

Abstract
Background
Ca2+-ATPases of endoplasmic reticulum (SERCAs) are responsible for maintenance of the micro- to millimolar Ca2+ ion concentrations within the endoplasmic reticulum (ER) of eukaryotic cells. This intralumenal Ca2+ storage is important for the generation of Ca2+ signals as well as for the correct folding and posttranslational processing of proteins entering ER after synthesis. ER perturbations such as depletion of Ca2+ or abolishing the oxidative potential, inhibition of glycosylation, or block of secretory pathway, activate the Unfolded Protein Response, consisting of an upregulation of a number of ER-resident chaperones/stress proteins in an effort to boost the impaired folding capacity.
Results
We show here that in PC12 cells, depletion of ER Ca2+ by EGTA, as well as inhibition of disulphide bridge formation within the ER by dithiotreitol or inhibition of N-glycosylation by tunicamycin, led to a 2- to 3-fold increase of the SERCA-mediated 45Ca2+ transport to microsomes isolated from cells exposed to these stress agents. The time course of this response corresponded to that for transcriptional upregulation of ER stress proteins, as well as to the increase in the SERCA2b mRNA, as we recently observed in an independent study.
Conclusions
These findings provide the first functional evidence for the increase of SERCA pumping capacity in cells subjected to the ER stress. Since at least three different and unrelated mechanisms of eliciting the ER stress response were found to cause this functional upregulation of Ca2+ transport into the ER, these results support the existence of a coupling between the induction of the UPR pathway in general, and the regulation of expression of at least one of the SERCA pump isoforms.
Background
Endoplasmic reticulum (ER) is a compartment simultaneously involved in the processes of protein synthesis and Ca2+ homeostasis in eukaryotic cells. Nascent polypeptide chains entering the secretory pathway, as well as extracytosolic portions of proteins destined for the external or internal cell membranes, translocate into the ER lumen. Within the ER, posttranslational processes including folding, glycosylation, subunit assembly and transfer to the Golgi compartment take place, assisted by chaperone proteins residing in the ER lumen [1,2]. In addition, proper protein folding and maturation depends on the maintenance of the oxidative milieu and high Ca2+ concentrations within the ER [3-5]. Reducing agents, depletion of ER lumenal Ca2+, inhibition of glycosylation or interference with the secretory pathway (by preventing transfer to the Golgi system), each lead to an accumulation of misfolded protein intermediates and increase the demand on the chaperoning capacity. Such conditions, collectively termed ER stress, have been shown to induce ER-specific stress response pathways. Two ER stress pathways concerned with protein processing are recognized today. The Unfolded Protein Response (UPR) was originally described in yeast [6], and has been more recently demonstrated in mammalian cells [7]. In mammals, the proximal ER stress-sensing element of this pathway appears to be Ire1p, a 110 kDa, highly conserved protein spanning the ER membrane. It is thought that when misfolded polypeptide chains accumulate within the ER causing a decrease in the level of free chaperones, the lumenal N-terminal portion of Ire1p undergoes dimerization. This in turn leads to a trans-autophosphorylation of the Ire1 cytosolic domains, triggering an ER-to-Nucleus (ERN) signaling [7]. The details of the ERN signaling are not understood at present. It appears to involve additional activities, in part residing in the cytosolic portion of the Ire1p molecule itself (RNA endonuclease activity), and in part contributed by proteins recruited through binding to Ire1p cytosolic domain (e.g. TRAF2, [8]). Overall, the major result of the UPR induction is a transcriptional upregulation of a number of stress proteins, including members of the family of Glucose Regulated Proteins (GRPs) such as GRP78 (BiP) and GRP94 (endoplasmin), as well as other chaperones (calreticulin, protein disulfide isomerase, ERp72) [7], thereby responding to the increased demand for the folding capacity within the ER.
Another ER-specific stress response has been termed Endoplasmic Reticulum Overload Response (EOR) [9]), and is triggered by certain of the same conditions known to activate UPR (e.g. glucose deprivation, glycosylation inhibition), as well as by heavy overexpression of proteins within the ER. The distinguishing feature of EOR is its association with the activation of the transcription factor NF-κB [9].
While necessary for the folding and maturation processes within the ER, the high concentration of Ca2+ ions in this compartment (1000-10,1000 times higher than in the cytosol [10,11]) is also a prerequisite for the universally employed Ca2+ signaling through ER Ca2+ channels (inositol 1,4,5-trisphosphate and ryanodine receptors) [12,13]. This high ER Ca2+ concentration is maintained by ER Ca2+ pumps, members of the family of Sarco-Endoplasmic Reticulum Calcium ATPases (SERCAs) [14-17]. Depletion of ER Ca2+ has been shown to cause a transcriptional induction of the mRNA for SERCA pumps [18]. However, the upregulation of an actual SERCA activity in such circumstances has not been demonstrated. In the present work, we provide the first functional evidence that depletion of ER Ca2+ during cell culture leads to an increase in the SERCA pumping capacity, as expressed by an enhancement of SERCA-mediated 45Ca2+ uptake to microsomes isolated from Ca2+ -depleted PC12 cells. In addition, we show that similar functional SERCA upregulation may also be elicited by conditions of ER stress previously shown not to be accompanied by Ca2+ depletion. Taken together with our earlier results [19], the present data support the existence of a linkage between the induction of UPR and the regulation of SERCA gene expression.
Results and Discussion
Determination of the SERCA-mediated 45Ca2+ uptake into the microsomes from PC12 cells
The family of SERCA ATPases comprises products of 3 genes, giving rise to a number of enzyme isoforms after differential splicing of primary RNA transcripts [16,17]. SERCA1a-1b enzymes are expressed exclusively in skeletal muscle fast-twitch fibers, SERCA2a occurs in slow-twitch fibers of skeletal muscle as well as in cardiac muscle, while SERCA2b is expressed ubiquitously. SERCA3 gene products comprise isoforms 3a, 3b and 3c in mouse and human [20] and 3a and 3 b/c in rat [15]. SERCA3 Ca2+ -ATPase appears to have a restricted tissue distribution, with the highest levels found in the intestine, lung, lymphatic and hematopoietic tissues. Both SERCA2b and SERCA3 isoforms have been identified in PC12 cells [21].
The sesquiterpene lactone thapsigargin has been found to have a uniformly high inhibitory potency towards all SERCA subtypes, while showing no activity against the plasma membrane (PMCA-type) Ca2+ pump at the comparable concentrations [22,23]. We therefore defined the SERCA-mediated 45Ca2+ uptake to PC12 cell microsomes as that portion of the total ATP-dependent 45Ca2+ accumulation which could be inhibited by 1 μM thapsigargin. In order to optimize the accuracy of the assay, we were seeking conditions under which this thapsigargin-sensitive fraction would be as large as possible. ER is known to be permeable to the oxalate anion, commonly employed in studies of 45Ca2+ uptake into the ER in order to increase the 45Ca2+ sequestering capacity of the vesicles and to prolong the linear course of 45Ca2+ accumulation [24,25]. However, the reported concentrations of oxalate appear to vary considerably. We therefore tested how a choice of a particular oxalate concentration would influence the size of the thapsigargin-sensitive fraction of 45Ca2+ uptake in our system. Fig. 1A shows that in the absence of thapsigargin, a strong linear correlation (P = 0.0002) existed between the amount of oxalate (up to 24 mM) in the assay and the amount of 45Ca2+ loaded to the microsomes. In contrast, in the presence of 1 μM thapsigargin, the regression line did not significantly differ from horizontal (P = 0.1696), showing that oxalate had a minimal, if any, augmenting effect on 45Ca2+ uptake to the thapsigargin-insensitive (SERCA-independent) microsomal compartments. These results suggested that the most advantageous conditions for measurements of the thapsigargin-sensitive component were present at the highest oxalate concentrations. However, we also observed an increasing non-linearity of the time dependence (steepening of the slope between 5-25 minutes) of 45Ca2+ uptake at high oxalate concentrations (not shown). Therefore, as a compromise 18 mM oxalate was chosen for the remaining experiments. (A minor non-linearity was still observed at this concentration, Fig. 1B, open circles). With 18 mM oxalate, the thapsigargin-sensitive component of 45Ca2+ uptake varied between 40-66% of the total uptake in non-stressed (control) cells (compare Figs. 1A and 1B).
Figure 1.

SERCA-mediated 45Ca2+ uptake into microsomes isolated from PC12 cells: oxalate dep endence and effect of endoplasmic reticulum 45Ca2+-store depletion by EGTA. In A, numbers of independent experiments (n) were 5,2,3,6 and 5 for oxalate concentrations 0, 6,12,18 and 24 mM, respectively. In B, n = 3 for all data points. Bars represent SEMs. Lines were fitted by linear regression. A. Oxalate selectively enhances 45Ca2+ uptake to the thapsigargin-sensitive (SERCA-mediated) portion of the total microsomal uptake. Microsomes were prepared from PC12 cells, and 45Ca2+ uptake was determined as described in Materials and Methods section, with 25 min incubation time and various concentrations of oxalate either in the absence (●) or presence (○) of 1 μM thapsigargin. B. Depletion of Ca2+ stores by exposure of cells to EGTA affects specifically the thapsigargin-sensitive (SERCA-mediated) component of microsomal 45Ca2+ uptake. Microsomes were prepared from cells cultured under control conditions (□,■) or in the presence of 2.5 mM EGTA for 7 h before harvesting (○,●). 45Ca2+ uptake was determined as a function of time in the presence of 18 mM oxalate either in the absence (○,□) or presence (●,■) of 1 μM thapsigargin.
The linearity of 45Ca2+ uptake with time argues against the 45Ca2+ accumulation in the vesicles being influenced by membrane leak. Since the rate of a passive leak would be increasing with the amount of 45Ca2+ accumulated, the slope of the uptake would be expected to decline with time. In contrast, the slope of the uptake time-dependence was essentially constant (or slightly increasing) even at the highest recorded rates (EGTA treatment of cells, Fig. 1B), indicating that the measurements closely approximated the initial velocity of SERCA pump, even with the absolute uptake rates well below Vmax due to the free 45Ca2+ held at 0.22-μM and the absence of DTT in the assay (see below). In addition, the following consideration argues strongly for the amounts of 45Ca2+ retained in the vesicles reflecting almost exclusively the formation of the insoluble precipitate, with no possibility of passive leak: given the solubility product of Ca2+-oxalate of approximately 2 × 10-9 M [24], already at 10 mM lumenal oxalate one would reach a maximal concentration of free 45Ca2+ in the lumen equal to the employed extravesicular free 45Ca2+ (0.2 μM), with no 45Ca2+ gradient available for a leak pathway. Since 18 mM oxalate was used, it appears highly unlikely that a leak gradient could be established, even allowing for an incomplete oxalate equilibration between the vesicles and the medium.
Increase of the SERCA-mediated 45Ca2+-uptake into the microsomes from PC12 cells subjected to depletion of ER Ca2+
In a number of systems, the ER stress response has been induced by manipulations depleting Ca2+ from the ER. Means of inducing this response have included Ca2+ ionophores [26-28] and Ca2+ chelating agents applied inside the cells (e.g. BAPTA, [29]). Alternatively, application of EGTA in the external medium has been shown to deplete ER Ca2+ in a number of cell types [30], including PC12 cells [31]. In order to test the effect of Ca2+depletion on the SERCA activity, we cultured PC12 cells for 7 hours in the presence of 2.5 mM EGTA, representing an excess of 0.7 mM over the total Ca2+ concentration in the DMEM medium. Such EGTA exposure would be expected to induce the UPR, but not the EOR [9] response. Recently we have confirmed this UPR-inducing effect of EGTA in PC12 cells by demonstrating an increase in GRP78 (BiP) mRNA [19]. Fig. 1B shows the time dependence of 45Ca2+ uptake measured in the presence or absence of 1 μM thapsigargin in microsomes prepared from the control cells and cells stressed by the presence of EGTA. A clear increase in the ATP-dependent 45Ca2+ uptake was found following the EGTA treatment. Importantly, Fig. 1B shows that this increase was due specifically to the thapsigargin-sensitive component of 45Ca2+ uptake: an identical activity was measured in the presence of thapsigargin in the microsomes from the stressed (filled circles) and non-stressed (filled squares) cells. (This unchanged activity in the presence of thapsigargin also indicated that the stress treatment of cells did not affect the membrane recovery or 45Ca2+ uptake in some gross, non-specific manner). Therefore, this upregulation of 45Ca2+ uptake was due to an increase in the SERCA activity in membrane vesicles derived from the ER.
Increase of the SERCA-mediated 45Ca2+ uptake following ER stress without Ca2+ depletion
The observed upregulation of the SERCA activity might be specifically due to Ca2+ depletion from the ER, or it might be linked to the activation of the UPR pathway, irrespective of the particular means of inducing ER stress. Tunicamycin is an inducer of UPR due to its inhibitory action on N-linked glycosylation [32]. Brefeldin A is a fungal toxin causing a collapse of the Golgi compartment through its absorption into the ER [33], and inducing the UPR in response to an overload of transport-arrested proteins in the ER lumen. The ER stress-inducing action of DTT is due to its perturbing of the ER lumen oxidative environment, thereby interfering with the protein disulfide bridge formation [5]. We have previously confirmed that tunicamycin, brefeldin A or DTT, while inducing the ER stress response, did not appear to deplete the ER Ca2+ in PC12 cells [19]. Fig. 2 shows the effects of the exposure of PC12 cells to brefeldin A, tunicamycin or DTT on the thapsigargin-sensitive component of 45Ca2+ uptake to the isolated microsomes. (For comparison, the effect of EGTA from Fig. 1B is also included). In Fig. 2, only the thapsigargin-sensitive 45Ca2+ uptake component is shown, as none of the indicated treatments affected the magnitude of 45Ca2+ uptake in the presence of 1 μM thapsigargin. It may be seen that while the increase in the SERCA-mediated 45Ca2+ uptake after EGTA exposure reached 326% (P < 0.01) of the value in non-stressed cells, increases were also observed with tunicamycin (341% of the control, P < 0.01) and DTT (227%, P < 0.02). Brefeldin A caused an increase to 150% of the level in the unstressed cells (not significant statistically).
Figure 2.
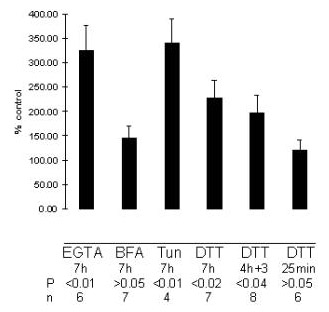
Upregulation of SERCA-mediated 45Ca2+ uptake into microsomes prepared after exposure of PC12 cells to various types of ER stress. EGTA (2.5 mM), brefeldin A (BFA) 5 μg/ml, tunicamycin (Tun) 10 g/ml, or DTT 1 mM were present in the culture medium for the indicated times prior to cell harvest (in "4h+3" condition, after 4 h in the presence of DTT, control medium was reintroduced for 3 h prior to cell harvest). The microsomes were prepared and 45Ca2+ uptake determined as described in Materials and Methods. For all conditions, the thapsigargin-sensitive portion of the uptake (see Figs. 1A,B) was calculated and expressed as % of the corresponding thapsigargin-sensitive portion for untreated cells, as determined in parallel for each separate condition. (None of the treatments affected the thapsigargin-insensitive portion of 45Ca2+ uptake). For each condition, statistical significance of the difference between these pairs of values was determined using 2-tailed student's t-test, with P values and numbers of independent experiments (n) as indicated. Bars represent SEMs.
When microsomes prepared from non-stressed cells were assayed for 45Ca2+ uptake in the presence of 1 mM DTT, an increase (524%, P < 0.01) in the SERCA-mediated uptake component was observed (not shown). This result suggested that an oxidation of ATPase on critical cysteine residues, with a consequent loss of activity, had occurred in the course of microsome preparation. Indeed, some studies have routinely included DTT in SERCA isolation buffers to prevent such loss of activity [34,35]. We therefore tested whether the observed increase of microsomal 45Ca2+ uptake following the exposure of PC12 cells to DTT for 7 hours did reflect an upregulation of Ca2+-pumping capacity in the intact cells, rather than representing an enzyme protection effect due to a carry-over of some DTT in the course of cell harvest and microsome preparation. However, the following results appear to rule out such an assay-related mode of action for DTT. First, an increase to 197% of the control (P < 0.04) (not significantly different from the 227% seen with 7 hours of DTT exposure) was obtained when DTT was added to the culture medium for a period of 4 hours, followed by a change to DTT-free medium for the remaining 3 hours prior to cell harvest (Fig. 2, "4h+3"). This result is consistent with the DTT-mediated induction of SERCA mRNA increase in the treated cells within the first 4 hours, similar to the known time course of the induction of mRNA for some of the ER stress proteins [19],[26-29]. Translation of SERCA protein from this increased amount of mRNA during the last 3 hours would not be expected to depend on the continuing presence of DTT. Secondly, in a reverse test, DTT was added to the medium for only 25 minutes (i.e. for the duration equal to the incubation time in the microsomal 45Ca2+ uptake assay) immediately before cell harvest. No change in the SERCA-mediated 45Ca2+ uptake to the microsomes was observed under these conditions (Fig. 2, "25min"). Thus, a mere 25 minutes exposure to 1mM DTT, while strongly enhancing SERCA activity at the stage of the 45Ca2+microsomal uptake assay, was not effective at the level of whole cells. Hence, the mechanisms by which DTT was able to enhance the SERCA-mediated microsomal 45Ca2+ accumulation were clearly different in these 2 modes of DTT action: the cell-dependent mode, observed after a prolonged (hours) presence of DTT in the cell culture, but not requiring DTT for the last 3 hours before cell harvesting, microsome preparation and assay; and the cell-independent mode, in which a relatively short (25 min) presence of DTT in the assay, but not during the cell culture, was sufficient.
Conclusions
This work shows for the first time an upregulation of ER Ca2+ -ATPase (SERCA) activity upon a disturbance of the internal ER environment by several agents known to evoke the ER stress response, or UPR. Since UPR may be activated by a variety of unrelated stimuli, e.g. Ca2+ depletion, inhibition of glycosylation, misfolding due to faulty amino acid precursors, energy deprivation or interference with the ER oxidative milieu [7,9], it may be thought of as a "final common pathway" for signaling ER stress caused by a range of factors. Therefore, we suggest that the UPR pathway is a likely candidate for the common mechanism to underlie the upregulation of SERCA activity following treatments with agents as different in their modes of action as EGTA, tunicamycin and DTT. Within this proposed common mechanism for SERCA activation due to UPR, some variation in the response amplitude between the individual agents appears to exist (Fig. 2). Further work will be required to understand the level (transcriptional and/or translational), and the exact mechanisms of such differences.
We have recently reported that treatment of PC12 cells with EGTA, tunicamycin, DTT or brefeldin A induced approximately 3- to 4-fold increases in the mRNA for SERCA2b, and approximately 2-fold increases in SERCA2b immunoreactive protein, with no corresponding changes for SERCA3 [19]. An upregulation of SERCA activity was also supported in this earlier work by an increase in the amount of SERCA phosphoenzyme. However, we would like to stress that despite these indications, the actual demonstration by the present paper of the stress-induced increase in the capacity of Ca2+transport into the ER was important for at least 2 reasons. First, as previously discussed [19, p.22371], technical difficulties precluded an accurate comparison of the extent of ATPase upregulation as measured at the immunoprotein and the phosphoenzyme level. Second, we were concerned with the possibility that ER stress, even if causing an increased synthesis of the immunoreactive protein, might at the same time by its very nature result in a faulty posttranslational processing of SERCA2b, with a degree of functional impairment. Since, as shown by the mutagenesis work [36], it is possible to disrupt the Ca2+ transport cycle of Ca2+ -ATPase without affecting at least some of the partial phosphorylation reactions, an actual full measurement in the present paper of the thapsigargin-sensitive Ca2+ transport into the ER was necessary to unequivocally demonstrate the validity of the ER stress-induced upregulation of SERCA2b activity.
Materials and Methods
PC12 cells were kindly provided by Drs. M. Grønborg and S. Gammeltoft, Glostrup County Hospital, Denmark. Dulbecco's modified Eagle's medium (DMEM;GlutaMAX-type), horse serum and fetal bovine serum were from GibcoBRL Life Technologies. 45CaCl2 (specific activity 0.2-2 Ci/mmole Ca2+) was from Pharmacia Amersham DK. Thapsigargin was a generous gift from Dr. S.B. Christensen, The Royal Danish School of Pharmacy.
Cell culture and stress treatments
Non-differentiated PC12 cells were grown in 88% DMEM containing HEPES 25 mM, glucose 4.5 mg/ml, penicillin 17.5 U/ml, streptomycin 17.5 μg/ml, and supplemented with horse serum 6%, fetal bovine serum 6% . Cells were grown at 37°C in the presence of 5% CO2 -atmosphere, and were passaged weekly. Before various experimental treatments, cells were plated onto rat tail collagen-coated culture dishes (100 mm diameter, 3.5 μg collagen/cm2) [37]. To elicit the stress response, agents (ethyleneglycolbis(β-aminomethyl ether)-N,N' -tetraacetic acid (EGTA) 2.5 mM, dithiotreitol (DTT) 1 mM, tunicamycin 10 g/ml or brefeldin A 5 μg/ml) were included in the medium for 7 hours prior to cell harvest. These agents were added to the culture medium as stock solutions in water concentrated 100-fold (EGTA) or 1000-fold (DTT), or as a 1000-fold concentrated solution in dimethyl sulfoxide (DMSO) (tunicamycin, brefeldin A). To neutralize the acidification of the medium upon addition of EGTA, the EGTA stock was adjusted to pH 9.16. Corresponding solvent volumes were added to the dishes with control cells for each of the treatments. Following the removal of the medium, the cells were harvested in cold phosphate-buffered saline . The cells were centrifuged and resuspended in the homogenization buffer (HB) consisting of sucrose 300 mM, N-tris(hydroxymethyl)methyl-2-aminomethanesulfonic acid (TES) 20 mM, pH 7.0, containing phenylmethyl sulfonyl fluoride (PMSF) 0.5 mM. Cell viability was monitored by Trypan Blue staining and was better than 90% in control and stress-agent treated cell batches.
Preparation of microsomes
Cells were broken on ice using the cell cracker from the European Molecular Biology Laboratory workshop [38], employing the clearance of 12 μm and 10 passages. The homogenates were centrifuged for 10 min at 1000 g, followed by a centrifugation of consecutive supernatants first for 10 min at 10,000 g, and then for 1 h at 100,000 g. The microsomal pellets were resuspended in HB and stored at -20°C. Approximately 30 μg of microsomal protein was obtained for each million cells fractionated.
45Ca2+ uptake
This was determined using 20-50 μg microsomal protein/ml in 1 ml final incubation volume by means of a filtration assay, as described earlier [39]. Briefly, the incubation medium (at 37°C) contained (in mM): TES 20 (pH 7.0), sucrose 60, KCl 114, MgCl2 5, EGTA 1 mM, 45CaCl2 (approximately 5.7 mCi/mmole) 0.35 mM (yielding free Ca2+ 0.22 μM [40], and various K+ -oxalate concentrations, as indicated for the individual experiments. Parallel determinations were run in the presence and absence of thapsigargin 1 μM, with an equivalent volume of DMSO solvent (1%) present in all incubations. The uptake was started by addition of ATP to the final 2.5 mM, and was terminated after 25 min, except when otherwise indicated. All determinations were carried out in triplicate.
Other methods
Protein was determined using fluorescamine assay [41]. Statistical significance of the results was estimated using student's two-tailed t-test.
Abbreviations
Ca2+-ATPase, Ca2+-dependent adenosine triphosphatase; DMSO, dimethyl sulfoxide; DTT, dithiotreitol; EGTA, ethyleneglycolbis(β-aminomethyl ether)-N,N'-tetraacetic acid; EOR, endoplasmic reticulum overload response; ER, endoplasmic reticulum; ERN signaling, ER-to-nucleus signaling; GRP, glucose-regulated protein; SERCA, sarco-endoplasmic reticulum calcium ATPase; UPR, unfolded protein response
Acknowledgments
Acknowledgements
This work was supported by the Danish Government Biotechnology Program, the Novo-Nordisk Foundation and the State Health Research Council (SHRC). AHL held a SHRC student scholarship. We are grateful to Ms. N. Buch Petersen for her excellent technical assistance during the completion stage of this work.
Contributor Information
Annette Højmann Larsen, Email: hojmann@telefona.dk.
Aase Frandsen, Email: aaf@dfh.dk.
Marek Treiman, Email: M.Treiman@mfi.ku.dk.
References
- Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Cooper GR, Brostrom CO, Brostrom MA. Analysis of the endoplasmic reticular Ca2+ requirement for alpha1-antitrypsin processing and transport competence. Biochem J. 1997;325:601–608. doi: 10.1042/bj3250601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish HF, Kong N, Wikstrom L. Calcium is required for folding of newly made subunits of the asialoglycoprotein receptor within the endoplasmic reticulum. J Biol Chem. 1992;267:12753–12760. [PubMed] [Google Scholar]
- Braakman I, Helenius J, Helenius A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J. 1992;11:1717–1722. doi: 10.1002/j.1460-2075.1992.tb05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething M-J, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose- regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes and Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. 1999;79:683–701. doi: 10.1152/physrev.1999.79.3.683. [DOI] [PubMed] [Google Scholar]
- Bygrave FL, Benedetti A. What is the concentration of calcium ions in the endoplasmic reticulum? Cell Calcium. 1996;19:547–551. doi: 10.1016/s0143-4160(96)90064-0. [DOI] [PubMed] [Google Scholar]
- Meldolesi J, Pozzan T. The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci. 1998;23:10–14. doi: 10.1016/S0968-0004(97)01143-2. [DOI] [PubMed] [Google Scholar]
- Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 1999;25:247–264. doi: 10.1054/ceca.1999.0021. [DOI] [PubMed] [Google Scholar]
- Mackrill JJ. Protein-protein interactions in intracellular Ca2+ release channel function. Biochem J. 1999;337:345–361. doi: 10.1042/0264-6021:3370345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk SE, Lytton J, MacLennan DH, Shull GE. cDNA cloning, functional expression, and mRNA tissue distribution of a third organellar Ca2+ pump. J Biol Chem. 1989;264:18561–18568. [PubMed] [Google Scholar]
- Martin V, Bredoux R, Corvazier E, Papp B, Enouf J. Platelet Ca2+ ATPases. A plural, species-specific, and multiple hypertension-regulated expression system. Hypertension. 2000;35:91–102. doi: 10.1161/01.hyp.35.1.91. [DOI] [PubMed] [Google Scholar]
- Lompre A. The sarco(endo)plasmic reticulum Ca2+-ATPases in the cardiovascular system during growth and proliferation. Trends in Cardiovascular Medicine. 1998;8:75–82. doi: 10.1016/S1050-1738(97)00130-810.1016/S1050-1738(97)00130-8. [DOI] [PubMed] [Google Scholar]
- Wuytack F, Raeymaekers L, De Smedt H, Eggermont JA, et al. Ca2+-transport ATPases and their regulation in muscle and brain. Annals of New York Academy of Sciences. 1992;671:82–91. doi: 10.1111/j.1749-6632.1992.tb43786.x. [DOI] [PubMed] [Google Scholar]
- Kuo TH, Liu B-F, Yu Y, Wuytack F, et al. Co-ordinated regulation of the plasma membrane calcium pump and the sarco(endo)plasmic reticular calcium pump gene expression by Ca2+. Cell Calcium. 1997;21:399–408. doi: 10.1016/s0143-4160(97)90051-8. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Pedersen PS, Treiman M. The Sarco-Endoplasmic Reticulum Calcium ATPase 2b is an endoplasmic reticulum stress-inducible protein. J Biol Chem. 2000;275:22363–22372. doi: 10.1074/jbc.M001569200. [DOI] [PubMed] [Google Scholar]
- Dode L, De Greef C, Mountian I, Attard M, et al. Structure of the human sarco/ endoplasmic reticulum Ca2+-ATPase 3 gene. J Biol Chem. 1998;273:13982–13994. doi: 10.1074/jbc.273.22.13982. [DOI] [PubMed] [Google Scholar]
- Rooney E, Meldolesi J. The endoplasmic reticulum in PC12 cells. J Biol Chem. 1996;271:29304–29311. doi: 10.1074/jbc.271.46.29304. [DOI] [PubMed] [Google Scholar]
- Inesi G, Sagara Y. Specific inhibitors of intracellular Ca2+ transport ATPases. J Membrane Biol. 1994;141:1–6. doi: 10.1007/BF00232868. [DOI] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–135. doi: 10.1016/S0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- Hasselbach W. Relaxing factor and the relaxation of muscle. Progress in Biophysics and Molecular Biology. 1964;14:169–222. [Google Scholar]
- Bian J, Ghosh TK, Wang JC, Gill DL. Identification of intracellular calcium pools. Selective modification by thapsigargin. J Biol Chem. 1991;266:8801–8806. [PubMed] [Google Scholar]
- Brostrom MA, Cade C, Prostko CR, Gmitter-Yellen D, Brostrom CO. Accomodation of protein synthesis to chronic deprivation of intracellular sequestered calcium. J Biol Chem. 1990;265:20539–20546. [PubMed] [Google Scholar]
- Li WW, Alexandre S, Cao XJ, Lee AS. Transactivation of the grp78 promoter by Ca2+ depletion. J Biol Chem. 1993;268:12003–12009. [PubMed] [Google Scholar]
- Resendez E, Ting J, Kim KS, Wooden SK, Lee AS. Calcium ionophore A23187 as a regulator of gene expression in mammalian cells. J Cell Biol. 1986;103:2145–2152. doi: 10.1083/jcb.103.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W, Gissel C, Linden T, Doutheil J. Erp72 expression activated by transient cerebral ischemia or disturbance of neuronal endoplasmic reticulum calcium stores. Metab Brain Dis. 1998;13:55–68. doi: 10.1023/A:1020631029168. [DOI] [PubMed] [Google Scholar]
- Brostrom CO, Brostrom MA. Regulation of translational initiation during cellular response to stress. Progress Nucl Acid Res Mol Biol. 1998;58:79–125. doi: 10.1016/s0079-6603(08)60034-3. [DOI] [PubMed] [Google Scholar]
- Bennett DL, Bootman MD, Berridge MJ, Cheek TR. Ca2+ entry into PC12 cells initiated by ryanodine receptors or inositol 1,4,5-trisphosphate receptors. Biochem J. 1998;329:349–357. doi: 10.1042/bj3290349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell. 1994;5:253–265. doi: 10.1091/mbc.5.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaky N, Presley J, Lippincott-Schwartz J, Smith C, et al. Golgi tubule traffic and effects of brefeldin A visualized in living cells. J Cell Biol. 1997;139:1137–1155. doi: 10.1083/jcb.139.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East JM, Lee AG. Lipid selectivity of the calcium and magnesium ion dependent adenosine trisphosphatase, studied with fluorescence quenching by a brominated phospholipid. Biochemistry. 1982;21:4144–4151. doi: 10.1021/bi00260a035. [DOI] [PubMed] [Google Scholar]
- Warren GB, Toon PA, Birsdall NJ, Lee AG, Metcalfe JC. Reconstitution of calcium pump using defined membrane components. Proc Natl Acad Sci USA. 1974;71:622–626. doi: 10.1073/pnas.71.3.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JP. Dissection of the functional domains of the sarcoplasmic reticulum Ca2+ -ATPase by site-directed mutagenesis. Bioscience Reports. 1995;15:243–261. doi: 10.1007/BF01788358. [DOI] [PubMed] [Google Scholar]
- Greene LA, Aletta JM, Rukenstein A, Green SH. PC12 pheochromocytoma cells: culture, nerve growth factor treatment, and experimental exploitation. Methods in Enzymol. 1987;147:207–216. doi: 10.1016/0076-6879(87)47111-5. [DOI] [PubMed] [Google Scholar]
- Balch WE, Dunphy WG, Braell WA, Rothman JE. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416. doi: 10.1016/0092-8674(84)90019-9. [DOI] [PubMed] [Google Scholar]
- Poulsen J-CJ, Caspersen C, Mathiasen D, East MJ, et al. Thapsigargin-sensitive Ca2+-ATPases account for Ca2+ uptake to inositol 1,4,5-trisphosphate-sensitive and caffeine-sensitive Ca2+ stores in adrenal chromaffin cells. Biochem J. 1995;307:749–758. doi: 10.1042/bj3070749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fohr KJ, Warchol W, Gratzl M. Calculation and control of free divalent cations in solutions used for membrane fusion studies. Methods in Enzymology. 1993;221:149–157. doi: 10.1016/0076-6879(93)21014-y. [DOI] [PubMed] [Google Scholar]
- Bohlen P, Stein S, Dairman W, Udenfriend S. Fluorometric assay of proteins in the nanogram range. Arch Biochem Biophys. 1973;155:213–220. doi: 10.1016/s0003-9861(73)80023-2. [DOI] [PubMed] [Google Scholar]