

Abstract
Type 3 secretion systems (T3SSs) are critical for the virulence of numerous deadly Gram-negative pathogens. T3SS translocator proteins are required for effector proteins to traverse the host cell membrane and perturb cell function. Translocator proteins include two hydrophobic proteins, represented in enteropathogenic Escherichia coli (EPEC) by EspB and EspD, which are thought to interact and form a pore in the host membrane. Here we adapted a sequence motif recognized by a host kinase to demonstrate that residues on the carboxyl-terminal side of the EspB transmembrane domain are localized to the host cell cytoplasm. Using functional internal polyhistidine tags, we confirm an interaction between EspD and EspB, and we demonstrate, for the first time, an interaction between EspD and the hydrophilic translocator protein EspA. Using a panel of espB insertion mutations, we describe two regions on either side of a putative transmembrane domain that are required for the binding of EspB to EspD. Finally, we demonstrate that EspB variants incapable of binding EspD fail to adopt the proper host cell membrane topology. These results provide new insights into interactions between translocator proteins critical for virulence.
INTRODUCTION
Type III secretion systems (T3SSs) are highly conserved multimolecular protein assemblies that allow Gram-negative bacterial pathogens and symbionts to deliver directly into eukaryotic cells effector proteins that usurp and subvert host processes (3, 14). T3SSs and the machinery that assembles the Gram-negative flagella have many similarities, indicating a common evolutionary origin. Significant progress has been made in recent years toward defining the architecture of T3SSs using prototypic microorganisms, including Yersinia spp., Shigella flexneri, enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively), and Salmonella enterica (40, 53, 54). EPEC and EHEC are important causes of infantile diarrhea in developing countries and of hemorrhagic colitis and hemolytic-uremic syndrome, respectively (34). In both EPEC and EHEC, a T3SS is required for characteristic attaching and effacing (A/E) activity, which consists of intimate attachment to host cells, effacement of microvilli, and accumulation of filamentous actin beneath attached bacteria (21, 29, 30).
The T3SS needle complex is composed of a basal body that spans the inner and outer membranes and a needle that projects from the bacterial surface. Null mutations in the genes that encode most of the T3SS components preclude protein secretion. Three proteins known as translocators are themselves secreted via the T3SS and are required, not for the secretion of effector proteins, but for their transit across the host cell membrane (32). One of these translocator proteins is hydrophilic, while the other two are hydrophobic and have predicted transmembrane helices. Although all T3SSs include such translocator proteins, the degree to which they are conserved is quite variable.
In the T3SSs of Yersinia spp. a protein known as LcrV, which represents a prototypic hydrophilic translocator, has been visualized by immunoelectron microscopy at the tip of the needle (33). IpaD, a distantly related homologue of LcrV from Shigella flexneri, is similarly located at the needle tip (11). The crystal structure of LcrV reveals two long alpha-helices that form a coiled-coil (8). Stoichiometric and molecular modeling studies suggest that LcrV forms a pentamer at the needle tip. In EPEC and EHEC, the hydrophilic translocon component EspA is only distantly related to LcrV. Although it shares the coiled-coil structure and binds to the needle protein EscF, EspA does not form a pentameric ring at the needle tip. Instead, EspA forms a sheath-like filament extending 75 to 260 nm from the bacterial surface and apparently tethering the bacterium to the host cell (24). The EspA filament is a helical tube with an outer diameter of 12 nm and an inner diameter of 25 Å (5). EspA filaments have 5.6 subunits per turn, but the axial rise per subunit ranges from about 3.6 to 5.6 Å, allowing the filament a certain longitudinal flexibility of unknown significance (49). A crystal structure of EspA in complex with its chaperone confirmed the alpha-helical coiled-coil organization of its termini, but the structure of the region between these domains remains unresolved (52).
YopB and YopD can be considered prototypes of the hydrophobic translocon components thought to form a pore in the host cell membrane through which effector proteins pass. Evidence for this function comes from experiments demonstrating contact-dependent hemolytic activity that requires these hydrophobic translocator proteins (2, 13, 16, 50). Furthermore, proteoliposomes containing YopB and YopD induce ion channels when fused to planar lipid bilayers (44). In the absence of YopB, no channel activity could be detected, whereas in the absence of YopD, fluctuations in conductance were evident. Upon exposure to liposomes at an acidic pH, Pseudomonas aeruginosa hydrophobic translocators PopB and PopD are both able to form oligomeric ring structures with an outer diameter of 80 Å and an inner diameter of 40 Å (39). While both proteins are able to partially lyse liposomes, an equimolar mixture of the two caused complete lysis. PopB caused more-rapid release of a self-quenching fluorescent dye from unilamellar vesicles than did PopD, and an equimolar mixture of the two proteins caused a synergistic increase in the release rate (12). While both hydrophobic translocators are required for hemolytic activity, the requirement for the YopB homologues appears to be more absolute than that for the YopD homologues. For example, an S. flexneri ipaB mutant is nonhemolytic, while an ipaC mutant retains detectable, albeit with reduced hemolytic activity (2). Similar results were reported for YopB and YopD and for EspD and EspB (28, 35, 41). Thus, YopB homologues, including PopB, IpaB, and EspD, seem to play a primary role in pore formation.
While they appear to play a secondary role in pore formation, some YopD homologues may have additional functions. IpaC can induce actin reorganization and filopodium formation when microinjected or expressed in host cells (47). A carboxyl-terminal domain of IpaC is required for recruitment of the Src kinase to the sites of bacterial invasion and for actin reorganization (31). When expressed in host cells, EspB induces a dramatic loss of actin stress fibers (45). EspB binds to myosin with a dissociation constant of 2.3 μM (20). Myosin binding requires a region of EspB that is distal to its putative transmembrane domain. A mutant with a deletion of this region retains translocation activity but is impaired in the ability to mediate the elongation of microvilli adjacent to attached bacteria and to inhibit phagocytosis by macrophages. Other authors have reported that EspB can interact with alpha-catenin (25) and with α1-antitrypsin and that α1-antitrypsin can inhibit pore formation (22).
To function as a pore and allow specific passage of effector proteins from the T3SS needle into host cells, hydrophobic translocon components should form a bridge physically linking the needle tip to host cell membranes. Indeed, IpaB can be found in association with the LcrV homologue IpaD at the tip of the T3SS needle when the bacteria are grown in the presence of the bile salt deoxycholate (37). Additional incubation with liposomes induces recruitment of the YopD homologue IpaC to the needle tip, perhaps simulating interactions with host cell membranes (10). The hydrophobic translocator proteins each possess one or more predicted alpha-helical transmembrane domains and are found closely associated with host cell membranes (2, 48). Consistent with its two predicted transmembrane domains, purified IpaB traverses model lipid membranes twice (18). EspD has been localized to a detergent-soluble fraction of infected epithelial cells, where it remains susceptible to proteases, suggesting a transmembrane configuration (48). EspD and EspB have also been found in association with erythrocyte membranes following hemolysis by wild-type bacteria (19, 41). Additionally, protein fusion studies, cell fractionation, and confocal microscopy indicate that EspB localizes both to the host cell membrane and to the cytoplasm of infected epithelial cells (46, 51). While several YopD homologues, such as EspB, have one predicted transmembrane domain beginning approximately 100 residues from the amino terminus, no direct evidence of host cell membrane topology has been reported previously. In this study, we exploited knowledge of permissive sites of EspB that could accept amino acid insertions without compromising function (28) to test the hypothesis that EspB adopts a specific host cell membrane topology and to identify domains of EspB required for interactions with EspD.
MATERIALS AND METHODS
Bacterial strains and genetic manipulations.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplementary material.
A mutant in which the espADB genes of wild-type EPEC strain E2348/69 were replaced with a so-called scar sequence was constructed using the lambda recombinase one-step PCR mutagenesis method (6) and primers Donne-1978 and Donne-1979 (see Table S2 in the supplemental material). Electroporation, selection of recombinants, plasmid curing, and excision of the cassette using pFT-A were performed as described previously (6). The correct insertion was confirmed by sequencing, and the resulting strain is called UMD867. A modification of the method was used to alter the espD gene in espB deletion mutant strain UMD864 to encode an in-frame polyhistidine tag. In this procedure, the antisense primer was modified and extended further into the chloramphenicol cassette to eliminate nonsense codons that would otherwise result after recombination. Sense primer Donne-1980 and antisense primer Donne-1981 were used to amplify the chloramphenicol cassette from pKD3. The resulting strain, UMD898, has an in-frame deletion of espB and an espD allele in which the codons for residues 32 to 46 are replaced with codons encoding the following amino acids (single-letter abbreviations): HHHHHHHHVKAGAASKFLYFLENRNFGIGTKED.
Using the QuikChange kit (Stratagene) and a previously published modification of the manufacturer's instructions (7), we introduced sequences encoding a 13-amino-acid glycogen synthase kinase (GSK-3β) tag (MSGRPRTTSFAES) (15) into the espB gene in plasmid pWSL-17 at two positions previously determined to be permissive for insertions (28). Primers Donne-2000 and Donne-2001 and primers Donne-2003 and Donne-2004 (see Table S2 in the supplemental material) were used to engineer EspB proteins with GSK insertions after residues 36 and 186, respectively. The correct changes were confirmed by sequencing, and the resulting plasmids, pEspB36GSK and pEspB186GSK, were transformed into espB mutant UMD864. Similar insertions were engineered into previously described plasmids containing espB alleles with insertions of 5 or 31 codons (28). Plasmids encoding internal polyhistidine-tagged EspA and EspB proteins were constructed using QuikChange and specific primers (see Table S2 in the supplemental material) as described previously (7). A plasmid encoding eight histidine residues in place of residues 32 to 46 of EspD was constructed by cloning a double-stranded oligonucleotide formed by annealing Donne-1976 and Donne-1977 (see Table S2) into the KpnI and SpeI sites of pQWD12 (7).
Phenotypic assays.
A previously described modification (28) of the fluorescence actin staining (FAS) test (23) was used to determine whether EPEC derivatives are capable of inducing dense accumulations of filamentous actin in HeLa cells (ATCC CCL2), a characteristic of the attaching and effacing effect. The stability and secretion of EspB were assessed as described previously (28).
Detection of GSK tag phosphorylation.
Approximately 4 × 106 HeLa cells were seeded in 10-cm-diameter dishes in Dulbecco's modified Eagle's medium-F-12 medium (DMEM-F12) supplemented with 10% fetal bovine serum and 50 μg/ml of gentamicin. Bacteria were grown overnight in LB containing tetracycline (10 μg/ml) and were diluted 1:50 into DMEM-F12 with antibiotics. Cultures were grown at 37°C and 225 rpm until the A600 reached 0.8 to 1.0. After overnight incubation and three washes in Hanks' balanced salt solution, HeLa cells were infected with bacteria at a multiplicity of infection (MOI) of 300:1 in DMEM-F12 without antibiotics and were incubated for 2 h at 37°C under an atmosphere of 95% air-5% CO2. Infected cells were washed three times with cold phosphate-buffered saline (PBS) containing 5 mM NaF, scraped, and resuspended in 5 ml cold PBS containing 5 mM NaF. Cells were collected by centrifugation (4 min, 500 × g, 4°C) and were lysed in 100 μl Triton X-100 buffer (1% Triton X-100, 50 mM Tris-HCl [pH 7.5], 5 mM NaF, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× Complete protease inhibitor cocktail [Roche]). After centrifugation (30 min, 16,000 × g, 4°C), supernatants were boiled for 10 min in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, separated by electrophoresis, and examined by immunoblotting using a phospho-GSK-specific (Cell Signaling) or EspB-specific (46) antibody.
Coelution of translocator proteins by nickel affinity chromatography.
EPEC bacteria expressing polyhistidine-tagged EspA, EspB, or EspD proteins were grown overnight in LB with tetracycline or chloramphenicol (20 μg/ml) as appropriate and were diluted 1:50 into 50 ml DMEM-F12 with antibiotics. Cultures were grown at 37°C and 225 rpm until the A600 reached 0.8 to 1.0. The supernatant was collected by centrifugation twice (15 min, 2,500 × g, 4°C) and was passed through a 0.45-μm-pore-size filter. One Complete protease inhibitor cocktail tablet, 0.60 g NaCl, 0.5 ml nickel-nitrilotriacetic acid (NTA) agarose bead slurry, and PMSF (final concentration, 1 mM) were added to the filtrate, and the suspension was incubated overnight at 4°C in a rotating tube holder. Afterward, the suspension was applied to a column, and the flowthrough was collected. The column was washed initially with 6 ml of wash buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 20 mM imidazole) eight times, and the combined wash fractions were pooled with the flowthrough. An additional wash using 2 ml of wash buffer was collected and defined as the final wash. Bound proteins were eluted in 1 ml elution buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 250 mM imidazole). Equal volumes of the original supernatant, the pooled flowthrough and washes, and the final wash, as well as the entire eluate, were precipitated on ice for 1 h by addition of trichloroacetic acid (final concentration, 10% [vol/vol]). After centrifugation (30 min, 16,000 × g, 4°C), pellets were allowed to air dry, dissolved in 45 μl SDS-PAGE loading buffer, boiled for 10 min, and separated by PAGE.
Quantitative immunoblotting.
Proteins separated by SDS-PAGE were transferred to PVDF-FL membranes (Millipore) using a semidry electroblotting apparatus. Primary antibodies were used at the following dilutions in PBS with 5% nonfat dry milk: anti-EspA, 1:20,000 (24); anti-EspB, 1:2,000 (46); anti-EspD, 1:2,000 (36); anti-phospho-GSK, 1:2,000 (Cell Signaling). Infrared fluorescent dye-conjugated goat anti-rabbit or anti-mouse secondary antibodies (LiCor) were used at a dilution of 1:10,000. Blots were developed using an Odyssey imaging system (LiCor), and quantification was performed using the manufacturer's software. An elution ratio for coeluted proteins was calculated as the intensity of the eluted protein divided by the intensity of the combined flowthrough and wash fractions. Comparisons were made by Student's t test (two sided), and P values of <0.05 were considered to be significant.
RESULTS
The carboxyl terminus of EspB is accessible to host cytoplasmic kinases.
Translocon components EspB and EspD, and their counterparts in other T3SSs, are thought to span the cytoplasmic membranes of host cells to form pores through which effectors are translocated. Bioinformatic analyses (“Prediction of transmembrane helices in proteins” program, TMHMM server, version 2.0 [http://www.cbs.dtu.dk/services/TMHMM/]) indicate a high likelihood of a single transmembrane domain from residues 97 to 119 of EspB (Fig. 1A). To resolve the membrane topology of EspB, we took advantage of a method developed to identify translocated proteins on the basis of the ability of host cell kinases to phosphorylate a 13-residue peptide sequence from GSK (15). In separate plasmids, we inserted sequences encoding the GSK tag after residues 36 and 186, on either side of the predicted transmembrane domain of espB. These sites were previously demonstrated to be permissive for insertions (28). The resulting plasmids were confirmed to restore A/E ability, as indicated by a positive FAS test when they were transformed into the espB mutant strain UMD864 (data not shown), and each protein was expressed and secreted as expected (Fig. 1B). To determine whether the GSK tag from each construct was accessible to host cell kinases, we infected HeLa cells and analyzed cell lysates by immunoblotting using an anti-phospho-GSK antibody (Fig. 1C, lanes 4 to 6). We were able to detect a specific band migrating at the expected position in lysates of cells infected with EPEC bacteria expressing EspB with a GSK tag after residue 186. In contrast, we were unable to detect a similar band in cells infected with EPEC expressing EspB with a GSK tag after residue 36. As expected, no band was detected in cells infected with the espB mutant complemented with a plasmid encoding wild-type, untagged EspB. These data confirm the cytoplasmic location of the C terminus of EspB and are consistent with a host cell membrane topology in which there is a single transmembrane domain and the N terminus is extracellular. Furthermore, these results do not confirm earlier suggestions that translocated EspB resides in the cytoplasm of host cells (46, 51). However, the possibility that the entire protein is cytoplasmic but the GSK tag at the amino terminus is inaccessible to host cell kinases cannot be excluded.
Fig. 1.

Predicted topology of EspB and secretion, phosphorylation, and cell association of EspB derivatives containing GSK tags. (A) Probability plot of EspB transmembrane domains generated using TMHMM. The calculated probability of transmembrane domains as a function of amino acid residue number is indicated by vertical lines. (B) Culture supernatants from bacteria grown in a tissue culture medium inducing type 3 secretion were concentrated, separated by SDS-PAGE, and subjected to immunoblotting with an antibody to EspB. Bacterial strains tested included escN mutant strain CVD452 (lane 1), wild-type EPEC strain E2348/69 (lane 2), espB mutant strain UMD864 containing the vector control plasmid pACYC184 (lane 3), and UMD864 containing a plasmid encoding either wild-type EspB (lane 4), EspB with a GSK tag inserted after residue 36 (lane 5) or after residue 186 (lane 6), or EspB with a GSK tag inserted after residue 186 but containing nonfunctional insertions after residue 75 (lane 7), 78 (lane 8), 166 (lane 9), or 305 (lane 10). (C) HeLa cells were infected for 2 h at an MOI of 300 with the same bacterial strains as those in the corresponding lanes in panel B. Cell lysates were separated by SDS-PAGE and were subjected to immunoblotting with an antibody to phosphorylated GSK. The asterisk indicates a cross-reactive protein, possibly endogenous phosphorylated GSK, which serves as a loading control. (D) The cell lysates described in the legend to panel C were probed with an antibody to EspB. The positions of molecular mass markers (in thousands) are indicated to the right of each panel.
Interactions among EspA, EspB, and EspD.
Data regarding interactions among the EspA, EspB, and EspD translocon components have been contradictory. One potential problem underlying this difficulty is that the insertion of tags at the termini to detect such interactions may have interfered with the function of the proteins. To overcome this limitation, we sought to insert a polyhistidine tag at permissive sites in EspA, EspB, and EspD based on our previous findings and published data. We had previously generated a series of in-frame insertion mutations in espB and in-frame insertion-deletion mutants of espD, and we had tested whether each allele could complement a corresponding null mutant to restore A/E activity (7, 28). Here we generated plasmids with polyhistidine tags inserted at permissive sites after residue 186 of EspB and replacing residues 32 to 46 of EspD to generate functional His-EspB and His-EspD. For the generation of functional His-EspA, we chose a site after residue 123 to avoid disrupting the terminal domains predicted by the partial crystal structure to be involved in EspA filament interactions (52). All constructs were transformed into corresponding null mutants and were confirmed to be functional by the FAS assay.
Next, we tested whether each translocon component is capable of interacting with the other two by examining eluates from nickel columns onto which we had loaded culture supernatants from a null mutant complemented with the plasmid encoding the corresponding tagged component. We found that EspA and EspB both coeluted with His-EspD (Fig. 2A), EspD coeluted with His-EspA (Fig. 2B), and EspD coeluted with His-EspB (Fig. 2C). In contrast, EspA was not coeluted with His-EspB, or vice versa. These data indicate that EspD interacts with both EspA and with EspB after secretion. However, we were unable to confirm an interaction between EspA and EspB. These data are consistent with prior observations regarding a direct interaction between EspB and EspD (19), and they provide the first experimental evidence that EspA, the main component of the filament, interacts directly with EspD, which is purported to form the main component of the pore. These data also suggest an ordered interaction of the filament needle EscF with the filament component EspA, of EspA with EspD, and of EspD with EspB.
Fig. 2.
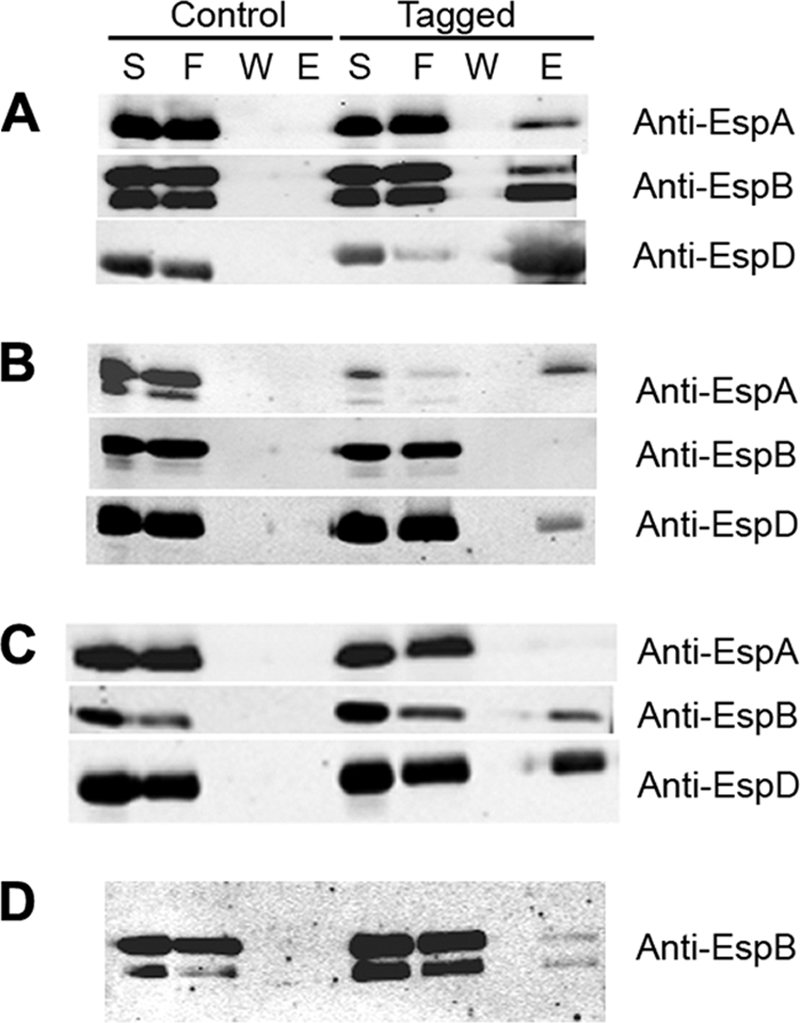
Coelution of EspA, EspB, and EspD translocation proteins from culture supernatants. (A) Supernatant proteins from espD mutant strain UMD870 complemented with a plasmid encoding either EspD (control) or functional, histidine-tagged EspD (tagged) were incubated with nickel-nitrilotriacetic acid agarose beads, and the supernatant (S), flowthrough (F), final wash (W), and eluate (E) fractions were collected and analyzed by immunoblotting using the indicated antibodies. (B and C) Fractions from the supernatants of espA mutant strain UMD872 complemented with a plasmid encoding either EspA (control) or functional, histidine-tagged EspA (tagged) (B) and fractions from the supernatants of espB mutant strain UMD864 complemented with a plasmid encoding either EspB (control) or functional, histidine-tagged EspB (tagged) (C) were similarly analyzed. (D) Fractions from the supernatants of espD mutant strain UMD870 containing empty plasmid pACYC184 cocultured with espADB deletion mutant UMD867 containing a plasmid encoding either wild-type EspD (control) or histidine-tagged EspD (tagged) were analyzed using an anti-EspB antibody. The degree of protein degradation varies from assay to assay.
T3SS translocator and effector proteins must adopt a largely unfolded state to transit the needle complex, given the size constraints imposed by the visualized needle conduit (42). To determine whether the interaction between EspB and EspD must take place concurrently with the emergence of the translocator proteins from the EscF needle prior to refolding, we tested whether EspB secreted by the espD mutant strain UMD870 could be coeluted with His-EspD expressed from a plasmid and secreted by a different strain, espADB mutant UMD867. We mixed overnight cultures from the two strains in equal amounts and grew them together in tissue culture medium to induce the T3SS. We found that EspB expressed from one strain could indeed coelute with His-EspD expressed from another (Fig. 2D). Thus, we conclude that a productive interaction between the two translocon components can occur subsequent to exit from the T3SS. Furthermore, these data suggest that the EspB-EspD interaction does not require a simultaneous interaction of EspD with the EspA filament.
Identification of EspB domains required for interaction with EspD.
To identify domains of EspB required for interaction with EspD, we first modified the chromosomal espD gene of the espB deletion mutant strain UMD864 by replacing codons 32 to 45 with sequences encoding an in-frame polyhistidine tag and recombination scar to generate strain UMD898. Next, we transformed into strain UMD898 plasmids encoding wild-type EspB, 2 EspB variants with in-frame insertions that do not disrupt function (EspB128 and EspB179), and 17 EspB variants with in-frame insertions that disrupt function. We verified that the strain transformed with the plasmid encoding wild-type EspB is positive in the FAS assay to confirm that the polyhistidine-tagged EspD is functional. As a negative control, we transformed the plasmid encoding wild-type EspB into strain UMD864, which has an in-frame espB deletion but encodes untagged EspD. We could not detect secretion of 5 EspB variants in the supernatant and did not pursue these further, while secretion of the 14 other variants was confirmed (data not shown). We then attempted to coelute the control and experimental variants of EspB with His-EspD by nickel affinity purification, and we analyzed the levels of the protein by quantitative immunoblotting. The large number of strains and replicates analyzed precludes presentation of these immunoblots. For analysis of these data, we divided the signal present in the eluate by that present in the flowthrough to compute the relative coelution intensity for each (Fig. 3). Our analysis revealed that EspB variants with insertions after residues 75, 78, and 166 were coeluted with His-EspD at significantly lower levels than wild-type EspB (P, 0.037, 0.024, and 0.044, respectively). Furthermore, there were no significant differences in the EspB eluate/flowthrough ratios between strains expressing these three variants and the negative-control strain encoding wild-type EspB and EspD without a tag. An additional EspB variant with an insertion after residue 260 appeared to have a reduced EspB eluate/flowthrough ratio. However, the values were not significantly different from that of either the positive control encoding wild-type EspB or the negative control encoding EspD without a tag (P, 0.330 and 0.108, respectively), and therefore, the ability of this EspB variant to interact with EspD remains ambiguous. An additional variant is worthy of attention. The eluate/flowthrough ratio of the functional EspB variant that has an insertion after residue 179 appeared to be reduced from that of the positive control, but this difference was not significant (P, 0.25), and the ratio for this variant was significantly greater than that for the negative control (P, 0.003). The eluate/flowthrough ratios of the remaining EspB variants, including the eight that are nonfunctional and the functional variant with an insertion after residue 128, were significantly greater than that of the negative control and equivalent to that of the positive control. In summary, our data suggest that domains on both sides of the EspB putative transmembrane domain near residues 75 to 78 and 166 are critical for binding to EspD. In addition, we cannot exclude a contribution of residues near 179 and 260 to the interaction between EspB and EspD.
Fig. 3.

Coelution of mutant forms of EspB with EspD. Supernatants from EPEC espB mutant strains lacking (UMD864) or containing (UMD898) a chromosomal espD gene encoding functional histidine-tagged EspD were incubated with nickel-NTA agarose beads, and the flowthrough and eluate fractions were analyzed by quantitative immunoblotting using an Odyssey infrared imaging system (LiCor). Each bar represents the mean ratio of the intensity of the eluate to that of the flowthrough from a minimum of four experiments. Error bars, standard errors of the means. Strains tested include UMD864 complemented with a plasmid encoding wild-type EspB (Neg); UMD898 complemented with a plasmid encoding wild-type EspB (Pos); and UMD898 containing plasmids that encode nonfunctional (bars 48, 68, 70, 75, 78, 98, 101, 166, 179, 239, 260, 289, and 305) or functional (bar 128) EspB proteins labeled according to the last residue prior to insertion mutations. The asterisks indicate values that are significantly different from that of the positive control but not significantly different from that of the negative control.
Mutations that disrupt EspB-EspD interactions are associated with failure of EspB translocation.
To explore the functional significance of the EspB-EspD interaction, we tested the hypothesis that espB mutations that interfere with EspB-EspD interactions would also interfere with the targeting of the C terminus of EspB to the host cell cytoplasm. For this purpose, we inserted codons for a GSK tag at the permissive carboxyl-terminal site after codon 186 in the three nonfunctional espB alleles encoding EspB variants that do not coelute with EspD. A GSK tag at this site was phosphorylated by host kinases in the context of an otherwise wild-type EspB protein (Fig. 1C, lane 6). As a positive control, we also inserted a GSK tag after codon 186 in an EspB variant that binds EspD, creating plasmids pEspB75-186GSK, pEspB78-186GSK, pEspB166-186GSK, and pEspB305-186GSK. These plasmids were transformed into espB mutant strain UMD864, and secretion of each EspB variant by the resulting transformants was verified (Fig. 1B). Next, HeLa cells were infected with each strain, and cell lysates were analyzed by immunoblotting with a phospho-GSK-specific antibody. Of note, these lysates were prepared with 1% Triton X-100, which solubilizes host membranes. We could not detect phosphorylation of the GSK tags of the three EspB variants with insertions after codons 75, 78, and 166, which are defective in binding to EspD (Fig. 1C, lanes 7 to 9), while the phosphorylated tag of the control EspB variant with an insertion after residue 305, which retains the ability to interact with EspD, was obvious (Fig. 1C, lane 10). When the blot was reprobed with an anti-EspB antibody, we were able to detect cell-associated EspB in the samples from strains expressing each functional protein (Fig. 1D, lanes 4 to 6) and the nonfunctional protein that retains its ability to bind to EspD (Fig. 1D lane 10), but not in those from strains expressing the nonfunctional proteins that cannot bind EspD (Fig. 1D, lanes 7 to 9). Thus, we conclude that in contrast to those of wild-type EspB and EspB variants that can bind EspD, the carboxyl termini of EspB variants unable to bind EspD are not exposed to the host cell cytoplasm, and the proteins are not associated with the host cell. These results suggest that the interaction between EspB and EspD is required for the recruitment of EspB to host cells and for EspB to assume its proper host cell membrane topology. Furthermore, since these EspB variants are defective in effector translocation (28), we conclude that the EspB-EspD interaction is required for effector translocation.
DISCUSSION
The virulence of some of the deadliest Gram-negative bacterial pathogens depends on the translocation of effector proteins into host cells by T3SSs. EspB, a member of a family of hydrophobic translocon components that includes YopD and IpaC, is an essential virulence factor in human and animal models of EPEC infection (1, 43). EspB and EspD, like their counterparts in other bacteria, are thought to interact with one another and to form a translocation pore in host cell membranes through which effector proteins pass. The results of this study shed light on both of these EspB functions. Using a novel assay, we demonstrate that the carboxyl terminus of EspB is in the host cytoplasm, and we suggest a specific host cell membrane topology, with the amino terminus exposed to the extracellular environment. Furthermore, we were able to confirm an interaction between EspB and EspD, and we provide the first evidence of an interaction between EspD and the hydrophilic transporter EspA. By quantitative immunoblot analysis, we determined that two regions of EspB, localized near residues 75 to 78 and 166, on either side of the transmembrane domain, are critical for EspD binding. Last, we provide evidence that these EspD binding regions are required for EspB to properly associate with the host cell membrane. Together, these data improve our understanding of the interactions and configurations of type 3 translocator proteins (Fig. 4).
Fig. 4.

Models of EspB. (A) Schematic of EspB indicating functional regions of the protein. This is a linear depiction of EspB, as indicated by the scale. The approximate locations of mutations relevant to this report are indicated by arrows. In the upper bar, regions of the protein that are required for interaction with EspD are shown in black, and those possibly involved in such interactions are shown in gray. A region predicted to form a transmembrane domain is shown in yellow. In the lower bar, regions previously shown to be tolerant to mutation are shown in green, and those required for function are shown in red. (B) Representation of the EPEC T3SS needle and translocation filament. The relatively short EscF needle is depicted joined end-to-end with the wider and much longer EspA filament. EspA makes direct contact with EspD, which, in turn, makes contact with other EspD molecules and with EspB. EspD molecules that bind EspA may not be able to bind EspB simultaneously. Both EspD and EspB are expected to cross the host cell membrane.
Previous studies have demonstrated that EspB associates with the host cell membrane after infection (19, 51), but the topology of EspB and related proteins in that environment had not been systematically studied previously. We took advantage of the 13-amino-acid GSK tag, developed to study the protein translocation of T3SS and type IV secretion system effectors (15), to determine the topology of EspB in the host membrane. By placing the GSK tag in sites of EspB that had previously been determined to tolerate insertions without loss of function (28), we were able to determine which side of the transmembrane domain is accessible to a cytoplasmic kinase. Our results unambiguously demonstrate that GSK inserted after EspB residue 186 is phosphorylated after infection of HeLa cells by EPEC. In contrast, no phosphorylation of GSK inserted after residue 36 could be detected. This result is consistent with studies that demonstrated that fusions of calmodulin-dependent adenylyl cyclase to the carboxyl terminus of EspB result in high levels of enzymatic activity (46, 51), a result that was originally interpreted as evidence that the entire protein is translocated to the cytoplasm. However, upon close reexamination, previous results showing focal EspB immunostaining are not consistent with diffuse cytoplasmic localization but rather suggest localization in insoluble or membrane-bound compartments. Of note, EspB staining of infected cells was observed by another group only in close association with EspA filaments, a result inconsistent with diffuse cytoplasmic localization (17). Our results are also consistent with studies demonstrating that a region of EspB on the carboxyl side of the transmembrane domain interacts with host cell myosin (20), as well as with studies of interactions between a carboxyl domain of IpaC and Src (31).
While our results are consistent with a topology in which EspB has one transmembrane domain and the amino terminus is extracellular, there are limitations to our study. We might have been unable to detect phosphorylation of the GSK tag after residue 36 because it was inaccessible to host cell kinases, rather than because it was extracellular. However, we believe this to be unlikely given the strong predictions of a single transmembrane domain by analytical software and previous experimental evidence that EspB is inserted into the host membrane (19, 51). Whether our topology results pertain to other members of the family to which EspB belongs remains an open question, since various algorithms for the prediction of transmembrane domains assign zero to 3 such features in these proteins (data not shown).
Using versions of each protein that have internal insertions of polyhistidine tags, which were confirmed to retain function, we were able to coelute EspD with tagged EspA, and vice versa, and EspD with tagged EspB, and vice versa. However, EspA did not coelute with tagged EspB, nor did EspB coelute with tagged EspA. These results provide evidence for a direct interaction between the hydrophilic translocon component EspA and the putative primary pore component EspD and for a direct interaction between the two hydrophobic translocon putative pore components EspD and EspB. Moreover, they suggest an order of interaction, with EspA interacting with the needle protein EscF and with EspD, EspD interacting with EspA and EspB, and EspB interacting only with EspD (Fig. 4B). It is noteworthy that this order is identical to that determined for distantly related Shigella translocon components by entirely different methods. In Shigella the hydrophilic translocator IpaD interacts with the needle protein MxiH and with the primary pore component IpaB, and IpaB interacts with IpaD and IpaC (10). Furthermore, our results confirm and extend those of an earlier study in which an interaction between EspB and EspD was detected by coeluting EspD from cultured supernatants with EspB tagged at the N terminus (19). These results are also consistent with the observation that EspA filaments are markedly shorter in espD mutants but not in espB mutants, suggesting a more direct interaction of EspA with EspD than with EspB (24). In contrast, we did not confirm a previously reported interaction between EspA and EspB (17). Those authors used multiple methods to investigate this interaction, including coimmunoprecipitation, solid-phase binding of tagged proteins, and coelution of EspB with N-terminally tagged EspA. Furthermore, a potential EspA-EspB interaction is also consistent with an interaction between N-terminally tagged LcrV and YopD (38). However, none of the tagged proteins were confirmed to retain function. It is possible that the affinity of the EspA-EspB interaction did not allow coelution under the conditions that we employed. Until binding affinities between functional proteins can be measured and assessed for biological relevance, a direct EspA-EspB interaction remains unconfirmed. The fact that no interactions among the translocon components EspA, EspB, and EspD could be detected using a yeast two-hybrid system (4) is another indication that caution should be exercised in interpreting such interactions observed using proteins that are modified but not confirmed to retain function. The fact that EspA did not coelute with tagged EspB, and vice versa, while both proteins coeluted with EspD suggests the hypothesis that the abilities of EspD to bind EspA and EspB are mutually exclusive. However, additional studies will be required to test this hypothesis, and alternative hypotheses are also possible.
While EspD and EspB might interact with one another immediately upon emergence in an unfolded state from the continuous EscF-EspA conduit, we provide evidence suggesting that such an immediate interaction is not necessary. We found that tagged EspD secreted by an espADB triple mutant was able to bind to EspB secreted by an espD mutant when both were cultured together. Thus, binding likely occurs between fully folded proteins.
We exploited quantitative immunoblotting and our previously reported collection of nonfunctional espB alleles that encode EspB variants with 5- or 31-amino-acid insertions (28) to provide the first comprehensive analysis of interaction domains between hydrophobic translocon components. It is possible that insertions that interfere with binding to EspD do so by causing dramatic changes in overall EspB structure rather than by disrupting a specific binding domain. However, the fact that the majority of insertions had no effect on binding is reassuring in this regard. We found at least two domains of EspB required for the EspD interaction, as defined by EspB variants with insertions at residues 75 and 78 and at residue 166 that fail to do so. In addition, an EspB variant with an insertion at residue 260 was coeluted with a value that was not significantly different from that of either the positive or the negative control, which may indicate that a third region of EspB is involved in EspD binding. These domains are located on both sides of the EspB transmembrane domain and surround insertions that are coeluted with EspD at levels indistinguishable from those of the wild type, indicating that separate domains of EspB interact with EspD. EspB-EspD interactions likely continue after insertion into the host cell membrane, as supported by the fact that they can both be extracted from the membrane (19, 41). Thus, these results suggest that the hydrophobic translocator proteins interact with each other on both sides of the host cell membrane. Our unpublished data indicate that the N terminus of EspD is localized inside the cytoplasm. Transmembrane domain predictions and experimental evidence indicate that EspD and related proteins traverse the host cell membrane twice (18). These considerations lead to specific hypotheses about regions of EspD that interact with particular domains of EspB. Experiments designed to test these hypotheses are ongoing.
We took advantage of the EspB-GSK fusion at residue 186 to determine whether mutations that disrupt EspD binding also affect the ability of EspB to properly associate with the host cell membrane. Accordingly, we generated versions of EspB that had both a GSK fusion after residue 186 and insertions at residues 75, 78, or 166 that interfere with EspD binding. In each case, no phosphorylation of the GSK tag was detected in EspB variants unable to bind to EspD. As an additional control, we also tested the ability of EspB with a GSK tag after residue 186 and an insertion after residue 305 to become phosphorylated. This EspB variant is nonfunctional with regard to the translocation of effectors but is capable of EspD binding and is phosphorylated to the same extent as EspB lacking additional insertions. Thus, the ability to bind EspD appears to be required for EspB itself to be inserted in its proper topology into the host cell membrane but insufficient to permit effector translocation.
We also note that the ability to bind EspD, as demonstrated in this study, is not sufficient for EspB to facilitate hemolytic activity, as previously demonstrated (28). Thus, EspB variants with insertions after residues 48, 60, 70, 98, 101, 289, and 305 are able to bind EspD but are nonfunctional and have significantly reduced hemolytic activity. EspB variants with insertions after residues 75, 78, and 166, which are defective in EspD binding, also have markedly reduced hemolytic activity and are nonfunctional. Interestingly however, EspB179 may have reduced binding to EspD and has reduced hemolytic activity, but these reduced activities are sufficient for function.
From this study and others, a model of EspB as a complex translocator-effector protein has begun to emerge (Fig. 4A). The first 38 amino acids of EspB constitute a domain that is required for secretion but highly tolerant of mutation. Residues 48 through 96 are essential for function and include a domain defined by residues 75 and 78 that is required for binding to EspD. We propose that these first 96 residues are exposed on the extracellular surface after the insertion of EspB into the host cell membrane via the transmembrane domain from residues 97 to 119. Residue 166 on the cytoplasmic side of the host cell membrane identifies another domain that is essential for function and EspD binding, while insertion at residue 179 may reduce EspD binding and hemolysis, but function is retained. Residues 179 through 203 are highly tolerant of insertions and can be deleted without loss of EspB attaching and effacing activity. However, the region between residues 158 and 219 is required and sufficient for binding to myosin (20). The region between residues 220 and the carboxyl terminus includes residues tolerant and intolerant of insertions, including an insertion at residue 239 that interferes with effector translocation but not with hemolysis (28).
While these and other studies have shed light on the interactions between EspB and EspD and between related hydrophobic translocator proteins, much remains to be learned about the assembly, architecture, and mechanism of action of these critical virulence determinants.
Supplementary Material
ACKNOWLEDGMENTS
We thank Li-Ching Lai for supplying pLCL134 and Gad Frankel for critical review of the manuscript.
This work was supported by Public Health Service award AI32074 from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 15 April 2011.
REFERENCES
- 1. Abe A., Heczko U., Hegele R. G., Finlay B. B. 1998. Two enteropathogenic Escherichia coli type III secreted proteins, EspA and EspB, are virulence factors. J. Exp. Med. 188:1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blocker A., et al. 1999. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147:683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cornelis G. R. 2006. The type III secretion injectisome. Nat. Rev. Microbiol. 4:811–825 [DOI] [PubMed] [Google Scholar]
- 4. Creasey E. A., Delahay R. M., Daniell S. J., Frankel G. 2003. Yeast two-hybrid system survey of interactions between LEE-encoded proteins of enteropathogenic Escherichia coli. Microbiology 149:2093–2106 [DOI] [PubMed] [Google Scholar]
- 5. Daniell S. J., et al. 2003. 3D structure of EspA filaments from enteropathogenic Escherichia coli. Mol. Microbiol. 49:301–308 [DOI] [PubMed] [Google Scholar]
- 6. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng Q., Luo W., Donnenberg M. S. 2007. Rapid site-directed domain scanning mutagenesis of enteropathogenic Escherichia coli espD. Biol. Proced. Online 9:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Derewenda U., et al. 2004. The structure of Yersinia pestis V-antigen, an essential virulence factor and mediator of immunity against plague. Structure 12:301–306 doi:10.1016/j.str.2004.01.010 [DOI] [PubMed] [Google Scholar]
- 9. Reference deleted.
- 10. Epler C. R., Dickenson N. E., Olive A. J., Picking W. L., Picking W. D. 2009. Liposomes recruit IpaC to the Shigella flexneri type III secretion apparatus needle as a final step in secretion induction. Infect. Immun. 77:2754–2761 doi:10.1128/IAI.00190-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Espina M., et al. 2006. IpaD localizes to the tip of the type III secretion system needle of Shigella flexneri. Infect. Immun. 74:4391–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faudry E., Vernier G., Neumann E., Forge V., Attree I. 2006. Synergistic pore formation by type III toxin translocators of Pseudomonas aeruginosa. Biochemistry 45:8117–8123 doi:10.1021/bi060452+ [DOI] [PubMed] [Google Scholar]
- 13. Frithz-Lindsten E., et al. 1998. Functional conservation of the effector protein translocators PopB/YopB and PopD/YopD of Pseudomonas aeruginosa and Yersinia pseudotuberculosis. Mol. Microbiol. 29:1155–1165 [DOI] [PubMed] [Google Scholar]
- 14. Galán J. E., Wolf-Watz H. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573 [DOI] [PubMed] [Google Scholar]
- 15. Garcia J. T., et al. 2006. Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect. Immun. 74:5645–5657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Håkansson S., et al. 1996. The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and displays a contact-dependent membrane disrupting activity. EMBO J. 15:5812–5823 [PMC free article] [PubMed] [Google Scholar]
- 17. Hartland E. L., et al. 2000. The type III protein translocation system of enteropathogenic Escherichia coli involves EspA-EspB protein interactions. Mol. Microbiol. 35:1483–1492 [DOI] [PubMed] [Google Scholar]
- 18. Hume P. J., McGhie E. J., Hayward R. D., Koronakis V. 2003. The purified Shigella IpaB and Salmonella SipB translocators share biochemical properties and membrane topology. Mol. Microbiol. 49:425–439 [DOI] [PubMed] [Google Scholar]
- 19. Ide T., et al. 2001. Characterization of translocation pores inserted into plasma membranes by type III-secreted Esp proteins of enteropathogenic Escherichia coli. Cell. Microbiol. 3:669–679 [DOI] [PubMed] [Google Scholar]
- 20. Iizumi Y., et al. 2007. The enteropathogenic E. coli effector EspB facilitates microvillus effacing and antiphagocytosis by inhibiting myosin function. Cell Host Microbe 2:383–392 [DOI] [PubMed] [Google Scholar]
- 21. Jarvis K. G., et al. 1995. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U. S. A. 92:7996–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knappstein S., Ide T., Schmidt M. A., Heusipp G. 2004. α1-Antitrypsin binds to and interferes with functionality of EspB from atypical and typical enteropathogenic Escherichia coli strains. Infect. Immun. 72:4344–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knutton S., Baldwin T., Williams P. H., McNeish A. S. 1988. New diagnostic test for enteropathogenic Escherichia coli. Lancet i:1337 (Letter.) [DOI] [PubMed] [Google Scholar]
- 24. Knutton S., et al. 1998. A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J. 17:2166–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kodama T., et al. 2002. The EspB protein of enterohaemorrhagic Escherichia coli interacts directly with alpha-catenin. Cell. Microbiol. 4:213–222 [DOI] [PubMed] [Google Scholar]
- 26. Reference deleted.
- 27. Reference deleted.
- 28. Luo W., Donnenberg M. S. 2006. Analysis of the function of enteropathogenic Escherichia coli EspB by random mutagenesis. Infect. Immun. 74:810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDaniel T. K., Jarvis K. G., Donnenberg M. S., Kaper J. B. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moon H. W., Whipp S. C., Argenzio R. A., Levine M. M., Giannella R. A. 1983. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41:1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mounier J., et al. 2009. The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog. 5:e1000271 doi:10.1371/journal.ppat.1000271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mueller C. A., Broz P., Cornelis G. R. 2008. The type III secretion system tip complex and translocon. Mol. Microbiol. 68:1085–1095 [DOI] [PubMed] [Google Scholar]
- 33. Mueller C. A., et al. 2005. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310:674–676 [DOI] [PubMed] [Google Scholar]
- 34. Nataro J. P., Kaper J. B. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neyt C., Cornelis G. R. 1999. Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol. Microbiol. 33:971–981 [DOI] [PubMed] [Google Scholar]
- 36. O'Connell C. B., et al. 2004. SepL, a protein required for enteropathogenic Escherichia coli type III translocation, interacts with secretion component SepD. Mol. Microbiol. 52:1613–1625 [DOI] [PubMed] [Google Scholar]
- 37. Olive A. J., et al. 2007. Bile salts stimulate recruitment of IpaB to the Shigella flexneri surface, where it colocalizes with IpaD at the tip of the type III secretion needle. Infect. Immun. 75:2626–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarker M. R., Neyt C., Stainier I., Cornelis G. R. 1998. The Yersinia Yop virulon: LcrV is required for extrusion of the translocators YopB and YopD. J. Bacteriol. 180:1207–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schoehn G., et al. 2003. Oligomerization of type III secretion proteins PopB and PopD precedes pore formation in Pseudomonas. EMBO J. 22:4957–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schraidt O., et al. 2010. Topology and organization of the Salmonella typhimurium type III secretion needle complex components. PLoS Pathog. 6:e1000824 doi:10.1371/journal.ppat.1000824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shaw R. K., Daniell S., Ebel F., Frankel G., Knutton S. 2001. EspA filament-mediated protein translocation into red blood cells. Cell. Microbiol. 3:213–222 [DOI] [PubMed] [Google Scholar]
- 42. Stebbins C. E., Galan J. E. 2001. Maintenance of an unfolded polypeptide by a cognate chaperone in bacterial type III secretion. Nature 414:77–81 [DOI] [PubMed] [Google Scholar]
- 43. Tacket C. O., et al. 2000. Role of EspB in experimental human enteropathogenic Escherichia coli infection. Infect. Immun. 68:3689–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tardy F., et al. 1999. Yersinia enterocolitica type III secretion-translocation system: channel formation by secreted Yops. EMBO J. 18:6793–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor K. A., Luther P. W., Donnenberg M. S. 1999. Expression of the EspB protein of enteropathogenic Escherichia coli within HeLa cells affects stress fibers and cellular morphology. Infect. Immun. 67:120–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taylor K. A., O'Connell C. B., Luther P. W., Donnenberg M. S. 1998. The EspB protein of enteropathogenic Escherichia coli is targeted to the cytoplasm of infected HeLa cells. Infect. Immun. 66:5501–5507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tran Van Nhieu G., Caron E., Hall A., Sansonetti P. J. 1999. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 18:3249–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wachter C., Beinke C., Mattes M., Schmidt M. A. 1999. Insertion of EspD into epithelial target cell membranes by infecting enteropathogenic Escherichia coli. Mol. Microbiol. 31:1695–1707 [DOI] [PubMed] [Google Scholar]
- 49. Wang Y. A., Yu X., Yip C., Strynadka N. C., Egelman E. H. 2006. Structural polymorphism in bacterial EspA filaments revealed by cryo-EM and an improved approach to helical reconstruction. Structure 14:1189–1196 [DOI] [PubMed] [Google Scholar]
- 50. Warawa J., Finlay B. B., Kenny B. 1999. Type III secretion-dependent hemolytic activity of enteropathogenic Escherichia coli. Infect. Immun. 67:5538–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wolff C., Nisan I., Hanski E., Frankel G., Rosenshine I. 1998. Protein translocation into host epithelial cells by infecting enteropathogenic Escherichia coli. Mol. Microbiol. 28:143–155 [DOI] [PubMed] [Google Scholar]
- 52. Yip C. K., Finlay B. B., Strynadka N. C. 2005. Structural characterization of a type III secretion system filament protein in complex with its chaperone. Nat. Struct. Mol. Biol. 12:75–81 [DOI] [PubMed] [Google Scholar]
- 53. Yip C. K., et al. 2005. Structural characterization of the molecular platform for type III secretion system assembly. Nature 435:702–707 [DOI] [PubMed] [Google Scholar]
- 54. Zarivach R., et al. 2008. Structural analysis of the essential self-cleaving type III secretion proteins EscU and SpaS. Nature 453:124–127 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.