

Abstract
We demonstrate that mutation of the staphylococcal accessory regulator (sarA) limits the accumulation of alpha-toxin and phenol-soluble modulins (PSMs) in Staphylococcus aureus isolates of the USA300 clonal lineage. Degradation assays and experiments done with protease inhibitors suggested that this was due to the increased production of extracellular proteases rather than differences associated with the impact of sarA on transcription of the target gene (hla) or the accessory gene regulator (agr). This was confirmed by demonstrating that concomitant mutation of the gene encoding aureolysin (aur) reversed the alpha-toxin and PSM-deficient phenotypes of a USA300 sarA mutant. Mutation of sarA had little impact on the alpha-toxin or PSM phenotypes of the commonly studied strain Newman, which is known to have a mutation in saeS that results in constitutive activation of the saeRS regulatory system, and we also demonstrate that repair of this defect resulted in the increased production of extracellular proteases and reversed both the alpha-toxin and PSM-positive phenotypes of a Newman sarA mutant.
INTRODUCTION
Staphylococcus aureus is an adaptive bacterial pathogen capable of causing both chronic, biofilm-associated infection and acute, life-threatening toxemia. Based on a specific interest in musculoskeletal infection, much of our research has been directed toward defining the mechanistic basis for S. aureus biofilm formation, and these studies have led us to conclude that the staphylococcal accessory regulator (sarA) plays a primary role in this regard. Specifically, we have demonstrated that mutation of sarA in diverse clinical isolates, including those of the USA300 lineage of community-associated methicillin-resistant S. aureus (CA-MRSA), limits biofilm formation to a degree that can be correlated with increased susceptibility to functionally distinct classes of antibiotics under both in vitro and in vivo conditions (1, 2, 45, 47, 48). This suggests that sarA would be a viable target for the development of therapeutic agents capable of overcoming the intrinsic resistance of biofilm-associated infections. However, sarA has a global regulatory impact in S. aureus (7, 15), and this makes it imperative to fully define the role of sarA in other clinically relevant contexts as a necessary prelude to the development of such agents.
One of the most important of these contexts is toxin production, a primary example being alpha-toxin. Indeed, alpha-toxin has been shown to be an important virulence factor in many forms of S. aureus infection (20, 21, 27, 28, 30, 32, 36). This is potentially problematic in that we previously demonstrated that mutation of sarA also results in an apparent increase in the production of alpha-toxin in many strains of S. aureus (3). In fact, the only exception among the strains we examined in our earlier study was the 8325-4 strain RN6390. It was subsequently suggested that this discrepancy was due to the combined effect of the rsbU and/or tcaR mutations that are present in all strains derived from NCTC8325 and the impact of these mutations on the expression of sigB and sarS, respectively (35). This would suggest that the targeted inhibition of sarA in isolates that lack these mutations would result in the increased production of alpha-toxin. This would be particularly important given the increasing clinical prominence USA300 isolates (43), many of which produce alpha-toxin at high levels owing to their high-level expression of the accessory gene regulator (agr) (22). Indeed, this is one factor thought to contribute to the enhanced virulence of these isolates (13).
At the same time, we recently demonstrated that mutation of sarA in a USA300 strain (UAMS-1625) isolated from a patient with a fatal brain abscess (42) not only resulted in a reduced capacity to form a biofilm but also reduced production of alpha-toxin and a reduced capacity to cause skin lesions (48). This was surprising in that, while isolates of the USA300 clonal lineage are closely related to 8325 strains (14), this isolate does not have the sigB or tcaR defects that are characteristic of 8325 strains (2). This suggests that other factors contribute to the strain-dependent impact of sarA on the production of alpha-toxin. However, these experiments were limited to a single isolate (48), and at present, it is unclear whether the same sarA-dependent alpha-toxin phenotype is conserved among other USA300 isolates and, if so, whether this distinguishes these isolates from those of other contemporary clonal lineages of S. aureus. We examined all of these issues by generating sarA mutants in diverse clinical isolates of S. aureus and examining the impact on alpha-toxin and other critical virulence factors, including phenol-soluble modulins.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The S. aureus strains examined in this study are listed in Table 1. To facilitate the generation of certain mutants and their complemented derivatives, the plasmid conferring resistance to both erythromycin (Erm) and kanamycin (Kan)/neomycin (Neo) was cured from each of three USA300 isolates by growth in half-strength tryptic soy broth (TSB) at 42°C followed by plating on selective and nonselective media to assess loss of the plasmid and to ensure a level of hemolytic activity in the cured derivative comparable to the isogenic parent strain. Generation of sarA, agr, hla, and protease mutants was done by Φ11-mediated transduction from existing mutants (2, 3, 38, 41). Complementation of the sarA mutation was done as previously described (3). The Newman saeRS mutant (ΔsaeRS) and saeS-repaired Newman derivative [saeS(P18L)] were constructed by using the pKOR1 system as previously described (25).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmida | Description | Source or reference |
|---|---|---|
| Strains | ||
| UAMS-1 | Methicillin-susceptible S. aureus, osteomyelitis isolate | 16 |
| UAMS-929 | sarA::kan | 3 |
| UAMS-155 | agr::tetM | 3 |
| UAMS-930 | agr::tetM sarA::kan | 3 |
| UAMS-969 | sarA::kan (pSARA) | 3 |
| UAMS-970 | agr::tetM sarA::kan (pSARA) | 3 |
| UAMS-7 | S6C | 3 |
| UAMS-985 | sarA::kan | 3 |
| UAMS-128 | RN6390 (8325-4) | 34 |
| UAMS-983 | sarA::kan | 3 |
| UAMS-240 | sarA::tetK | 3 |
| UAMS-982 | agr::tetM | 3 |
| UAMS-959 | sarA::kanagr::tetM | 3 |
| UAMS-979 | sarA::kan (pSARA) | 3 |
| UAMS-980 | sarA::kanagr::tetM (pSARA) | 3 |
| UAMS-1639 | scpA::erm | 41 |
| UAMS-1640 | aur::erm | 41 |
| UAMS-1974 | spl::erm | 38 |
| UAMS-1975 | sspABC::erm | 41 |
| UAMS-200 | Newman | 29 |
| UAMS-988 | sarA::kan | 3 |
| UAMS-974 | agr::tetM | 3 |
| UAMS-975 | sarA::kan agr::tetM | 3 |
| UAMS-1907 | sarA::kan (pSARA) | This study |
| UAMS-1909 | sarA::kan agr::tetM (pSARA) | This study |
| UAMS-2189 | hla::erm | This study |
| UAMS-2190 | sarA::kanhla::erm | This study |
| UAMS-2167 | saeS(P18L) (CYL11481) | 25 |
| UAMS-2170 | saeS(P18L) sarA::kan | This study |
| UAMS-2200 | saeS(P18L) hla::erm | This study |
| UAMS-2212 | saeS(P18L) hla::erm sarA::kan | This study |
| UAMS-2225 | saeS(P18L) aur::erm | This study |
| UAMS-2226 | saeS(P18L) aur::erm sarA::kan | This study |
| UAMS-2166 | ΔsaeRS (CYL11771) | 25 |
| UAMS-2169 | ΔsaeRS sarA::kan | This study |
| UAMS-261 | SH1000 | 18 |
| UAMS-1762 | sarA::kan | This study |
| UAMS-732 | SC-01 | 44 |
| UAMS-310 | sarA::tetK | 3 |
| UAMS-1625 | USA300 isolate | 42 |
| UAMS-1653 | sarA::tetK | 48 |
| UAMS-1782 | USA300 isolate FPR3757 | 2 |
| UAMS-1804 | sarA::kan | 2 |
| UAMS-1819 | agr::tetM | 2 |
| UAMS-1837 | agr::tetMsarA::kan | 2 |
| UAMS-1901 | sarA::kan (pSARA) | 2 |
| UAMS-1904 | agr::tetMsarA::kan (pSARA) | 2 |
| UAMS-1790 | USA300 isolate | 2 |
| UAMS-1796 | sarA::tetK | 2 |
| UAMS-1794 | UAMS-1782, Erm-sensitive | This study |
| UAMS-1802 | sarA::kan | This study |
| UAMS-2192 | hla::erm | This study |
| UAMS-2193 | sarA::kanhla::erm | This study |
| UAMS-2205 | scpA::erm | This study |
| UAMS-2206 | scpA::erm sarA::kan | This study |
| UAMS-2218 | spl::erm | This study |
| UAMS-2219 | spl::erm sarA::kan | This study |
| UAMS-2220 | aur::erm | This study |
| UAMS-2221 | aur::erm sarA::kan | This study |
| UAMS-2222 | sspABC::erm | This study |
| UAMS-2223 | sspABC::erm sarA::kan | This study |
| UAMS-1893 | USA100 isolate, NRS642 | 2 |
| UAMS-1941 | sarA::tetK | 2 |
| UAMS-1898 | USA800 isolate, NRS653 | 2 |
| UAMS-1944 | sarA::tetK | 2 |
| UAMS-1899 | USA1000 isolate, NRS676 | 2 |
| UAMS-1930 | sarA::kan | 2 |
| UAMS-1900 | USA1100 isolate, NRS484 | 2 |
| UAMS-1931 | sarA::kan | 2 |
| UAMS-2172 | USA500, BD02-25 | 6 |
| UAMS-2174 | sarA::kan | This study |
| Plasmid | ||
| pSARA | 4 |
The prefix “UAMS” refers to strain designations in the corresponding author's culture collection. All UAMS strains are designated with the prefix “U” in the figures and figure legends.
All strains were maintained as stock cultures at −80°C in tryptic soy broth (TSB) containing 25% (vol/vol) glycerol. For each experiment, each strain was retrieved from cold storage by plating on tryptic soy agar (TSA) with appropriate antibiotic selection. Antibiotics were used at the following concentrations: erythromycin (Erm; 10 μg per ml), tetracycline (Tet; 5 μg per ml), kanamycin (Kan; 50 μg per ml), and neomycin (Neo; 50 μg per ml). Kanamycin and neomycin were always used together to avoid the spontaneous generation of resistant strains. For phenotypic assays, each strain was grown in TSB without antibiotic selection at 37°C with constant aeration and a medium-to-flask volume ratio of 0.40. Where appropriate, the postexponential growth phase was defined as an optical density at 560 nm (OD560) of 3.0, while stationary-phase samples were defined by overnight (16-h) growth.
In experiments employing the protease inhibitors E-64, 1-10-phenanthroline, and dichloroisocoumarin (DIC), which are specific inhibitors of cysteine, serine, and metalloproteases, respectively, each inhibitor was dissolved in dimethyl sulfoxide (DMSO) and added to TSB at final concentrations of 1 mM, 10 μM, and 0.1 mM (protease inhibitor formulation 1 [PI]) or 1 mM, 10 μM and 0.2 mM (protease inhibitor formulation 2 [PI2]), respectively. As a control, cultures were also grown in TSB containing the equivalent concentration of DMSO (0.7% [vol/vol]) without inhibitors. With the exception of RN6390, which did not grow in the presence of PI2, growth was unaffected by the inclusion of DMSO with or without protease inhibitors (data not shown).
Western blotting.
The production of alpha-toxin was assessed by Western blotting of standardized cell-free supernatants using rabbit polyclonal anti-alpha-toxin IgG antibody (Sigma Chemical Co., St. Louis, MO) as previously described (48). For quantitative comparisons, 50 ng of purified alpha-toxin (List Biological Laboratories, Campbell, CA) was included as an internal standard on each gel. The amount of signal observed with this standard was assessed with an Alpha Innotech Flourochem FC2 gel documentation system (Cell Biosciences, Santa Clara, CA) and Image J software analysis. Results observed with this standard were then set to 1.0, with all other results shown relative to this value. Western blots were blocked with 0.5% skim milk containing 0.1 mg/ml human IgG (Sigma Chemical Co., St. Louis, MO).
Transcriptional analysis.
To assess the levels of hla and RNAIII expression, total bacterial RNA was isolated using the Qiagen RNeasy minikit as previously described (3). Quantitative, real-time reverse transcription-PCR (qRT-PCR) was then performed using hla- or RNAIII-specific primers and a corresponding TaqMan probe (Table 2). Results were calibrated by comparison to the results obtained with the same RNA samples using primers and a TaqMan probe corresponding to a 16S rRNA gene (Table 2). Results are reported as relative units by comparison to the results observed with the lowest sample in any given experiment, with the latter being set to a value of 1.0.
Table 2.
PCR primers and probes used in this study
| Primer or probe | Oligonucleotide sequence (5′→3′) |
|---|---|
| RNAIII-F | AGC CAT CCC AAC TTA ATA ACC ATG T |
| RNAIII-R | GAT GTT GTT TAC GAT AGC TTA CAT GCT AGA |
| RNAIII-Probe | AGT AGA GTT AGT TTC CTT GGA CTC AGT GCT |
| hla-F | ACA ACA CTA TTG CTA GGT TCT ATA |
| hla-R | TGT AGT ATT GCT TCC TAT ATC TGT A |
| hla-Probe | ATG AAT CCT GTC GCT AAT GCC GCA |
| 16S-F | TGA GAT GTT GGG TTA AGT CCC GCA |
| 16S-R | CGG TTT CGC TGC CCT TTG TAT TGT |
| 16S-Probe | AGC GCA ACC CTT AAG CTT AGT TGC CA |
| saeR7 | GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC ACG TCA TCT TCG GCG GCG CT |
| saeR8-1 | TCC AGA ACC ACC CGG CGG CCG CTT CAT CAT CCA CGA TCA GT |
| saeS9KO | ATC GTG GAT GAT GAA GCG GCC GCC GGG TGG TTC TGG ATT AGG C |
| saeS8 | GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC AAA GCG TGA TAG AAG TGG TGA |
| saeS1 | GGG GAC AAG TTT GTA CAA AAA AGC AGG CTA ACG ACA ACT AGC GGT AAA GAA G |
| saeS2 | GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ACG CTC AAG TGG CGT TCG ATA |
| saeS3 | ATT GGC GTC GTA TCG AGT ATA CC |
| saeS4 | GCA ATT GCT AAA ATA GTT GAA GTT AAT A |
Production of extracellular proteases.
Protease activity was assessed by zymography as previously described (2) using both 4 to 16% Zymogram Blue casein and 10% Zymogram gelatin gels (Invitrogen, Carlsbad, CA).
Analysis of phenol-soluble modulins.
High-pressure liquid chromatography-mass spectrometry (HPLC-MS) was used to detect and quantify phenol-soluble modulins (PSMs) in bacterial culture supernatants as previously described (46). Briefly, 100-μl samples from stationary-phase cultures were injected onto an analytical reversed-phase column and eluted with a gradient from 0.1% trifluoroacetic acid (TFA) in 50% acetonitrile-50% water to 0.1% TFA in 90% acetonitrile-10% water. Using this method, all PSMs can be detected in both their formylated and deformylated forms, but not with equal efficiency (46). Thus, quantitative comparisons can only be made within each class of PSM. Unless otherwise noted, results are presented as the amounts relative to those observed for each class of PSM in a designated parent strain included in each experiment.
RESULTS
Impact of sarA on the alpha-toxin phenotype of USA300 isolates.
When supernatants from stationary-phase cultures were examined by Western blotting with anti-alpha-toxin antibody, mutation of sarA was found to result in an apparent decrease in the production of alpha-toxin in each of three isolates of the USA300 clonal lineage (Fig. 1). In contrast, mutation of sarA resulted in an apparent increase in the production of alpha-toxin in the commonly studied strain Newman (Fig. 1). The level of alpha-toxin in early postexponential-phase supernatants (OD560 of 3.0) was low (generally less than 10% of that observed in stationary-phase cultures), and mutation of sarA had little impact on the alpha-toxin phenotype of any strain (Fig. 2A). This is consistent with the current S. aureus regulatory paradigm concluding that production of most exotoxins increases as cultures transition from the exponential to stationary growth phases. This transition was apparent when comparing supernatants from postexponential and stationary-phase cultures from each of the wild-type strains, including Newman (Fig. 2B). However, when the same comparison was done with the isogenic sarA mutants, this increase was not apparent in any of the USA300 isolates but remained apparent in Newman (Fig. 2B). This demonstrates that sarA function is required for the growth-phase strain-dependent transition to alpha-toxin accumulation in USA300 isolates but not in Newman.
Fig. 1.

Impact of sarA on the alpha-toxin phenotype of stationary-phase cultures. (A) Western blot using alpha-toxin antibody and stationary-phase supernatants from the indicated wild-type (WT) strains and their isogenic sarA (S) mutants. UAMS-1625 (U1625), U1782, and U1790 are isolates of the USA300 clonal lineage (Table 1). STD, alpha-toxin standard. (B) Production of alpha-toxin was assessed by Western blotting of stationary-phase supernatants. Results obtained with each wild-type strain (black) and its isogenic sarA mutant (gray) were quantified with Image J and are shown relative to the signal observed with 50 ng of purified alpha-toxin, the amount of which was set to a value of 1.0. Results are shown as the average ± standard deviation from three independent experiments.
Fig. 2.

Impact of sarA on the alpha-toxin phenotype of post-exponential-phase cultures. (A) Alpha-toxin production was assessed by Western blotting of supernatants from post-exponential-phase cultures (OD560 of 3.0) and quantified by using Image J. Results are shown for the wild-type strains (black) and their isogenic sarA mutants (gray) relative to the signal observed with 50 ng of a purified alpha-toxin standard. Results are shown as the average ± standard deviation from three independent experiments. (B) Results of Western blots using supernatants from post-exponential-phase (solid bars [data from panel A]) and stationary-phase supernatants (cross-hatched bars [data from Fig. 1]) supernatants were replotted to illustrate the impact of mutating sarA on growth-phase-dependent changes in the alpha-toxin phenotype in different strains of S. aureus.
Impact of sarA on transcription of agr and hla.
sarA is known to modulate gene expression in S. aureus via both agr-dependent and agr-independent pathways (7, 10–12, 15). To address these alternative possibilities, we examined the impact of mutating sarA on transcription of both hla- and the agr-associated regulatory molecule RNAIII, and the results confirmed that mutation of sarA had an impact in both contexts. However, these changes were not consistent with the alpha-toxin phenotype. Specifically, the level of both RNAIII and hla mRNA was decreased in stationary-phase cultures of a Newman sarA mutant (Fig. 3), while the amount of alpha-toxin was increased (Fig. 1). Conversely, the level of hla mRNA was increased in USA300 sarA mutants, while the amount of both RNAIII (Fig. 3) and alpha-toxin was decreased (Fig. 1). While these results are consistent with a scenario in which sarA modulates the phenotype of S. aureus at the transcriptional level via both agr-dependent and agr-independent pathways, the disparity between the transcriptional impact of mutating sarA on agr and/or hla and its impact on the alpha-toxin phenotype suggests the existence of a posttranscriptional component that ultimately defines the sarA-dependent alpha-toxin phenotype.
Fig. 3.

Impact of sarA on the production of RNAIII and transcription of hla. Relative levels of the hla (A) and RNAIII (B) transcripts in the stationary growth phase were determined for the indicated wild-type (WT) strains (black) and their isogenic sarA mutants (gray) by qRT-PCR. Results are shown as the mean ± standard deviation of triplicate samples.
Impact of sarA in the context of agr. In USA300 isolates, mutation of sarA resulted in increased expression of hla but decreased expression of agr at least as defined by relative levels of RNAIII (Fig. 3). Because RNAIII is required for efficient translation of hla mRNA (31), one possible explanation for the disparity between the increased levels of the hla transcript and decreased levels of alpha-toxin is reduced translation of hla mRNA owing to the relative absence of RNAIII. To further investigate this issue, we generated sarA, agr, and sarA agr mutants and examined the impact on the production of alpha-toxin with and without complementation of the sarA defect. In RN6390 and the USA300 isolate UAMS-1782, mutation of sarA or agr resulted in an apparent decrease in the production of alpha-toxin, and complementation of the sarA defect restored alpha-toxin production in a sarA mutant but not in the isogenic sarA agr mutant (Fig. 4). This is consistent with the hypothesis that the impact of sarA on the production of alpha-toxin is mediated through posttranscriptional changes associated with the reduced production of RNAIII. However, these results must be interpreted with caution because mutation of sarA or agr had the same impact on the alpha-toxin phenotype, and it is possible that in these strains the impact of any sarA-dependent, agr-independent effect would be masked by the impact of mutating agr on the transcription and/or translation of hla. Further support for this hypothesis comes from the observation that the same complementation results were observed in Newman despite the fact that mutation of sarA and agr had opposite effects on the alpha-toxin phenotype (Fig. 4). This is consistent with the hypothesis that sarA has an impact on the alpha-toxin phenotype that is independent of both agr-mediated transcriptional regulation and RNAIII-mediated posttranscriptional effects.
Fig. 4.
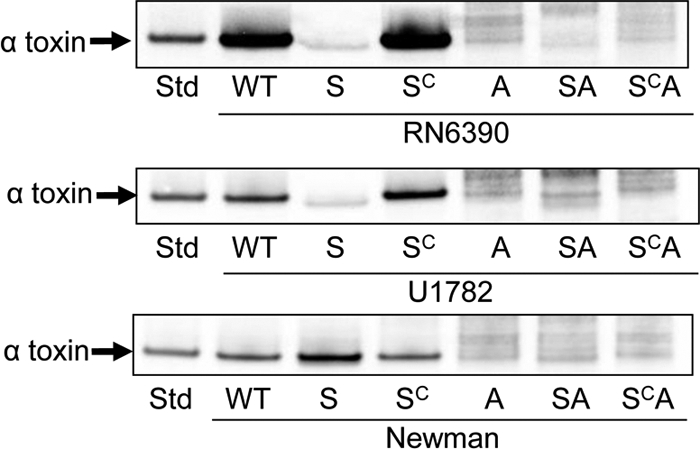
Production of alpha-toxin in sarA and agr mutants. Western blots of culture supernatants from stationary-phase cultures were done using anti-alpha-toxin antibody after blocking with skim milk alone. Alpha-toxin was identified by comparison to a purified alpha-toxin standard (Std). Parent strains are indicated below each panel. U1782 is the designation in the corresponding authors' culture collection for the USA300 isolate FPR3757. Lane designations: WT, wild-type strain; S, sarA mutant; SC, complemented sarA mutant; A, agr mutant; SA, sarA agr double mutant; SCA, sarA agr double mutant complemented for sarA.
Role of extracellular proteases in defining the sarA-dependent alpha-toxin phenotype.
In previous reports, we demonstrated that mutation of sarA results in a dramatic increase in the production of extracellular proteases and that this plays an important role in defining the biofilm-deficient phenotype of S. aureus sarA mutants (2, 45). Additionally, Newman is one of the few strains we have examined in which mutation of sarA had relatively little impact on biofilm formation (2). Based on this, we next examined whether the impact of sarA on extracellular proteases may also play a role in defining the sarA-dependent alpha-toxin phenotype. The inclusion of a protease inhibitor cocktail in the growth medium restored the alpha-toxin phenotype in a UAMS-1782 sarA mutant (Fig. 5 A), but it was difficult to assess the comparative effect in Newman owing to the differential impact of mutating sarA on the production and/or accumulation of endogenous alpha-toxin. To overcome this limitation, we repeated the experiment by adding exogenous alpha-toxin to growing cultures of sarA hla double mutants generated in both Newman and the USA300 isolate erythromycin-sensitive UAMS-1782 derivative UAMS-1794, and the results confirmed the degradation of purified alpha-toxin in a growing culture of the UAMS-1794 sarA hla mutant but not in a Newman sarA hla mutant (Fig. 5B).
Fig. 5.

Impact of extracellular proteases on the alpha-toxin phenotype of sarA mutants. Western blots were done with anti-alpha-toxin antibody after blocking with both skim milk and human IgG. (A) Blots were done with stationary-phase supernatants from the USA300 isolate U1782 (WT) and its isogenic sarA mutant (S) complemented for the sarA defect (SC) or grown in the presence of DMSO (SD) or DMSO containing increasing concentrations of protease inhibitor cocktail (SPI versus SPI2). (B) Blots were done after adding purified alpha-toxin (8 μg per ml) to growing cultures of hla (H) and sarA hla (SH) mutants generated in strain Newman and the USA300 isolate U1794, the latter being derived from U1782 by curing the plasmid conferring resistance to erythromycin. This was necessary to allow generation of the sarA hla double mutant. After overnight incubation, supernatants were harvested for Western blotting using anti-alpha-toxin antibody. Controls included an equivalent amount of purified alpha-toxin standard (Std) incubated at 37°C overnight in sterile tryptic soy broth (TSB). (C) Blots were done with stationary-phase supernatants from U1794 (WT) and isogenic derivatives carrying mutations in sarA (S) with or without the indicated genes encoding extracellular proteases.
All of the results discussed above are consistent with the hypothesis that the strain-dependent impact of sarA on the alpha-toxin phenotype of S. aureus is defined by the impact of sarA on the production of specific extracellular proteases. However, these results do not prove a cause-and-effect relationship. To address this issue, we generated a USA300 sarA mutant of UAMS-1794 (UAMS-1802) that carries mutations in the genes encoding different extracellular proteases (scpAB, splABCDEF, sspABC, or aur) and examined the impact on the alpha-toxin phenotype. As assessed by casein and gelatin zymography, mutation of scpAB or splABCDEF had no impact on the protease phenotype (see Fig. S1 in the supplemental material). In contrast, mutation of sspABC essentially eliminated all proteases, while mutation of aur resulted in the appearance of multiple proteolytic bands in gelatin (but not casein) zymograms. Because aureolysin is an activator of other proteases, including those encoded within the sspABC operon (41), we attribute these additional bands to alternative forms of SspA and/or SspB. More importantly, while mutation of scpAB, splABCDEF, or sspABC had no impact on the alpha-toxin phenotype, mutation of the gene encoding aureolysin restored the alpha-toxin-deficient phenotype of a UAMS-1794 sarA mutant (Fig. 5C). This confirms that the sarA-dependent alpha-toxin phenotype of a USA300 sarA mutant is defined by the increased production of extracellular proteases rather than transcriptional changes associated with hla or agr.
To the extent that mutation of scpAB, splABCDEF, or sspABC individually had no discernible impact on the sarA-dependent alpha-toxin phenotype, these results also suggest that the impact of aureolysin is mediated through something other than its role as an activator of other extracellular proteases. However, we did observe a greater effect with the PI2 inhibitor cocktail than the PI formulation (Fig. 5A). These two formulations differ only in the concentration of DIC, which was higher in PI2 than in PI. To the extent that DIC is a specific inhibitor of serine proteases, including sspA, and the spl proteases, these results suggest a role for proteases other than aureolysin itself.
Impact of sarA on phenol-soluble modulins.
Although we placed a primary focus in this report on alpha-toxin, the important role of phenol-soluble modulins (PSMs) in defining the virulence of contemporary CA-MRSA led us to assess the impact of the increased production of extracellular proteases in sarA mutants in this context. PSMs are not easily detectable by SDS-PAGE, but HPLC analysis demonstrated that both the alpha and beta classes of PSMs were almost undetectable in sarA mutants generated in isolates of the USA300 clonal lineage (Fig. 6). This was also true in agr mutants, and it was not possible to complement the sarA defect in a sarA agr double mutant. This suggests that the impact of sarA on PSM production occurs in an agr-dependent manner. However, while mutation of sarA also limited the PSM phenotype in Newman, as with alpha-toxin, the impact was limited by comparison to USA300 isolates (Fig. 7 A). Moreover, while the inclusion of protease inhibitors had no impact on the PSM-negative phenotype of a USA300 sarA mutant, it partially restored the PSM-positive phenotype of a Newman sarA mutant. This suggests that, as with alpha-toxin, the differential impact of sarA on the production of extracellular proteases may also contribute to the sarA-dependent PSM phenotype. This was also confirmed by demonstrating that concomitant mutation of aur partially restored the PSM-deficient phenotype of a USA300 sarA mutant (Fig. 7B).
Fig. 6.

Impact of sarA on phenol-soluble modulins. The PSM phenotype was assessed in stationary-phase cultures by HPLC as previously described (46). Strain designations: WT, wild-type strain; S, sarA mutant; SC, complemented sarA mutant; A, agr mutant; SA, sarA agr double mutant; SCA, sarA agr double mutant complemented for sarA. Results for each individual class (alpha- and beta-toxins versus delta-toxin) of PSM in both their formylated and unformylated forms were combined, and the amounts of each class observed in the USA300 isolate U1782 were set to 100%. Results observed in each of the indicated U1782 mutants and in U1 and its derivatives are shown relative to these amounts.
Fig. 7.

Role of sarA and extracellular proteases in the agr-dependent PSM phenotype of USA300 isolates. The PSM phenotype was assessed in stationary-phase cultures by HPLC. (A) The amount of each individual class (alpha- and beta-toxins versus delta-toxin) of PSM in stationary-phase cultures was assessed in both their formylated and unformylated forms by HPLC. The results observed with strain Newman were set to 100%, with the amounts observed in U1782 and isogenic sarA mutants generated in both strains (S) and grown with (SPI versus SPI2) and without protease inhibitors shown relative to these amounts. (B) The amounts of each individual class (alpha- and beta-toxin versus delta-toxin) of PSM in both their formylated and unformylated forms were combined, and the amount of each class observed in the USA300 isolate U1794 (WT) was set to 100%. The results observed in each of the indicated U1794 mutants are shown relative to these amounts.
Impact of saeRS on the sarA-dependent alpha-toxin and PSM phenotypes.
Although Newman is one of the S. aureus strains most frequently used in studies focusing on both pathogenesis and regulation of gene expression, it is known to carry a defect in the saeS sensor kinase that results in constitutive expression of the saeRS regulatory locus (40). To determine whether this might contribute to the sarA-dependent alpha-toxin and PSM phenotypes of Newman, we both repaired the saeS defect to restore the regulated expression of saeRS and generated an saeRS null mutant. It was difficult to detect any extracellular proteases by zymography in strains derived from Newman itself, but it was possible to detect altered protease production in the isogenic sarA mutants. Specifically, both repair and deletion of saeRS in isogenic sarA mutants resulted in the increased production of multiple extracellular proteases (see Fig. S2 in the supplemental material). This is consistent with a previous report demonstrating that mutation of saeRS results in the increased expression of multiple protease genes, including aur and sspA (39). The observation that these changes were apparent only in sarA mutants also suggests that the impact of saeRS on protease production is independent of sarA.
As expected based on a previous report demonstrating that transcription of hla is dramatically reduced in an saeRS mutant (33), mutation of saeRS eliminated alpha-toxin production in both strain Newman and its sarA mutant (Fig. 8 A). However, repair of the saeS defect reversed the alpha-toxin-positive phenotype of a Newman sarA mutant. We also demonstrated that the alpha-toxin-deficient phenotype of a sarA mutant generated in the saeS-repaired derivative of Newman was reversed by concomitant mutation of aur (Fig. 8B). Finally, both repair of the saeS defect or deletion of saeRS in Newman resulted in a sarA-dependent PSM-deficient phenotype comparable to that observed in USA300 (Fig. 9A). Taken together, these results confirm that the impact of sarA on the production of extracellular proteases is responsible for the strain-dependent impact on both the alpha-toxin and PSM phenotypes of S. aureus and that, at least in Newman, this is a function of the saeS defect and its impact on the production of these proteases. Although we have not yet defined the mechanism by which the constitutive activation of saeRS impacts the sarA-defined alpha-toxin and PSM phenotypes, neither repair nor mutation of saeRS had an impact on the production of SarA itself (Fig. 9B), and this also suggests that the constitutive activation of saeRS either represses protease production in a sarA-independent manner or alters the functional status rather than the production of SarA.
Fig. 8.

Impact of saeRS on the sarA-dependent alpha-toxin phenotype of strain Newman. (A) Western blot with alpha-toxin antibody examining the impact of mutating sarA in Newman, its saeS-repaired [saeS(P18L)] derivative (P18L), and an isogenic saeRS mutant on the alpha-toxin phenotype. (B) Impact of mutating aur and sarA alone and in combination with each other in the saeS-repaired derivative of strain Newman (P18L).
Fig. 9.

Impact of saeS on the sarA-dependent PSM phenotype of strain Newman. (A) The PSM phenotype was assessed by HPLC in Newman, its saeS-repaired [saeS(P18L)] derivative (P18L), and a Newman saeRS mutant. Results observed with strain Newman for each class of PSM were set to 100%, with results observed in each of the other strains shown relative to this amount. (B) The amount of SarA was assessed in Newman, its saeRS mutant, an saeS-repaired derivative (P18L), and an saeS(P18L) aureolysin mutant (P18Laur) by Western blotting as previously described (4).
Impact of sarA on the alpha-toxin phenotype of other clinical isolates.
To determine whether differences like those observed between USA300 isolates and Newman exist among other clinical isolates, we carried out quantitative Western blot analysis using supernatants from isolates of other clonal lineages. Alpha-toxin was undetectable in isolates of the USA200 (UAMS-1 and UAMS-601), USA400 (MW2), and USA600 clonal lineages irrespective of the functional status of sarA. This is consistent with the observations that many USA200 isolates, including UAMS-1 and UAMS-601, have a nonsense mutation in hla (6), while MW2 expresses hla at very low levels (46). Mutation of sarA also had comparatively little impact on the alpha-toxin phenotype of S6C, SC-01, or a single isolate of the USA100 clonal lineage (see Fig. S3 in the supplemental material). In all other strains tested, mutation of sarA resulted in an alpha-toxin-deficient phenotype comparable to that observed in USA300. These included RN6390, its rsbU-repaired derivative SH1000, and individual isolates of the USA500, USA800, USA1000, and USA1100 clonal lineages (Fig. S3). Thus, the phenotype observed in USA300 sarA mutants appears to be representative of most contemporary clonal lineages of S. aureus.
DISCUSSION
The sarA locus was originally identified in the S. aureus strain DB based in part on its increased hemolytic activity owing to its increased production of alpha-toxin (9). This report also noted the reduced production of extracellular protein A in a strain DB sarA mutant, and it was suggested that one possible explanation for this was the increased production of extracellular proteases. However, it was concluded that this could not account for the alpha-toxin phenotype due to its increased rather than decreased production. Given its apparently opposing regulatory role in comparison to agr, it was also proposed that sarA may serve as a “counterregulatory system” to agr. However, the sarA mutation was subsequently generated in the commonly studied 8325-4 laboratory strain RN6390, and in this background, the alpha-toxin phenotype was opposite to that observed in DB (10, 11). Based on this, the current regulatory paradigm in S. aureus is that mutation of sarA results in the reduced production of alpha-toxin due to reduced transcription of both hla and agr (3, 12).
Given the contrasting reports describing DB and RN6390 sarA mutants, we examined in an earlier report (3) the phenotypic impact of mutating sarA on hemolytic activity in each of seven strains of S. aureus (UAMS-1, UAMS-601, DB, SC-01, S6C, Newman, and the 8325-4 strain RN6390). In six of these strains, hemolytic activity was increased, the only exception being RN6390 (3). On the basis of Northern blots examining transcription of hla in these strains and Western blots examining the production of alpha-toxin in DB, we concluded that the increased hemolytic activity observed in strains other than RN6390 was a function of the increased transcription of hla and consequently the increased production of alpha-toxin (3). More directly, we concluded that the strain-dependent difference in alpha-toxin phenotype was mediated at the transcriptional level and that the phenotype observed in RN6390 was not representative of clinical isolates of S. aureus.
A subsequent report concurred with this conclusion and suggested that the phenotypic disparity between 8325-4 strains like RN6390 and other S. aureus isolates was due to the combined effects of the rsbU and tcaR mutations that are present in all 8325-derived strains (35). Specifically, mutation of sarA was found to result in increased rather than decreased hemolytic activity in the rsbU-repaired 8325-4 strain SH1000, which suggests that the rsbU defect in RN6390 plays an important role in defining the sarA-dependent alpha-toxin phenotype in S. aureus. However, it was also concluded that this was not the only relevant factor since mutation of sarA in strain V8 resulted in increased hemolytic activity and increased transcription of hla despite the fact that this strain is also an rsbU mutant (35). Because SH1000 contains a defect in tcaR while V8 does not, and because tcaR modulates the expression of sarS (26), a model was proposed in which SarA and SarS act cooperatively to repress transcription of hla, with the end result of mutating sarA being a function of the impact on the total amount of repressor (e.g., SarA plus SarS) (35). Specifically, it was proposed that sarA represses the transcription of both hla and sarS, with SarS in turn acting as a repressor of hla transcription. In 8325-4 strains, which already express sarS at low levels due to the tcaR mutation, mutation of sarA results in a paradoxical increase in the overall amount of repressor because of the increased expression of sarS, and this would presumably account for the reduced production of alpha-toxin in these strains. In contrast, mutation of sarA in strains that do not have the tcaR mutation would result in a net decrease in the total amount of repressor due to the loss of SarA and a comparatively modest effect on expression of sarS, and this would presumably account for the increased transcription of hla and increased production of alpha-toxin (35).
While we have begun to investigate this model directly, the more important consideration is that, based on this model, it would be anticipated that mutation of sarA in contemporary clinical isolates that lack both the rsbU and tcaR mutations would result in the increased production of alpha-toxin. However, we found that mutation of sarA in the USA300 isolate UAMS-1625 resulted in reduced hemolytic activity and an apparent reduction in the production of alpha-toxin (48), despite the fact that this isolate expresses both asp23 and sarS, which are indicators of the functional status of sigB and tcaR, respectively, at levels that are comparable to those of other clinical isolates and significantly higher than those observed in RN6390 (2). In fact, of the strains included in the experiments reported here, all but one expressed both asp23 and sarS at levels that were comparable to each other and considerably higher than those observed in RN6390 (data not shown). The exception was S6C, which expressed sarS at levels comparable to those of other clinical isolates but asp23 at levels comparable to those of RN6390.
This suggests that S6C also has a deficiency in the sigB regulatory pathway, and in this respect, it is important to note that S6C and RN6390 exhibited disparate sarA-dependent alpha-toxin phenotypes, despite the fact that they share this common defect. Similarly, different sarA-dependent alpha-toxin phenotypes were observed among strains that expressed sarS at comparable levels. In fact, our SH1000 isolate expressed both asp23 and sarS at levels that exceeded those observed in RN6390, although the latter was lower than the levels observed in contemporary clinical isolates, but in contrast to the earlier report cited above (35), we found that this did not alter the sarA-dependent alpha-toxin phenotype. More directly, mutation of sarA in both RN6390 and SH1000 resulted in an alpha-toxin-deficient phenotype, the only difference being the overall levels in the parent strains and their isogenic sarA mutants. While indirect, all of these results collectively provide support for the hypothesis that the strain-dependent impact of sarA on the alpha-toxin phenotype of S. aureus involves something other than the rsbU and/or tcaR defects present in 8325-4 strains.
This brings up two important questions, the first being whether there is in fact a predominant sarA-dependent alpha-toxin phenotype among S. aureus clinical isolates and the second being the mechanistic basis for the strain-dependent phenotypes that do exist. With respect to the first, we conclude based on the results presented here that the most common phenotype of an S. aureus sarA mutant, particularly among contemporary clinical isolates, is a reduction in the level of alpha-toxin. In this respect, it is important to note that of the six strains that exhibited the opposite phenotype in our earlier study (3), two (UAMS-1 and UAMS-601) do not produce alpha-toxin due to a nonsense mutation in hla (7, 46), one (Newman) is known to carry a defect in saeS (17, 40), and, based on transcription levels of asp23, another (S6C) appears to carry a defect in the sigB pathway. SC-01 was described in a report comparing different strain typing techniques (44), but to date neither this strain nor DB (9) has been extensively characterized. Whether any of these strains also have defects related to saeRS that contribute to their sarA-dependent alpha-toxin phenotypes remains unknown. Nevertheless, these results collectively suggest that it is in fact these strains that represent the anomaly by comparison to contemporary clinical isolates and that the most prominent and clinically relevant phenotype of S. aureus sarA mutants is a decrease in the production and/or accumulation of alpha-toxin. In this respect, it is also worth noting that, in all of the strains other than Newman in which mutation of sarA did not result in an alpha-toxin-deficient phenotype, the overall impact was modest by comparison to that in strains that exhibited the opposite phenotype.
With respect to the mechanistic basis for these differences, the current S. aureus regulatory paradigm indicates that the sarA-encoded DNA binding protein SarA induces the production of alpha-toxin at a transcriptional level via both agr-dependent and agr-independent pathways (8, 11, 12), and our results demonstrate that the impact of sarA on the transcription of both RNAIII and hla is a strain-dependent phenotype. In most strains, mutation of sarA generally resulted in reduced transcription of agr, as measured by the levels of RNAIII, and this is consistent with the hypothesis that sarA is required for maximum activation of agr transcription (10, 11). However, with the exception of RN6390, SH1000, and Newman, mutation of sarA resulted in decreased rather than increased transcription of hla, and this suggests that the more important consideration in the context of alpha-toxin is the agr-independent impact of sarA on hla transcription. However, even in this context, a disparity exists between hla transcription and the resulting alpha-toxin phenotype, and our results demonstrate that the strain-dependent impact of sarA on the production of extracellular proteases is a primary reason for this disparity. This is consistent with a previous report concluding that the primary impact of sarA in defining the alpha-toxin-deficient phenotype even in 8325-4 strains is in fact the impact of sarA on the production of extracellular proteases (24). Interestingly, while they did not consider sarA, there is also a report demonstrating that the strain-dependent production of both alpha-toxin and protein A is generally consistent with the level of RNAIII but that the amount of alpha-toxin produced by one clinical isolate was unexpectedly high given its relatively low levels of agr expression, and it was suggested that this may be a function of the reduced production of extracellular proteases (23).
Our results are also the first to demonstrate that sarA is required for maximum PSM production and confirm that strain-dependent differences in protease production are also relevant in this context. Whether this is true with other exoproteins has not yet been assessed. It is known that sarA mutants produce increased amounts of extracellular nuclease and that this contributes to some degree to their reduced capacity to form a biofilm (2, 45). Additionally, the results we present do not preclude other regulatory roles for sarA; indeed, genome-scale transcriptional profiling experiments have confirmed that mutation of sarA limits the production and/or stability of multiple transcripts, important examples including all of the genes encoding extracellular proteases (7, 15). Nevertheless, our results emphasize the need to consider the phenotype of S. aureus sarA mutants rather than focus on transcriptional changes alone.
Based on all of these considerations, we propose that the primary phenotypic impact of sarA on the phenotype of at least some critical exotoxins produced by S. aureus is not transcriptional but rather is mediated by the impact of sarA on the production of extracellular proteases. In fact, taken together with our previous reports demonstrating that the increased production of extracellular proteases is a primary factor in the biofilm-deficient phenotype of S. aureus sarA mutants (2, 45), we would propose a model in which the sarA-mediated repression of protease production serves the important purposes of both promoting biofilm formation (2, 45) and allowing agr to “fine-tune” gene expression patterns in a manner that simultaneously promotes dispersal from an established biofilm (5) and the production of the extracellular toxins, including alpha-toxin and PSMs, that promote the ability of S. aureus to survive outside the protective environment of the biofilm. By analogy, we would propose that sarA is the “dam” that represses the “flow” of proteases and thereby allows the “lake” to develop in the form of a biofilm and that agr is the “outlet” that allows the controlled release of S. aureus and induction of the virulence factors necessary to survive the downstream trip through the bloodstream to a new site of colonization. In this scenario, mutation of sarA would represent a failure of the dam such that transcriptional events within defined regions of the biofilm, including those associated with agr, are essentially washed away.
In this context it should be noted that a primary motivation for the experiments we describe was the observation that mutation of sarA results in a reduced capacity to form a biofilm due to the increased production of extracellular proteases and that this limitation can be correlated with increased antibiotic susceptibility in the context of a biofilm-associated infection (2, 45, 47, 48). This suggests that inhibitors of sarA could be used to limit S. aureus biofilm formation to a therapeutically relevant degree. However, our earlier results suggested that any therapeutic benefit derived from such inhibitors would potentially be compromised by the adverse consequence of increasing the production of alpha-toxin (3). Thus, the results we report not only demonstrate that this is not the case in most clonal lineages of contemporary S. aureus clinical isolates but also that an inhibitor of sarA production and/or function would in several important respects be an inhibitor of agr-mediated phenotypes, including the “production” of at least some critical exotoxins.
Whether this effect is evident under in vivo conditions and therefore therapeutically relevant remains to be determined, but many serious S. aureus infections do occur in the relatively localized environment of a biofilm and/or abscess, and in this context, it might be expected that extracellular products, including proteases and exotoxins, would remain in close proximity to each other. In this respect, we would also note that a second important motivation behind the experiments we describe was our demonstration that mutation of sarA in the USA300 isolate UAMS-1625 limited the development of skin lesions in our in vivo model of biofilm-associated infection (48). To the extent that the results reported here confirm that the alpha-toxin phenotype observed in this isolate is not unique among contemporary clinical isolates of S. aureus suggests that sarA may in fact be a viable therapeutic target in the context of both chronic, biofilm-associated S. aureus infection and acute, toxin-mediated infections, including those caused by CA-MRSA isolates of the USA300 clonal lineage.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants AI074935 (M.S.S.), AI090250 (L.N.S.), and AI37027 (C.Y.L. and T.T.L.) and by The Intramural Research Program of the National lnstitute of Allergy and Infectious Diseases (M.O.). Support was also obtained from resources provided through the Clinical and Translational Sciences Award (RR0298884) to the University of Arkansas for Medical Sciences. Bacterial isolates were obtained from the NIAID-Supported Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA).
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 8 April 2011.
REFERENCES
- 1. Beenken K. E., Blevins J. S., Smeltzer M. S. 2003. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect. Immun. 71:4206–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beenken K. E., et al. 2010. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS One 5:e10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blevins J. S., Beenken K. E., Elasri M. O., Hurlburt B. K., Smeltzer M. S. 2002. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect. Immun. 70:470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blevins J. S., Gillaspy A. F., Rechtin T. M., Hurlburt B. K., Smeltzer M. S. 1999. The staphylococcal accessory regulator (sar) controls expression of the Staphylococcus aureus collagen adhesin gene (cna) in an agr-independent manner. Mol. Microbiol. 33:317–326 [DOI] [PubMed] [Google Scholar]
- 5. Boles B. R., Horswill A. R. 2008. agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 4:e1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cassat J. E., et al. 2005. Comparative genomics of Staphylococcus aureus musculoskeletal isolates. J. Bacteriol. 187:576–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cassat J. E., et al. 2006. Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiology 152:3075–3090 [DOI] [PubMed] [Google Scholar]
- 8. Cheung A. L., Bayer A. S., Zhang G., Gresham H., Xiong Y.-Q. 2004. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus. FEMS Immunol. Med. Microbiol., 40:1–9 [DOI] [PubMed] [Google Scholar]
- 9. Cheung A. L., Koomey J. M., Butler C. A., Projan S. J., Fischetti V. A. 1992. Regulation of exoprotein expression in Staphylococcus aureus by a locus (sar) distinct from agr. Proc. Natl. Acad Sci. U. S. A. 89:6462–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung A. L., Projan S. J. 1994. Cloning and sequencing of sarA of Staphylococcus aureus, a gene required for the expression of agr. J. Bacteriol. 176:4168–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chien Y., Manna A. C., Cheung A. L. 1998. SarA level is a determinant of agr activation in Staphylococcus aureus. Mol. Microbiol. 30:991–1001 [DOI] [PubMed] [Google Scholar]
- 12. Chien Y., Manna A. C., Projan S. J., Cheung A. L. 1999. SarA, a global regulator of virulence determinants in Staphylococcus aureus, binds to a conserved motif essential for sar-dependent gene regulation. J. Biol. Chem. 274:37169–37176 [DOI] [PubMed] [Google Scholar]
- 13. DeLeo F. R., Otto M., Kreiswirth B. N., Chambers H. F. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diep B. A., Carleton H. A., Chang R. F., Sensabaugh G. F., Perdreau-Remington F. 2006. Roles of 34 virulence genes in the evolution of hospital- and community-associated strains of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 193:1495–1503 [DOI] [PubMed] [Google Scholar]
- 15. Dunman P. M., et al. 2001. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 183:7341–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gillaspy A. F., et al. 1995. Role of the accessory gene regulator (agr) in the pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 63:3373–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herbert S., et al. 2010. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 78:2877–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horsburgh M. J., et al. 2002. σB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J. Bacteriol. 184:5457–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reference deleted.
- 20. Kernodle D. S., Voladri R. K., Menzies B. E., Hager C. C., Edwards K. M. 1997. Expression of an antisense hla fragment in Staphylococcus aureus reduces alpha-toxin production in vitro and attenuates lethal activity in a murine model. Infect. Immun. 65:179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kielian T., Cheung A., Hickey W. F. 2001. Diminished virulence of an alpha-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect. Immun. 69:6902–6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobayashi S. D., DeLeo F. R. 2009. An update on community-associated MRSA virulence. Curr. Opin. Pharmacol. 9:545–551 [DOI] [PubMed] [Google Scholar]
- 23. Li S., Arvidson S., Möllby R. 1997. Variation in the agr-dependent expression of alpha-toxin and protein A among clinical isolates of Staphylococcus aureus from patients with septicaemia. FEMS Microbiol. Lett. 152:155–161 [DOI] [PubMed] [Google Scholar]
- 24. Lindsay J. A., Foster S. J. 1999. Interactive regulatory pathways control virulence determinant production and stability in response to environmental conditions in Staphylococcus aureus. Mol. Gen. Genet. 262:323–331 [DOI] [PubMed] [Google Scholar]
- 25. Luong T. T., et al. 2011. Staphylococcus aureus ClpC divergently regulates capsule via sae and codY in strain Newman but activates capsule via codY in strain UAMS-1 and in strain Newman with repaired saeS. J. Bacteriol. 193:686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCallum N., Bischoff M., Maki H., Wada A., Berger-Bachi B. 2004. TcaR, a putative MarR-like regulator of sarS expression. J. Bacteriol. 186:2966–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCormick C. C., Caballero A. R., Balzli C. L., Tang A., O'Callaghan R. J. 2009. Chemical inhibition of alpha-toxin, a key corneal virulence factor of Staphylococcus aureus. Invest. Ophthalmol. Vis. Sci. 50:2848–2854 [DOI] [PubMed] [Google Scholar]
- 28. McElroy M. C., et al. 1999. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect. Immun. 67:5541–5544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller K. D., Hetrick D. L., Bielefeldt D. J. 1977. Production and properties of Staphylococcus aureus (strain Newman D2C) with uniform clumping factor activity. Thromb. Res. 10:203–211 [DOI] [PubMed] [Google Scholar]
- 30. Montgomery C. P., et al. 2008. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes USA300 and USA400 in a rat model of pneumonia. J. Infect. Dis. 198:561–570 [DOI] [PubMed] [Google Scholar]
- 31. Morfeldt E., Taylor D., von Gabain A., Arvidson S. 1995. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 14:4569–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nilsson I. M., Hartford O., Foster T., Tarkowski A. 1999. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 67:1045–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Novick R. P., Jiang D. 2003. The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing. Microbiology 149:2709–2717 [DOI] [PubMed] [Google Scholar]
- 34. Novick R. P., et al. 1993. Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J. 12:3967–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oscarsson J., Kanth A., Tegmark-Wisell K., Arvidson S. 2006. SarA is a repressor of hla (alpha-hemolysin) transcription in Staphylococcus aureus: its apparent role as an activator of hla in the prototype strain NCTC 8325 depends on reduced expression of sarS. J. Bacteriol. 188:8526–8533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phillips J. R., Tripp T. J., Regelmann W. E., Schlievert P. M., Wangensteen O. D. 2006. Staphylococcal alpha-toxin causes increased tracheal epithelial permeability. Pediatr. Pulmonol. 41:1146–1152 [DOI] [PubMed] [Google Scholar]
- 37. Reference deleted.
- 38. Reed S. B., et al. 2001. Molecular characterization of a novel Staphylococcus aureus serine protease operon. Infect. Immun. 69:1521–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rogasch K., et al. 2006. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J. Bacteriol. 188:7742–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schafer D., et al. 2009. A point mutation in the sensor hisitidine kinase SaeS of Staphylococcus aureus strain Newman alters the response to biocide exposure. J. Bacteriol. 191:7306–7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shaw L., Golonka E., Potempa J., Foster S. J. 2004. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology 150:217–228 [DOI] [PubMed] [Google Scholar]
- 42. Sifri C. D., Park J., Helm G. A., Stemper M. E., Shukla S. K. 2007. Fatal brain abscess due to community-associated methicillin-resistant Staphylococcus aureus strain USA300. Clin. Infect. Dis. 45:e113–e117 [DOI] [PubMed] [Google Scholar]
- 43. Tenover F. C., Goering R. V. 2009. Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J. Antimicrob. Chemother. 64:441–446 [DOI] [PubMed] [Google Scholar]
- 44. Tenover F. C., et al. 1994. Comparison of traditional and molecular methods of typing isolates of Staphylococcus aureus. J. Clin. Microbiol. 32:407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsang L. H., Cassat J. E., Shaw L. N., Beenken K. E., Smeltzer M. S. 2008. Factors contributing to the biofilm-deficient phenotype of Staphylococcus aureus sarA mutants. PLoS One 3:e3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang R., et al. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 13:1510–1514 [DOI] [PubMed] [Google Scholar]
- 47. Weiss E. C., Spencer H. J., Daily S. J., Weiss B. D., Smeltzer M. S. 2009. Impact of sarA on antibiotic susceptibility of Staphylococcus aureus in a catheter-associated in vitro model of biofilm formation. Antimicrob. Agents Chemother. 53:2475–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weiss E. C., et al. 2009. Impact of sarA on daptomycin susceptibility of Staphylococcus aureus biofilms in vivo. Antimicrob. Agents Chemother. 53:4096–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.