by the residual activity after exposure to the air at room temperature. It was unexpected that the enzyme was oxygen sensitive, although it was much more resistant to oxidation than iron-containing ADHs. The time required to decrease 50% of the full activity upon exposure to the air (t1/2) was about 4 h, and such inactivation was slightly decreased in the presence of 2 mM dithiothreitol. The thermostability of the purified enzyme was investigated by determining its residual activities when the enzyme samples were incubated at 80 and 95°C. The t1/2 values at 95 and 80°C were determined to be 24 and 70 h, respectively, revealing its hyperthermostable feature.
The substrate specificity of the enzyme was determined using a set of alcohols, aldehydes, and ketones (Table 2). In the oxidation reactions, T. guaymasensis ADH was able to transform a broad range of primary alcohols but not oxidize methanol. Moreover, the enzyme showed higher activities with the use of secondary alcohols such as 2-butanol and 2-pentanol as substrates, suggesting that the enzyme is a primary-secondary ADH. T. guaymasensis ADH could oxidize diols but not primary diols such as ethanediol, 1,4-butanediol, and 1,5-pentanediol as tested. The enzyme showed a very low activity toward polyols such as glycerol and had no activity on l-serine and l-threonine. In the reduction reactions, the enzyme exhibited the ability to reduce various aldehydes and ketones. In particular, T. guaymasensis ADH could not oxidize acetoin to diacetyl, indicating that the reduction of diacetyl to acetoin is irreversible.
Table 2.
Substrate specificity of T. guaymasensis ADH
| Substrate | Relative activity (%) |
|---|---|
| Alcohols (50 mM) | |
| Methanol | 0 |
| Ethanol | 5.7 ± 0.3 |
| 1-Propanol | 15.1 ± 0.3 |
| 1-Butanol | 5.1 ± 0.2 |
| 1-Pentanol | 0.8 ± 0.2 |
| Glycerol | 0.2 ± 0.1 |
| 2-Propanol | 88 ± 1.1 |
| 2-Butanol | 100 ± 2.2a |
| 2-Pentanol | 66.3 ± 1.1 |
| 1,2-Butanediol | 25.6 ± 2.3 |
| 1,3-Butanediol | 35.4 ± 1.1 |
| 1,4-Butanediol | 0 |
| 2,3-Butanediol | 78.7 ± 3.4 |
| 1,2-Pentanediol | 4.7 ± 0.3 |
| 1,5-Pentanediol | 0 |
| 2,4-Pentanediol | 9.2 ± 0.1 |
| Ethanediol | 0 |
| Phenylethanol | 0 |
| l-Serine | 0 |
| l-Threonine | 0 |
| Acetoin | 0 |
| Aldehydes or ketones (6 mM) | |
| Acetone | 149.4 ± 2.8 |
| 2-Butanone | 100 ± 3.4b |
| 2-Pentanone | 86.2 ± 3.1 |
| Acetoin | 93 ± 3.2 |
| Diacetyl | 134.8 ± 5.9 |
| Acetaldehyde | 36 ± 4.2 |
| Butyraldehyde | 112.4 ± 11.9 |
A relative activity of 100% in alcohol oxidation is 1,144 ± 24 U mg−1.
A relative activity of 100% in aldehyde/ketone reduction is 223 ± 7.6 U mg−1.
The apparent Km value for the coenzyme NADPH was over 40 times lower than that for the coenzyme NADP+ (Table 3). The specificity constant kcat/Km for NADPH as an electron donor in the ketone reduction (14,363,000 s−1 M−1) was about 4.3 times higher than that of the NADP+ electron acceptor in the oxidation of corresponding alcohol (3,333,000 s−1 M−1). These catalytic properties suggest that the enzyme could play an important role in the oxidation of NADPH rather than the reduction of NADP+ in vivo. However, the apparent Km value for 2-butanone (0.31 mM) was close to that of 2-butanol (0.38 mM), and the specificity constant kcat/Km for 2-butanone (613,000 s−1 M−1) was about one-third of that for 2-butanol (2,192,000 s−1 M−1). To catalyze the reduction of diacetyl to 2,3-butanediol via acetoin, the enzyme had higher catalytic efficiency for diacetyl and lower catalytic efficiency for 2,3-butanediol, suggesting its possible roles involving the reduction of diacetyl to acetoin or 2,3-butanediol (Table 3).
Table 3.
Kinetic parameter values for T. guaymasensis ADH
| Substrate (mM) | Cosubstrate (mM) | Apparent Km (mM) | Apparent Vmax (U/mg) | kcat (s−1) | kcat/Km (s−1 M−1) |
|---|---|---|---|---|---|
| 2-Butanol | NADP+ (0.4) | 0.38 | 1,250 | 833 | 2,192,000 |
| (2R,3R)-(−)-2,3-Butanediola | NADP+ (0.4) | 15.2 | 1,111 | 740 | 49,000 |
| (2S,3S)-(+)-2,3-Butanediola | NADP+ (0.4) | 246 | 769 | 512 | 2082 |
| meso-2,3- Butanediola | NADP+ (0.4) | 19.3 | 1,428 | 952 | 49,000 |
| NADP+ | 2-butanol (11) | 0.4 | 2,000 | 1,333 | 3,333,000 |
| 2-Butanone | NADPH (0.2) | 0.31 | 285 | 190 | 613,000 |
| R/S-Acetoin | NADPH (0.2) | 0.32 | 213 | 142 | 444,000 |
| Diacetyl | NADPH (0.2) | 0.21 | 303 | 202 | 962,000 |
| NADPH | 2-Butanone (3) | 0.011 | 237 | 158 | 14,363,000 |
Various concentrations for (2R,3R)-(−)-2,3-butanediol (0, 2.7, 5.4, 8.1, 10.7, 16, 21.2, 26.5, and 51.7 mM), (2S,3S)-(+)-2,3-butanediol (0, 11, 22, 27.5, 44, 55, 82.5, and 110 mM), and meso-2,3-butanediol (0, 2.7, 5.4, 8.1, 10.7, 16, 21.2, 26.5, and 51.7 mM) were used for the determination of kinetic parameter values.
Effect of metals on the activity of T. guaymasensis ADH and its metal content.
Since the enzyme was a medium-chain ADH rather than a short-chain ADH lacking metal, it was speculated that the enzyme could contain either zinc or iron. The ICP-MS analyses showed that the purified enzyme contained 0.9 ± 0.03 g-atoms of zinc per subunit. Therefore, each subunit of the enzyme could contain 1 g zinc per subunit. In addition, the enzyme contained a trace amount of nickel (<0.01 g-atom per subunit) and 0.1 g-atom of iron per subunit, which could be exogenous.
The effect of various cations on the enzyme activity was examined by adding selected chemical (1 mM) into each enzyme assay mixture. EDTA slightly decreased the enzyme activity, indicating that zinc was resistant to the chelation by EDTA. The cations such as Ni2+ and Fe2+ decreased the enzyme activity by 10 to 20% while Co2+, Ca2+, Mg2+, and Mn2+ slightly increased the activity (not more than 15%). The enzyme was completely inhibited by 1 mM Zn2+, Cu2+, Hg2+, and Cd2+ in assay mixtures. It was unexpected that the zinc-containing ADH was inhibited by zinc ion. Thus, the inhibition by zinc was further confirmed by reducing the concentration of zinc in the assay mixture. When the zinc concentrations were 10, 25, and 100 μM in the assay mixtures, the corresponding activities were approximately 90%, 85%, and 3% of the full activity, respectively.
Reduction of 2-butanone coupled with NADPH regeneration.
Enzymes resistant to solvent inactivation are of great interest from scientific and practical points of view. The solvent tolerance of T. guaymasensis ADH on the oxidation of 2-butanol and the reduction of 2-butanone was examined by adding methanol into the assay mixtures. The purified enzyme did not oxidize methanol. The enzyme retained almost its full activity when the concentration of methanol was up to 5% (vol/vol). When the concentration of methanol was 40% (vol/vol) in the assay mixture, the enzyme activities remained at 30% and 15% of the full activity on both oxidation and reduction, respectively, indicating that the enzyme had outstanding solvent tolerance.
The solvent-tolerant feature of T. guaymasensis ADH made it feasible to regenerate the coenzyme NADPH using the cosubstrate isopropanol in excess amounts (500 mM). A gas chromatography method was developed to determine the reactants which had the following retention times: 1.33 min for isopropanol, 2.26 min for 2-butanone, and 2.51 min for 2-butanol. Driven by isopropanol (500 mM), the addition of 50 mM 2-butanone could result in the production of 45.6 mM 2-butanol, while the control without the addition of isopropanol produced 2-butanol in a low concentration similar to that of the coenzyme added (1 mM). The yield was about 91%. Similar results were also obtained when 2-pentanone was used to replace 2-butanone in the same reaction system.
Stereoselectivity of T. guaymasensis ADH.
To investigate the stereoselectivity of T. guaymasensis ADH, two methods based on a GC equipped with a chiral column were developed to efficiently separate the substrates and products of the reactions and the transformations between acetoin and 2,3-butanediol or between 2-butanone and 2-butanol. In the interconversion between acetoin and 2,3-butanediol, the retention times of their isomers were 3.6 min for (R)-2-acetoin, 4.2 min for (S)-2-acetoin, 9.1 min for (2S,3S)-(+)-2,3-butanediol, 9.4 min for (2R,3R)-(−)-2,3-butanediol, and 10.2 min for meso-2,3-butanediol (24). The enzyme showed higher oxidation activities on the (2R,3R)-(−)-2,3-butanediol and meso-2,3-butanediol than on the (2S,3S)-(+)-2,3-butanediol (Table 4), indicating that the enzyme predominantly oxidized the R-hydroxyl group of 2,3-butanediol and minorly functioned on the S-hydroxyl group. When meso-2,3-butanediol was oxidized, (S)-2-acetoin was the predominant product, with an enantiomeric excess (ee) of 88%, while oxidation of (2R,3R)-2,3-butanediol resulted in the production of (R)-2-acetoin with higher stereoselectivity (94% ee). With respect to the reduction reaction, the racemic R/S-acetoin was used since neither R- nor S-acetoin was commercially available. Consistently, the reduction of racemic R/S-acetoin formed (2R,3R)-2,3-butanediol (presumably from R-acetoin) and meso-2,3-butanediol (presumably from S-acetoin) with extremely high stereoselectivity [>99% ee over (2S,3S)-(+)-2,3-butanediol]. In addition, the kinetic parameters also confirmed that the enzyme had a higher Km value for (2S,3S)-(+)-2,3-butanediol than for (2R,3R)-(−)-2,3-butanediol or meso-2,3-butanediol (Table 3).
Table 4.
Stereoselectivity of T. guaymasensis ADH in 2,3-butanediol/acetoin interconversion
| Direction and substrate | Sp act (U/mg) | Product(s) | Stereoselectivity (eepa) (%) | Yield (%) |
|---|---|---|---|---|
| Oxidation | ||||
| meso-2,3-Butanediol | 926 ± 25 | (S)-2-Acetoin | 84 ± 3 | 69 ± 2 |
| (2R,3R)-(−)-2,3-Butanediol | 905 ± 30 | (R)-2-Acetoin | 94 ± 3 | 73 ± 2 |
| (2S,3S)-(+)-2,3-Butanediol | 158 ± 5 | NDb | ND | ND |
| Reduction [racemic (R/S)-acetoin] | 93 ± 3.2 | (2R,3R)-2,3-Butanediol, meso-2,3-butanediolc | >99d | 80 ± 1 |
eep, product enantiomeric excess value.
ND, not determined.
The ratio of (2R,3R)-2,3-butanediol to meso-2,3-butanediol was approximately 1:1.
The amount of (2S,3S)-(+)-2,3-butanediol was not detectable.
With respect to the reduction of 2-butanone to 2-butanol, the retention times of products on gas chromatography profiles were 6.5 min for R-2-butanol and 6.85 min for S-2-butanol. The products from reduction of 2-butanone showed the equal compositions of R-2-butanol and S-2-butanol, and T. guaymasensis ADH showed almost identical activities on the oxidation of R-2-butanol and S-2-butanol, indicating that the enzyme does not possess stereoselectivity on 2-butanol or 2-butanone as the substrate.
Cloning and sequence analyses of the gene encoding T. guaymasensis ADH.
To sequence the gene encoding the ADH, the amino-terminal (N-terminal) sequence was determined using the Edman degradation, SKMRGFAMVDF, which starts from S (serine), indicating the presence of N-terminal methionine excision after translation. Three internal sequences were successfully identified using mass spectrometry. Those amino acid sequences were aligned to determine conserved regions where the primers were fitted in. With a combination of PCR and inverse PCR strategies, the gene encoding T. guaymasensis ADH was fully sequenced. The structural gene encoding T. guaymasensis ADH consisted of 1,095 bp and ended at two consecutive stop codons (TGA and TAA). One putative archaeal terminator sequence, TTTTTCT, was found 24 bases farther downstream of the stop codon of TGA (54).
The deduced amino acid sequence of T. guaymasensis ADH contained 365 amino acid residues; however, the native enzyme had only 364 amino acid residues due to the lack of the initial methionine, corresponding to a calculated size of 39.4 kDa. The deduced amino acid sequence of T. guaymasensis ADH showed high overall identities to threonine dehydrogenase (TDH) or zinc-containing ADHs from thermophilic bacteria, e.g., ADHs from T. tengcongensis MB4 (77% identity; AAM23957), T. brockii (77% identity; CAA46053), T. ethanolicus ATCC 33223 (77% identity; EAO63648), T. ethanolicus X514 (76% identity; EAU57308), Thermosinus carboxydivorans Nor1 (72% identity; EAX46383), and the mesophile C. beijerinckii (67% identity; EAX46383). No hits (>30% identity) that correspond to T. guaymasensis ADH have been found in other Thermococcales genomes to date.
T. guaymasensis ADH and its homologues harbored the highly conserved amino acid residues (Fig. 4). The putative active site and NADP+ binding motifs were identified to be G63H64E65X2G68X5G74X2V77 and G184XG186XXG189, respectively (34). Consistent with metal analyses, no structural zinc-binding motif was observed. The three-dimensional (3-D) modeling of the structure indicated that the monomer of T. guaymasensis ADH folded into two domains, one catalytic domain closing to the amino-terminal end and one NADP+-binding domain closing to the C-terminal end. Both domains were separated by the cleft where the active site of the enzyme might be situated.
Fig. 4.

Alignment of sequence of T. guaymasensis ADH and other related zinc-containing ADHs. The sequences were aligned using Clustal W (64). Highlights in shadow, putative binding sites of catalytic zinc; highlights in box, putative motif of cofactor binding. TgADH, T. guaymasensis ADH; TbADH, T. brockii ADH; CbADH, C. beijerinckii ADH. Symbols indicate residues or nucleotides that are identical in all sequences in the alignment (*), conserved substitutions (:), semiconserved substitutions (.), or no corresponding amino acid (–).
DISCUSSION
For hyperthermophilic archaea, a few zinc-containing ADHs have recently been purified and characterized. They are either ADHs from the aerobic hyperthermophilic archaea S. solfataricus and A. pernix or TDHs from the anaerobic hyperthermophiles P. furiosus, T. kodakaraensis, and P. horikoshii (Table 5). Similar to other zinc-containing ADHs or TDHs, ADH from the anaerobic hyperthermophile T. guaymasensis contained 364 amino acid residues and thereby was a member of medium-chain ADHs. The native T. guaymasensis ADH was in the quaternary structure of the homotetramer, which is a usual structural characteristic of zinc-containing ADHs in archaea and bacteria. The hyperthermophilic ADHs, including T. guaymasensis ADH, showed that the optimum pH for the oxidation reaction was more alkaline than that for the reduction reaction. T. guaymasensis ADH was specific for NADP+ as a coenzyme, whereas other known hyperthermophilic zinc-containing ADHs or TDHs preferred NAD+ as a coenzyme. The monomer of zinc-containing ADHs from hyperthermophiles contained catalytic and structural zinc atoms, except that T. guaymasensis ADH contained 1 g-atom of zinc per subunit. Its amino acid sequence possessed no binding motif of structural zinc but catalytic zinc, thus indicating that its zinc atom was highly likely to play a catalytic role. Its sequence alignment also showed that the enzyme had high similarities to those NADP+-dependent ADHs containing catalytic zinc only, e.g., ADHs from T. brockii and T. ethanolicus.
Table 5.
Properties of zinc-containing ADHs from hyperthermophilic archaea
| Species | Enzyme | arCOG no.a | Structure | Polypeptide length (aa) | Coenzyme | Sp act (U/mg) | Optimal temp (°C) | Optimal pH(s) | Types of zinc | Source or reference(s) |
|---|---|---|---|---|---|---|---|---|---|---|
| T. guaymasensis | ADH | Homotetramer | 364 | NADP+ | 1,149 | >95 | 10.5e/7.5f | Putatively catalytic | This work | |
| S. solfataricus | ADH | arCOG01455 | Homotetramerb | 347 | NAD+ | 5.3 | >95 | 9.0e/5.5f | Catalytic and structural | 2, 55 |
| A. pernix | ADH | arCOG01455 | Homotetramer | 359 | NAD+ | 1.1 | >95 | 10.5e/8.0f | Catalytic and structural | 25, 28 |
| P. furiosus | TDH | arCOG01459 | Homotetramer | 348 | NAD+ | 10.3 | >100 | 6.6e | Putatively catalytic and structural | 45 |
| P. horikoshii | TDH | arCOG01459 | Homotetramerb | 348 | NAD+ | 1,750c | NAd | 7.5e | Catalytic and structural | 26, 27, 30 |
| T. kodakaraensis | TDH | arCOG01459 | Homotetramerb | 350 | NAD+ | 7.26 | 90 | 12e | Putatively catalytic and structural | 5, 11 |
The enzyme from T. guaymasensis possesses several outstanding features to be a competitive biocatalyst. The enzyme was active within a broad temperature range from 30 to 95°C as tested while the optimal temperature was over 95°C, which feature is common for ADHs originated from hyperthermophiles. The thermoactivity with 1,149 U mg−1 at 80°C was remarkably higher than that for other zinc-containing ADHs characterized except the TDH from P. horikoshii. The activity of the butanediol dehydrogenase (BDH) from Saccharomyces cerevisiae was reported to be 968 U mg−1 (24), and this dehydrogenase was obviously not as thermostable as T. guaymasensis ADH. The latter was hyperthermostable, and its t1/2 at 95°C was about 24 h, so this dehydrogenase is the most thermostable among the family of zinc-containing ADHs. The enzyme had broad substrate specificity. In the oxidation direction, the enzyme transformed various alcohols, including primary and secondary alcohols, polyols, and diols, while it reduced various aldehydes and ketones in the reduction direction. In the substrate spectrum of T. guaymasensis ADH, some of the chiral alcohols have served as valuable intermediates for synthesis of biologically or pharmacologically active compounds. (R)-1,3-Butanediol is the important intermediate in the synthesis of penem and carbapenem antibiotics (47), while acetoin and related α-hydroxy ketones have been used as building blocks for the synthesis of various pharmaceuticals, such as antidepressants, antifungal agents, and antitumor antibiotics (49). When the methanol was used to test the solvent tolerance, the methanol concentration at which half of the full activity remained was about 24% (vol/vol) in the assay mixture. The solvent tolerance of T. guaymasensis ADH is of great interest in that it offers an option to regenerate NADPH with the cheaper cosubstrate isopropanol instead of enzymes such as formate dehydrogenase or glucose dehydrogenase. The NADPH regeneration system led to production of 45.6 mM butanol from 50 mM butanone with the use of only 1 mM NADPH, with a yield up to 91%, indicating that this is a successful example for the NADPH regeneration system.
Coupled to the NADPH regeneration using isopropanol, the enzyme showed asymmetric reduction of racemic acetoin, in which only (2R,3R)-2,3-butanediol and meso-butanediol were produced. The enzyme also showed asymmetric oxidation of 2,3-butanediol isomers, in which it had much lower specificity constant on (2S,3S)-(+)-2,3-butanediol than (2R,3R)-2,3-butanediol and meso-2,3-butanediol. Regarding the stereoselectivity of T. guaymasensis ADH, the highly similar example was the butanediol dehydrogenase from S. cerevisiae (21). Selective oxidation of vicinal diols could be exploited to produce enantiopure α-hydroxy ketones. A diacetyl reductase from Bacillus stearothermophilus could selectively oxidize vicinal diols to α-hydroxy ketones with high enantioselectivity (>99%), but the yields were quite low (<23%) (10). Obeying the anti-Prelog rule, T. guaymasensis ADH might undergo transference of a hydride ion from an R-configured alcohol to the pro-R face of NADP+ or transference of a hydride ion from the pro-R face of NADPH to the si face of a carbonyl group of a ketone (51). The anti-Prelog ADHs were of greater interest since they are not as abundant as Prelog ADHs like those in horse liver, T. brockii, P. furiosus, and S. solfataricus. Since T. guaymasensis ADH shared high similarity to the ADHs from T. brockii and T. ethanolicus, it was not expected that the enzyme had no stereoselectivity on the reduction of 2-butanone, which property has been well characterized for ADHs from T. brockii and T. ethanolicus (32, 69). In addition, the enzyme could not catalyze the oxidation of l-threonine and l-serine. It was recently noted that the ADH from P. furiosus showed higher enantioselectivity on phenyl-substituted keto esters than the substrates lacking phenyl groups (70). The stereoselectivity of T. guaymasensis ADH might be also affected by side groups of carbonyl group, in particular, a larger side group.
The enzyme was oxygen sensitive. The reports of ADH oxygen inactivation were usually associated with iron-containing ADHs but not zinc-containing ADHs. The well-known example is the zinc-containing ADH from mesophilic S. cerevisiae whose inactivation was due to the oxidation of the SH group (13). Since the zinc ion cannot be oxidized further, the inactivation of the enzyme may also be a consequence of the damage of amino acid residues such as cysteine. The enzyme of T. guaymasensis ADH had 4 cysteine residues per subunit (Cys39, Cys56, Cys213, and Cys306). Except Cys56, all of these residues were conserved in T. brockii ADH. Cys39 was highly conserved in zinc-containing ADHs and a putative active site residue, which has been proved to coordinate the binding of catalytic zinc in T. brockii ADH. The residue Cys56 was unique in T. guaymasensis ADH and did not exist in the same location of any other zinc-containing ADH sharing high similarity, so site-directed mutagenesis at the Cys56 residue would shed light on the role of Cys56 in oxygen sensitivity.
The N-terminal amino acid sequence determination showed that serine was the initial amino acid of mature T. guaymasensis ADH. N-terminal methionine was excised in the mature enzyme of T. guaymasensis ADH, which might be governed by the side chain length of the penultimate amino acid (29). As observed in bacteria and yeasts, N-terminal methionine excision of an enzyme could be critical for its function and stability (18), but its role for archaeal enzymes is not clear yet. Amino acid composition and its substitution patterns between mesophilic and hyperthermophilic proteins shed light on the understanding of common features of thermostability (56, 63). T. guaymasensis ADH showed 77% identity to T. brockii ADH and 65% identity to C. beijerinckii ADH, suggesting that gains in stabilization might be achieved in regions that are less conserved (35). The uncharged polar residues Gln, Asn, and Ser decreased in T. guaymasensis ADH, in which the first two are prone to deamidation and known to be the most temperature sensitive (14, 67). In contrast, hyperthermophilic and thermophilic proteins showed increases of charged amino acid residues, especially Arg and Glu. The equal increases of oppositely charged residues (Arg and Glu) in hyperthermophiles most likely led to the increased amounts of ion pairs already observed on their proteins (14). As the best helix-forming residue, alanine increased, similarly to what was found in previous observations (66). On the other hand, proline composition increased significantly, which might be the structural base of the rigidity of hyperthermophilic enzymes (23). The difference between hyperthermophilic and mesophilic proteins/enzymes would provide clues for increasing the thermal stability of mesophilic enzymes (8, 9).
The physiological role of T. guaymasensis ADH seems not to be clearly ascertained, simply relying on its conserved domain homology to threonine dehydrogenase. T. guaymasensis ADH did not catalyze the oxidation of threonine as tested at different pHs (pH 7.5, 8.8, and 10.5). The possible physiological role of T. guaymasensis ADH might arise from its coenzyme preference and ability of interconversion between alcohols and corresponding ketones or aldehydes. T. guaymasensis ADH utilized NADP(H) as coenzymes but not NAD(H). In T. kodakaraensis, the generation of NADPH was coupled with oxidation of glutamate and reduced ferredoxin (21), whereas T. guaymasensis ADH had higher catalytic efficiency on NADPH than on NADP+ as a coenzyme, suggesting that its role was more likely to be associated with NADPH-oxidizing metabolisms. Furthermore, T. guaymasensis ADH reversibly catalyzed the oxidation of 2,3-butanediol to acetoin, which cannot be oxidized to diacetyl. Regarding this feature, the properties of T. guaymasensis ADH, including its stereoselectivity, were very similar to those observed on the (2R,3R)-(−)-butanediol dehydrogenase (BDH) from S. cerevisiae (24). S. cerevisiae can grow on 2,3-butanediol as the sole carbon and energy source, in which BDH content had >3-fold increases. The role of BDH in S. cerevisiae was suggested to be required for oxidation and formation of 2,3-butanediol. However, no production of 2,3-butanediol was observed in the spent culture medium of T. guaymasensis, implying that T. guaymasensis ADH might be more likely to be involved in the formation of acetoin from diacetyl, which may be formed either enzymatically or spontaneously from α-acetolactate (Fig. 5). The production of α-acetolactate can be catalyzed by α-acetolactate synthase, which was found to be present in T. guaymasensis (M. S. Eram and K. Ma, unpublished results). There was no homolog of α-acetolactate decarboxylase identified in any Thermococcus genome sequence available, nor was such activity reported for these organisms (Eram and Ma, unpublished). It is intriguing to investigate whether and how diacetyl would be produced from acetolactate in the absence of oxygen at high temperatures.
Fig. 5.

Schematic pathways of alcohol formation in T. guaymasensis. ADH, alcohol dehydrogenase (this study); ALDC, α-acetolactate decarboxylase (7); ALDH, CoA-dependent aldehyde dehydrogenase (68); ALS, α-acetolactate synthase (Eram and Ma, unpublished); PDC, pyruvate decarboxylase (41); POR, pyruvate ferredoxin oxidoreductase (Eram and Ma, unpublished). The dashed arrows represent speculated steps, while the solid arrows represent the steps approved by activities of purified enzymes.
In addition, T. guaymasensis produced ethanol at the mM level, which would be the result from the reduction of acetaldehyde. The production of acetaldehyde would be catalyzed by either pyruvate decarboxylase (PDC) or the combination of pyruvate ferredoxin reductase (POR) and coenzyme A (CoA)-dependent aldehyde dehydrogenase (ALDH). Activities of both PDC and POR but not ALDH were found to be present in T. guaymasensis (Eram and Ma, unpublished). Thus, the physiological role of the enzyme was proposed to be concurrently responsible for ethanol and acetoin formation concomitant with the NADPH oxidation during glucose fermentation (Fig. 5), where NADP+ reduction to NADPH may be catalyzed by a ferredoxin NADP+ oxidoreductase (40) from reduced ferredoxin whose production is catalyzed by POR and glyceraldehyde-3-phosphate ferredoxin oxidoreductase (48).
ACKNOWLEDGMENTS
This work was supported by research grants from the Natural Sciences and Engineering Research Council (Canada) and the Canada Foundation for Innovation to K.M.
We thank Liangliang Hao for assistance in sequencing the gene.
Footnotes
Published ahead of print on 22 April 2011.
REFERENCES
- 1. Altschul S. F., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ammendola S., et al. 1992. Thermostable NAD+-dependent alcohol dehydrogenase from Sulfolobus solfataricus: gene and protein sequence determination and relationship to other alcohol dehydrogenases. Biochemistry 31:12514–12523 [DOI] [PubMed] [Google Scholar]
- 3. Antoine E., Rolland J. L., Raffin J. P., Dietrich J. 1999. Cloning and over-expression in Escherichia coli of the gene encoding NADPH group III alcohol dehydrogenase from Thermococcus hydrothermalis. Eur. J. Biochem. 264:880–889 [DOI] [PubMed] [Google Scholar]
- 4. Balch W. E., Fox G. E., Magrum L. J., Woese C. R., Wolfe W. S. 1979. Methanogens: re-evaluation of a unique biological group. Microbiol. Rev. 43:260–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bashir Q., Rashid N., Jamil F., Imanaka T., Akhtar M. 2009. Highly thermostable l-threonine dehydrogenase from the hyperthermophilic archaeon Thermococcus kodakaraensis. J. Biochem. 146:95–102 [DOI] [PubMed] [Google Scholar]
- 6. Bertoldo C., Antranikian G. 2006. The order Thermococcales. Prokaryotes 3:69–81 [Google Scholar]
- 7. Blomqvist K., et al. 1993. Characterization of the genes of the 2,3-butanediol operons from Klebsiella terrigena and Enterobacter aerogenes. J. Bacteriol. 175:1392–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bogin O., et al. 2002. Structural basis for the enhanced thermal stability of alcohol dehydrogenase mutants from the mesophilic bacterium Clostridium beijerinckii: contribution of salt bridging. Protein Sci. 11:2561–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bogin O., et al. 1998. Enhanced thermal stability of Clostridium beijerinckii alcohol dehydrogenase after strategic substitution of amino acid residue with prolines from the homologous thermophilic Thermoanaerobacter brockii alcohol dehydrogenase. Protein Sci. 7:1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bortolini O., et al. 1998. Kinetic resolution of vic-diols by Bacillus stearothermophilus diacetyl reductase. Tetrahedron Asymmetry 9:647–651 [Google Scholar]
- 11. Bowyer A., et al. 2009. Structure and function of the l-threonine dehydrogenase (TkTDH) from the hyperthermophilic archaeon Thermococcus kodakaraensis. J. Struct. Biol. 168:294–304 [DOI] [PubMed] [Google Scholar]
- 12. Bradford M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 13. Bühner M., Sund H. 1969. Yeast alcohol dehydrogenase: SH groups, disulfide groups, quaternary structure, and reactivation by reductive cleavage of disulfide groups. Eur. J. Biochem. 11:73–79 [DOI] [PubMed] [Google Scholar]
- 14. Cambillau C., Claverie J. M. 2000. Structural and genomic correlates of hyperthermostability. J. Biol. Chem. 275:32383–32386 [DOI] [PubMed] [Google Scholar]
- 15. Canganella F., Jones W. J., Gambacorta A., Antranikian G. 1998. Thermococcus guaymasensis sp. nov. and Thermococcus aggregans sp. nov., two novel thermophilic archaea isolated from the Guaymas Basin hydrothermal vent site. Int. J. Syst. Bacteriol. 48:1181–1185 [DOI] [PubMed] [Google Scholar]
- 16. Chong P. K., Burja A. M., Radianingtyas H., Fazeli A., Wright P. C. 2007. Translational and transcriptional analysis of Sulfolobus solfataricus P2 to provide insights into alcohol and ketone utilization. Proteomics 7:424–435 [DOI] [PubMed] [Google Scholar]
- 17. Delano W. L. 2002. The PyMOL molecular graphics system. Delano Scientific, Palo Alto, CA [Google Scholar]
- 18. Eichler J., Adams M. W. W. 2005. Posttranslational protein modification in Archaea. Microbiol. Mol. Biol. Rev. 69:393–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Esposito L., et al. 2003. Structural study of a single-point mutant of Sulfolobus solfataricus alcohol dehydrogenase with enhanced activity. FEBS Lett. 539:14–18 [DOI] [PubMed] [Google Scholar]
- 20. Esposito L., et al. 2002. Crystal structure of the alcohol dehydrogenase from the hyperthermophilic archaeon Sulfolobus solfataricus at 1.85 Å resolution. J. Mol. Biol. 318:463–477 [DOI] [PubMed] [Google Scholar]
- 21. Fukui T., et al. 2005. Complete genome sequence of the hyperthermophilic archaeon Thermocococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res. 15:352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gasteiger E., et al. 2005. Protein identification and analysis tools on the ExPASy Server, p. 571–607 In Walker J. M. (ed.), The proteomics protocols handbook. Humana Press, Totowa, NJ [Google Scholar]
- 23. Goihberg E., et al. 2007. A single proline substitution is critical for the thermostabilization of Clostridium beijerinckii alcohol dehydrogenase. Proteins 66:196–204 [DOI] [PubMed] [Google Scholar]
- 24. González E., et al. 2000. Characterization of a (2R,3R)-2,3-butanediol dehydrogenase as the Saccharomyces cerevisiae YAL060W gene product. J. Biol. Chem. 275:35876–35885 [DOI] [PubMed] [Google Scholar]
- 25. Guy J. E., Isupov M. N., Littlechild J. A. 2003. The structure of an alcohol dehydrogenase from the hyperthermophilic archaeon Aeropyrum pernix. J. Mol. Biol. 331:1041–1051 [DOI] [PubMed] [Google Scholar]
- 26. Higashi N., Fukada H., Ishikawa K. 2005. Kinetic study of thermostable l-threonine dehydrogenase from an archaeon Pyrococcus horikoshii. J. Biosci. Bioeng. 99:175–180 [DOI] [PubMed] [Google Scholar]
- 27. Higashi N., Matsuura T., Nakagawa A., Ishikawa K. 2005. Crystallization and preliminary X-ray analysis of hyperthermophilic l-threonine dehydrogenase from the archaeon Pyrococcus horikoshii. Acta Crystallogr. F 61:432–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hirakawa H., Kamiya N., Kawarabayashi Y., Nagamune T. 2004. Properties of an alcohol dehydrogenase from the hyperthermophilic archaeon Aeropyrum pernix K1. J. Biosci. Bioeng. 97:202–206 [DOI] [PubMed] [Google Scholar]
- 29. Hirel P. H., Schmitter J. M., Dessen P., Fayat G., Blanquet S. 1989. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. U. S. A. 86:8247–8251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishikawa K., Higashi N., Nakamura T., Matsuura T., Nakagawa A. 2007. The first crystal structure of l-threonine dehydrogenase. J. Mol. Biol. 366:857–867 [DOI] [PubMed] [Google Scholar]
- 31. Karakashev D., Thomsen A. B., Angelidaki I. 2007. Anaerobic biotechnological approaches for production of liquid energy carriers from biomass. Biotechnol. Lett. 29:1005–1012 [DOI] [PubMed] [Google Scholar]
- 32. Keinan E., Seth K. K., Lamed R. 1986. Organic synthesis with enzymes. 3. TBADH-catalyzed reduction of chloro ketones. Total synthesis of (+)-(S,S)-(cis-6-methyltetrahydropyran-2-yl)-acetic acid: a civet constituent. J. Am. Chem. Soc. 108:3473–3480 [Google Scholar]
- 33. Kopp J., Schwede T. 2004. The SWISS-MODEL repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res. 32:D230–D234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Korkhin Y., et al. 1998. NADP-dependent bacterial alcohol dehydrogenases: crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol. 278:967–981 [DOI] [PubMed] [Google Scholar]
- 35. Kumar S., Tsai C. J., Nussinov P. 2000. Factors enhancing protein thermostability. Protein Eng. 13:179–191 [DOI] [PubMed] [Google Scholar]
- 36. Laemmli U. K. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 37. Larroy C., Fernández M. R., González E., Parés X., Biosca J. A. J. A. 2002. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: relevance in aldehyde reduction. Biochem. J. 361:163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee H. S., et al. 2008. The complete genome sequence of Thermococcus onnurineus NA1 reveals a mixed heterotrophilic and carboxydotrophic metabolism. J. Bacteriol. 190:7491–7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li D., Stevenson K. J. 1997. Purification and sequence analysis of a novel NADP(H)-dependent type III alcohol dehydrogenase from Thermococcus strain AN1. J. Bacteriol. 179:4433–4437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma K., Adams M. W. W. 1994. Sulfide dehydrogenase from the hyperthermophilic archaeon, Pyrococcus furiosus: a new multifunctional enzyme involved in the reduction of elemental sulfur. J. Bacteriol. 176:6509–6517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma K., Hutchins A., Sung S. J. S., Adams M. W. W. 1997. Pyruvate ferrodoxin oxidoreductase from the hyperthermophilic archaeon, Pyrococcus furiosus, functions as a CoA-dependent pyruvate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 94:9608–9613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma K., Loessner H., Heider J., Johnson M. K., Adams M. W. W. 1995. Effects of elemental sulfur on the metabolism of the deep-sea hyperthermophilic archaeon Thermococcus strain ES-1: characterization of a sulfur-regulated, non-heme iron alcohol dehydrogenase. J. Bacteriol. 177:4748–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma K., Robb F. T., Adams M. W. W. 1994. Purification and characterization of NADP+-specific alcohol dehydrogenase and glutamate dehydrogenase from the hyperthermophilic archaeon Thermococcus litoralis. Appl. Environ. Microbiol. 60:562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Machielsen R., Uria A. R., Kengen S. W. M., van der Oost J. 2006. Production and characterization of a thermostable alcohol dehydrogenase that belongs to the aldo-keto reductase superfamily. Appl. Environ. Microbiol. 72:233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Machielsen R., van der Oost J. 2006. Production and characterization of a thermostable l-threonine dehydrogenase from the hyperthermophilic archaeon Pyrococcus furiosus. FEBS J. 273:2722–2729 [DOI] [PubMed] [Google Scholar]
- 46. Makarova K. S., Sorokin A. V., Novichkov P. S., Wolf U. I., Koonin E. V. 2007. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol. Direct 2:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matsuyama A., Yamamoto H., Kawada N., Kobayashi Y. 2001. Industrial production of (R)-1,3-butanediol by new biocatalysts. J. Mol. Catal. B Enzym. 11:513–521 [Google Scholar]
- 48. Mukund S., Adams M. W. W. 1995. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase, a novel tungsten-containing enzyme with a potential glycolytic role in the hyperthermophilic archaeon Pyrococcus furiosus. J. Biol. Chem. 270:8389–8392 [DOI] [PubMed] [Google Scholar]
- 49. Nestl B. M., et al. 2007. Biocatalytic racemization of synthetically important functionalized α-hydroxyketones using microbial cells. Tetrahedron Asymmetry 18:1465–1474 [Google Scholar]
- 50. Pikuta E. V., et al. 2007. Thermococcus thioreducens sp. nov., a novel hyperthermophilic, obligately sulfur-reducing archaeon from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 57:1612–1618 [DOI] [PubMed] [Google Scholar]
- 51. Prelog V. 1964. Specification of the stereospecificity of some oxido-reductases by diamond lattice sections. Pure Appl. Chem. 9:119–130 [Google Scholar]
- 52. Raia C. A., Giordano A., Rossi M. 2001. Alcohol dehydrogenase from Sulfolobus solfataricus. Methods Enzymol. 331:176–195 [DOI] [PubMed] [Google Scholar]
- 53. Reid M. F., Fewson C. A. 1994. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 20:13–56 [DOI] [PubMed] [Google Scholar]
- 54. Reiter W. D., Palm P., Zillig W. 1988. Transcription termination in the archaebacterium Sulfolobus: signal structures and linkage to transcription initiation. Nucleic Acids Res. 16:2445–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rella R., et al. 1987. A novel archaebacterial NAD+-dependent alcohol dehydrogenase: purification and properties. Eur. J. Biochem. 167:475–479 [DOI] [PubMed] [Google Scholar]
- 56. Saelensminde G., Halskau Ø., Helland R., Willassen N. P., Jonassen I. 2007. Structure-dependent relationships between growth temperature of prokaryotes and the amino acid frequency in their proteins. Extremophiles 11:585–596 [DOI] [PubMed] [Google Scholar]
- 57. Sambrook J., Fritsch E. F., Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, NY [Google Scholar]
- 58. Schwede T., Kopp J., Guex N., Peitsch M. C. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Selig M., Xavier K. B., Santos H., Schönheit P. 1997. Comparative analysis of Embden—Meyerhof and Entner—Doudoroff glycolytic pathways in hyperthermophilic archaea and the bacterium Thermotoga. Arch. Microbiol. 167:217–232 [DOI] [PubMed] [Google Scholar]
- 60. Shevchenko A., Wilm M., Vorm O., Mann M. 1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 61. Sokolova T. G., et al. 2004. The first evidence of anaerobic CO oxidation coupled with H2 production by a hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent. Extremophiles 8:317–323 [DOI] [PubMed] [Google Scholar]
- 62. Stekhanova T. N., et al. 2010. Characterization of a thermostable short-chain alcohol dehydrogenase from the hyperthermophilic archaeon Thermococcus sibiricus. Appl. Environ. Microbiol. 76:4096–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sterner R., Liebl W. 2001. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 36:39–106 [DOI] [PubMed] [Google Scholar]
- 64. Thompson J. D., Higgins D. G., Gibson T. J. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Triglia T., Peterson M., Kemp D. 1988. A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res. 16:8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vieille C., Zeikus G. J. 2001. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65:1–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wright H. T. 1991. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in proteins. Crit. Rev. Biochem. Mol. Biol. 26:1–52 [DOI] [PubMed] [Google Scholar]
- 68. Yan R., Chen J. 1990. Coenzyme A-acylating aldehyde dehydrogenase from Clostridium beijerinckii NRRL B592. Appl. Environ. Microbiol. 56:2591–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zheng C. S., Pham V. T., Phillips R. S. 1992. Asymmetric reduction of ketoesters with alcohol dehydrogenase from Thermoanaerobacter ethanolicus. Bio. Med. Chem. Lett. 2:619–622 [Google Scholar]
- 70. Zhu D., Malik H. T., Hua L. 2006. Asymmetric ketone reduction by a hyperthermophilic alcohol dehydrogenase: the substrate specificity, enantioselectivity and tolerance of organic solvents. Tetrahedron Asymmetry 17:3010–3014 [Google Scholar]