

Abstract
The Saccharomyces cerevisiae Rmi1 protein is a component of the highly conserved Sgs1-Top3-Rmi1 complex. Deletion of SGS1, TOP3, or RMI1 is synthetically lethal when combined with the loss of the Mus81-Mms4 or Slx1-Slx4 endonucleases, which have been implicated in Holliday junction (HJ) resolution. To investigate the causes of this synthetic lethality, we isolated a temperature-sensitive mutant of the RMI1 strain, referred to as the rmi1-1 mutant. At the restrictive temperature, this mutant phenocopies an rmi1Δ strain but behaves like the wild type at the permissive temperature. Following a transient exposure to methyl methanesulfonate, rmi1-1 mutants accumulate unprocessed homologous recombination repair (HRR) intermediates. These intermediates are slowly resolved at the restrictive temperature, revealing a redundant resolution activity when Rmi1 is impaired. This resolution depends on Mus81-Mms4 but not on either Slx1-Slx4 or another HJ resolvase, Yen1. Similar results were also observed when Top3 function was impaired. We propose that the Sgs1-Top3-Rmi1 complex constitutes the main pathway for the processing of HJ-containing HRR intermediates but that Mus81-Mms4 can also resolve these intermediates.
INTRODUCTION
The homologous recombination repair (HRR) repair pathway involves the transfer of genetic information between two identical, or highly similar, sequences (59). An important function of the pathway is in the restart of stalled or broken DNA replication forks, but it is also required for double-strand-break (DSB) and interstrand cross-link (ICL) repair (7, 47). The first step in HRR repair is 5′-3′ DNA end resection, resulting in 3′ single-stranded DNA (ssDNA) that becomes coated by Rad51 (7, 76). This Rad51 presynaptic filament invades the sister chromatid or the homologous chromosome to form a displacement loop (D-loop), and DNA synthesis then occurs to restore the missing sequence from the invading strand. Then, either the D-loop is dismantled, allowing completion of repair by the synthesis-dependent strand-annealing (SDSA) pathway, or the DNA gaps are ligated, to form a double Holliday junction (DHJ). DHJs comprise two adjacent, mobile, four-way DNA junctions and can be resolved by two different pathways. The first is via conventional resolution catalyzed by specialized nucleases known as HJ resolvases. HJ resolvases theoretically produce a 1:1 mixture of crossover products (where the flanking DNA is exchanged) and noncrossover products (77). An alternative pathway is DHJ dissolution, in which convergent branch migration of the two HJs produces a hemicatenane structure that is decatenated to form exclusively noncrossover products (86). The criteria used by cells to determine whether mitotic DHJs are processed by resolution or by DHJ dissolution are currently unclear.
In human cells, DHJ dissolution is catalyzed by the heteromeric BLM complex. The BLM complex is composed of BLM (a RecQ helicase), hTOPOIIIα (a type IA topoisomerase), hRMI1, and hRMI2 (69, 73, 85–87). The interactions between BLM, hTOPOIIIα, and hRMI1 are conserved with their S. cerevisiae orthologs, Sgs1, Top3, and Rmi1, although an hRMI2 ortholog appears to be absent from yeast (6, 19, 20, 37, 38, 62, 65). The S. cerevisiae Sgs1-Top3-Rmi1 complex, which has been termed the RTR (RecQ helicase-topoisomerase III-Rmi1) complex (2), appears to play a role similar to that of the human BLM complex in HRR, because the RTR complex is also able to catalyze DHJ dissolution (17). Study of these proteins is important not only because of their involvement in the HRR pathway but also because mutations in the human genes cause syndromes associated with chromosomal instability, premature aging, and cancer predisposition (9, 22, 67). The human BLM gene is orthologous to the Escherichia coli RecQ helicase and has four paralogs, designated WRN, RECQ1, RECQ4, and RECQ5 (22, 82). Mutations in BLM cause the rare autosomal recessive disorder Bloom's syndrome (BS), and mutations in WRN cause the progeroid disorder Werner's syndrome (29, 90). RECQ4 mutations can cause three distinct disorders, namely, Rothmund-Thomson syndrome, Baller-Gerold syndrome, and RAPADILINO syndrome (46, 72, 81). Recent studies have shown that a polymorphism in hRMI1, leading to a Ser455Asn substitution, confers an increased risk of acute myeloid leukemia/myelodysplastic syndrome and malignant melanoma (13, 14). Although the frequency of this allele in the general population is unknown, these findings suggest that impairment of hRMI1 function leads to an increased cancer risk.
hRMI1 contains a DUF1767 domain at the N terminus and two OB fold domains, OB1 at the N terminus and OB2 at the C terminus (87, 89). DUF1767 is required for the proper folding of hRMI1 and has high sequence similarity with Mycobacterium tuberculosis MtRuvA domain III, which binds MtRuvB and is required for HJ branch migration (64, 68, 84). OB1 interacts with BLM and hTOPOIIIα and is essential for the stimulation of DHJ dissolution by hRMI1 (70, 84). OB2 interacts directly with both FANCM1027-1362 and hRMI2 but is dispensable for stability of the BLM complex (26, 41, 73, 84, 87). Although hRMI1 and the hRMI1/hRMI2 complex have very weak ssDNA- and double-strand DNA (dsDNA)-binding activities in vitro, hRMI1 specifically stimulates hTOPOIIIα in DHJ dissolution and also stimulates hTOPOIIIα to relax negatively supercoiled plasmid DNA and to decatenate a single-stranded catenane substrate (70, 85, 88). S. cerevisiae Rmi1 lacks the C-terminal region, which in hRMI1 contains the OB2 domain. Nevertheless, Rmi1 is required for Top3 to bind to Sgs1 and enhances both binding of Top3 to HJs and the ability of Top3 to relax negatively supercoiled plasmid DNA (20). Detailed genetic analysis of RMI1 has been severely hampered because rmi1Δ deletion mutants exhibit poor growth, reduced viability, and a rapid accumulation of second-site suppressor mutations (19, 62).
HJ resolution via an E. coli RuvC-like mechanism involves symmetrical HJ cleavage on opposing strands to generate products that can be ligated together (28). Human GEN1 and SLX1-SLX4 and the S. cerevisiae GEN1 ortholog, Yen1, were recently characterized as eukaryotic HJ resolvases (33, 36, 44, 63, 77, 78). HJ resolution activity has also been observed with S. cerevisiae Slx1-Slx4, although this complex cleaves an HJ asymmetrically (36). Another heterodimer capable of HJ resolution is the human 3′-flap endonuclease, MUS81-EME1, and its S. cerevisiae ortholog, Mus81-Mms4 (25, 80). It is well documented that MUS81-EME1 and Mus81-Mms4 can efficiently cleave nicked HJs, D-loops, and partially regressed replication forks in vitro (21, 23, 24, 35), although it has been demonstrated that these endonucleases are also able to process intact HJs in vitro and in vivo (25, 79, 80). It was also demonstrated recently that DHJs form during HRR of DSBs in mitotic cells in vivo (15). However, it is not clear which of these HJ resolvases are important for cleavage of DHJs arising during HRR in vivo, and whether different mechanisms are invoked in different organisms to process HJs. For example, there appears to be no Yen1/GEN1 homolog in Schizosaccharomyces pombe.
Due to their synthetic lethal interactions, it has not previously been possible to investigate the genetic interactions between the RTR complex and the putative HJ resolvase genes MUS81, MMS4, SLX1, and SLX4 (3, 4, 19, 31, 61, 62). In order to conduct such analyses, we isolated a novel temperature-sensitive mutant of the RMI1 strain, referred to as the rmi1-1 strain. Here, we used this mutant to show that methyl methanesulfonate (MMS)-induced, Rad51-dependent, RusA-sensitive, X-shaped DNA molecules persist during a perturbed mitotic S phase when Rmi1 is impaired, and that these structures are eventually removed by Mus81-Mms4 in vivo.
MATERIALS AND METHODS
S. cerevisiae strains and plasmids.
All strains were isogenic derivatives of T344 (42) or BY4741. All strains carrying gene deletions were either obtained from the yeast deletion collection (EUROSCARF, University of Frankfurt, Germany), constructed using a PCR-based gene disruption method (83), or obtained by tetrad dissection. The rmi1Δ haploid strain was obtained by sporulating a heterozygous RMI1+/− diploid strain. The pYES2-TOP3, pYES2-TOP3Y356F, pYES2-NLS-GFP, pYES2-NLS-RusA-GFP, and pYES2-NLS-RusAD70N plasmids were described previously (53, 55, 65). The pRS415 plasmid is available from Stratagene (United Kingdom). The construction details of pADE2-RMI1 are available upon request. The strains used in this study are listed in Table 1.
Table 1.
Strains used in this study
| Strain or mutation | Genotype |
|---|---|
| BY4741 | MATahis3Δ1leu2Δ0met15Δ0ura3Δ0 |
| BY4743 | MATa/α his3Δ1/his3Δ1leu2Δ0/leu2Δ0LYS2/lys2Δ0met15Δ0/MET15ura3Δ0/ura3Δ0 |
| rmi1-1 | BY4741 + rmi1E69K |
| RMI1+/− | BY4743 + RMI1/RMI1::KanMX4 |
| rad51Δ | BY4741 + RAD51::KanMX4 |
| sgs1Δ | BY4741 + SGS1::KanMX4 |
| rmi1-1 rad51Δ | rmi1-1 + RAD51::KanMX4 |
| rmi1-1 sgs1Δ | rmi1-1 + SGS1::KanMX4 |
| RMI1-HA | BY4741 + RM1-HA-KanMX4 |
| rmi1-1-HA | BY4741 + rmi1E69K-HA-KanMX4 |
| slx1Δ | BY4741 + SLX1::KanMX4 |
| slx4Δ | BY4741 + SLX4::KanMX4 |
| rmi1-1 slx1Δ | rmi1-1 + SLX1::KanMX4 |
| rmi1-1 slx4Δ | rmi1-1 + SLX4::KanMX4 |
| mus81Δ | BY4741 + MUS81::KanMX4 |
| mms4Δ | BY4741 + MMS4::KanMX4 |
| rmi1-1 mus81Δ | rmi1-1 + MUS81::KanMX4 |
| rmi1-1 mms4Δ | rmi1-1 + MMS4::KanMX4 |
| T344 | MATapep4-3 prb1-1122 ura3-52 leu2-3112 reg1-501 gal1 Δtrp1::hisG |
| mus81Δ | T344 + MUS81::KanMX4 |
| slx1Δ | T344 + SLX1::KanMX |
| yen1Δ | T344 + YEN1::KanMX4 |
To isolate a temperature-sensitive mutant of RMI1, a mutagenized RMI1 plasmid library was created by cloning the products of an error-prone PCR into the pRS415 vector. The mutagenized RMI1 plasmid library was transformed into a BY4741 rmi1Δ ade2Δ strain containing a pADE2-RMI1 plasmid expressing the wild-type RMI1, ADE2 and URA3 genes. The resulting strain was grown at 35°C, producing a mixture of white and red colonies in the absence of selection for ADE2. We assumed that the white colonies contained pADE2-RMI1 and a nonfunctional RMI1 gene on pRS415-RMI1, because these colonies were under selective pressure to retain pADE2-RMI1 and were significantly larger than those made by rmi1Δ cells. In contrast, we assumed that the red colonies would contain a functional RMI1 gene on pRS415-RMI1 but no pADE2-RMI1 plasmid. Consequently, the white colonies were chosen and grown at 25°C on yeast-peptone-dextrose (YPD) plates containing 0.005% MMS in order to select for cells containing an RMI1 allele that is functional at 25°C. The resulting candidates were plated onto agar containing 5-fluoroorotic acid (5-FOA) to force loss of the pADE2-RMI1 plasmid, and then a spot dilution assay was performed to identify strains that grew normally at 25°C but poorly at 35°C. One such pRS415-RMI1 plasmid candidate was identified, and DNA sequencing revealed that this RMI1 allele had a codon 69 mutation, altering Glu-69 to Lys (E69K). A PCR-based allele replacement method was then used to integrate this allele into the genome in a BY4741 background in order to create the rmi1-1 mutant (30).
Growth conditions, cell synchronization, and flow cytometry analysis.
For synchronization in G1, strains were grown to early log phase at 25°C and incubated with α-factor mating pheromone at either 25°C or 35°C, as indicated in the figure legends. Where the G1 arrest was performed at 35°C, the cells were incubated at 35°C for a minimum of 1 h. For synchronization in G2/M, strains were incubated with 15 μg/ml nocodazole (Sigma-Aldrich, United Kingdom). To analyze recovery from MMS treatment, the cells were harvested, washed, and then resuspended in fresh medium containing α-factor, which was added in order to prevent a second round of replication. Cell cycle progression was monitored using flow cytometry, as described previously (55, 57).
Two-dimensional (2D) gel electrophoresis.
The hexadecyltrimethylammonium bromide (CTAB) method of DNA extraction was used (51). The DNA extracts were digested with NcoI and were resolved using two-dimensional gel procedures that were described previously (12, 48, 55, 57). The DNA was hybridized to a probe that binds to the early-firing ARS305 replication origin.
Pulsed-field gel electrophoresis.
Analysis of global replication was performed using clamped homogeneous electrophoretic field (CHEF) gels to separate intact chromosomes. Cells (6 × 107) were harvested by centrifugation, and chromosomal DNA was prepared in gel molds according to the manufacturer's instructions (Bio-Rad). Proteinase K incubation was conducted at 37°C to prevent chromosomal fragmentation, which occurs at 50°C (52). The following parameters were used for electrophoresis: duration, 24 h; interval, 100 to 10 s log; angle, 120° to 110° linear; voltage, 200 to 150 V log; temperature, 13°C; rotor speed, 5. An ARS305 probe was used to detect chromosome III by Southern hybridization (75).
Western blot analysis.
Protein extraction, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis were performed as described previously (55). The primary antibodies used were rat antihemagglutinin (anti-HA) (clone 3F10; Roche, United Kingdom) at a 1:200 dilution, mouse antiactin at a 1:1,500 dilution (Abcam, United Kingdom), and Rad53 mouse monoclonal (EL7) at a 1:10 dilution, which was a gift from Marco Foiani (Institute of Molecular Oncology Foundation, Milan, Italy).
Microscopy.
Yeast immunofluorescence was performed as described previously (1). To visualize mitotic spindles, the primary antibody used was rat anti-alpha tubulin (ABD Serotech, United Kingdom), and the secondary antibody was Alexa Fluor 555-conjugated goat anti-rat immunoglobulin (Invitrogen, United Kingdom). Cells were stained with DAPI mounting medium (Vectashield, United Kingdom) prior to microscopic analysis. Images were captured with a Nikon Eclipse 80i fluorescence microscope, using Lucia G/F software. Samples in which 60%, 40%, or 20% of cells were in G2/M on the flow cytometry trace were scored, which represented the 60-, 70-, and 80-min samples for wild-type cells and the 80-, 90-, and 120-min samples for rmi1-1 cells.
RESULTS
Characterization of the temperature-sensitive rmi1-1 mutant.
S. cerevisiae rmi1Δ deletion mutants have proven exceptionally difficult to study because they exhibit poor growth and reduced viability and rapidly accumulate second-site suppressor mutations (19, 62). To further investigate the phenotypic consequences of acute inactivation of Rmi1, we isolated a temperature-sensitive mutant of the RMI1 strain, referred to as the rmi1-1 strain (described in Materials and Methods). DNA sequencing revealed that this RMI1 allele had a codon 69 mutation, altering Glu-69 to Lys (E69K) (Fig. 1A). The rmi1-1 mutant exhibited wild-type growth at the permissive temperature (25°C) but showed severe growth inhibition at the restrictive temperature (35°C) (Fig. 1B). Furthermore, rmi1-1 cells were highly sensitive to the alkylating agent methyl methanesulfonate (MMS) and the replication inhibitor hydroxyurea (HU) at 35°C but not at 25°C (Fig. 1B). The MMS sensitivity of rmi1-1 cells at 35°C was suppressed by mutation of RAD51 or SGS1 (Fig. 1C). These phenotypes exhibited by the rmi1-1 temperature-sensitive mutant at 35°C mimic rmi1Δ phenotypes seen at all temperatures (19, 62).
Fig. 1.

rmi1-1 encodes a temperature-sensitive allele of RMI1. (A) RMI1 homologs aligned by the OB-fold domain OB1. The position of the E69K point mutation in the rmi1-1 allele is indicated. (B) rmi1-1 cells exhibit normal growth at 25°C but growth inhibition at 35°C. Growth of wild-type (BY4741), rmi1-1, and rmi1Δ strains was compared on plates in the absence of DNA-damaging agents (YPD) and on plates containing either 0.0005% MMS or 20 mM HU. Tenfold serial dilutions of each strain were spotted onto plates, and plates were incubated at 25°C for 3 days or at 35°C for 2 days. (C) Mutation of RAD51 or SGS1 suppresses rmi1-1 phenotypes. Growth of the indicated strains was compared on plates in the absence of DNA damaging agents (YPD) and on plates containing either 0.002% MMS or 20 mM HU. (D) Rmi1 is not degraded at 35°C in rmi1-1 cells. Protein extracts were prepared from asynchronous wild-type (C), RMI1-HA, and rmi1-1-HA strains grown at 25°C. The RMI1-HA and rmi1-1-HA strains were incubated at 37°C for 180 min. Levels of Rmi1 were analyzed by Western blotting. Actin was used as a loading control. (E) Degradation of Rmi1 is slightly faster in rmi1-1 cells when de novo protein synthesis is inhibited. RMI1-HA and rmi1-1-HA strains were arrested in G1 at 25°C and released into fresh medium. Once the cells had reached S phase, cycloheximide was added, and the cells were incubated at 35°C for 180 min. (F) Mutation of SLX1, SLX4, MUS81, and MMS4 causes synthetic lethality in rmi1-1 cells at 35°C.
To further characterize the phenotype of the rmi1-1mutant, we examined whether Rmi1 protein levels are altered in rmi1-1 cells at 35°C. The 3′ end of RMI1 in both wild-type and rmi1-1 strains was modified to encode an influenza virus hemagglutinin (HA) epitope tag (50). There was no significant decrease in Rmi1 protein levels in the resulting RMI1-HA and rmi1-1-HA strains after incubation for 180 min at 35°C (Fig. 1D). To exclude the possibility that a high level of de novo protein synthesis maintains Rmi1 protein levels in rmi1-1-HA cells at 35°C, Rmi1 levels were examined in S-phase cells in the presence of cycloheximide to inhibit de novo protein synthesis (Fig. 1E). The Rmi1 protein had a half-life of approximately 30 min in RMI1-HA cells but only 15 min in rmi1-1-HA cells. A possible reason for this discrepancy may be that the level of RMI1 transcription in rmi1-1 cells is slightly higher than that in wild-type cells in order to maintain equivalent Rmi1 protein levels at 35°C. From these experiments, we conclude that the Rmi1 protein is still present after an extended incubation at 35°C in rmi1-1 cells and at levels similar to those detectable in wild-type cells.
The rmi1-1 temperature-sensitive mutant was then used to investigate the genetic interactions between RMI1 and the MUS81, MMS4, SLX1, SLX4, and YEN1 genes, each of which has been implicated in HJ resolution (25, 36, 44). Previous studies showed that deletion of MUS81-MMS4 or SLX1-SLX4 in sgs1Δ, top3Δ, and rmi1Δ mutants causes synthetic lethality (3, 4, 19, 31, 61, 62). Accordingly, synthetic lethality was observed at 35°C when these resolvase genes were deleted in the rmi1-1 strain (Fig. 1F). Deletion of YEN1 in the rmi1-1 strain did not cause synthetic lethality, and rmi1-1 yen1Δ mutants exhibited a level of sensitivity to MMS similar to that of rmi1-1 cells. This phenotype is consistent with that observed following deletion of YEN1 in an sgs1Δ strain (8). These synthetic lethality profiles and the sensitivity of rmi1-1 cells to MMS and HU at 35°C indicate that the rmi1-1 strain phenocopies loss of RMI1 function at the restrictive temperature.
rmi1-1 cells have an extended G2/M delay after completing a perturbed S phase but are proficient in DNA damage checkpoint activation.
Asynchronous rmi1Δ cells accumulate in G2/M, as indicated by the development of large-budded cells with a nucleus at the neck between the mother and daughter cell (19). However, it is not clear whether the G2/M delay in rmi1Δ cells is caused by defects that occur in one cell cycle or if they accumulate as the cells progress through successive cell cycles. We therefore utilized the rmi1-1 strain to investigate the consequences of acute Rmi1 impairment. Wild-type and rmi1-1 cells were grown at 25°C, arrested in G1 at 35°C, and then released into fresh medium (Fig. 2A). α-Factor was added after 50 min to hold cells that were able to successfully traverse mitosis in G1, thus preventing a second round of DNA replication. Both rmi1-1 and wild-type cells progressed through S phase with similar kinetics and completed bulk DNA replication by 50 min. However, there was a delay in the progression from G2/M to G1 in rmi1-1 cells compared to wild-type cells (Fig. 2A). To further examine the rmi1-1 strain's G2/M delay, microscopic analysis was conducted, and the positions of the nucleus and the mitotic spindle in budding cells were scored at various time points (Fig. 2B). An average of 30% of rmi1-1 cells had DNA at the neck with the mitotic spindle passing through the neck, compared to only 12% of wild-type cells (Fig. 2B). Correspondingly, in the wild type there was a higher proportion of cells with DNA in both the mother and daughter (42% in the wild type and 21% in the rmi1-1 strain). We propose that mitotic DNA segregation defects arise after the first S phase in which Rmi1 is impaired.
Fig. 2.

rmi1-1 cells have an extended G2/M delay after completing a perturbed S phase but are proficient in DNA damage checkpoint activation. (A) rmi1-1 cells exhibit a mild G2/M delay. Wild-type and rmi1-1 strains were arrested in G1 (1N) at 35°C, washed, and resuspended in fresh medium. Cell cycle progression was followed by flow cytometry. α-Factor was added to the cultures at 50 min to prevent cells from entering a second S phase. The shaded peaks represent experimental data, and the unshaded peaks indicate a normal G2/M peak (2N). (B) Mitotic DNA segregation defects arise after traversing the first S phase in which functional Rmi1 is absent. Fifty cells were scored blindly in three independent experiments from the samples in which 60%, 40%, or 20% of the cells were in G2/M (60, 70, and 80 min in wild-type cells and 80, 90, and 120 min in rmi1-1 cells, respectively). The position of the nucleus was scored as follows: at the neck of the mother cell (at neck), elsewhere in the mother cell (in mother only), in the daughter cell only, or in both the mother and the daughter cells. The spindle was stained using an anti-α-tubulin antibody, and its position in each cell was also scored. (C) MMS exacerbates the G2/M delay in rmi1-1 cells. Wild-type and rmi1-1 cells were grown at 25°C, arrested in G1 at either 25°C or 35°C, and then released into medium containing 0.0167% MMS at the indicated temperature. After 1.5 h the MMS was washed off, and the cells were allowed to recover for 4 h in drug-free medium. α-Factor was added to prevent any cells traversing mitosis and entering a second round of DNA replication. (D) DNA damage checkpoint activation is proficient in rmi1-1 cells. Protein extracts were prepared from the cultures in Fig. 2C. Rad53 phosphorylation status was monitored by Western blotting. The positions of unphosphorylated Rad53 and slower-migrating phosphorylated forms of Rad53 are shown on the right. α, α-factor-arrested sample; M, sample taken after 1.5 h exposure to 0.0167% MMS. The numbers 1, 2, 3, and 4 refer to the number of hours after the MMS was removed from the cultures.
MMS causes DNA lesions that lead to stalling of DNA replication forks. Deletion of SGS1, TOP3, or RMI1 confers MMS sensitivity (18, 19, 34, 60, 62). MMS sensitivity is also observed in the absence of BLM or RMI2 in human cells (73, 74). To investigate if exposure to MMS exacerbates the G2/M delay observed in rmi1-1 cells, wild-type and rmi1-1 cells were transiently exposed to 0.0167% MMS at either 25°C or 35°C (Fig. 2C). After 1.5 h, the MMS was removed and the cells were allowed to recover for 4 h in drug-free medium. Both wild-type and rmi1-1 cells traversed S phase with similar kinetics, suggesting that bulk DNA replication is largely unaffected in rmi1-1 cells. The finding that rmi1-1 cells are eventually able to complete replication at 35°C is unsurprising, because top3Δ cells released from a G1 arrest are also able to do so (18). After 4 h of recovery from MMS treatment, the majority of rmi1-1 cells at 25°C and of wild-type cells at both 25°C and 35°C had traversed mitosis and accumulated in G1 (Fig. 2C). However, approximately 50% of rmi1-1 cells at 35°C remained arrested in G2/M at this time.
One possible explanation for the G2/M delay is that a DNA damage checkpoint may be hyperactivated in rmi1-1 cells, preventing them from entering mitosis. The Rad53 serine/threonine kinase is a DNA damage checkpoint effector protein that is hyperphosphorylated in response to both DNA damage and replication defects (66, 71). However, in rmi1Δ and top3Δ cells, there is attenuated phosphorylation of Rad53 following exposure to MMS (18, 19). To determine the phosphorylation status of Rad53 in rmi1-1 cells treated with MMS, samples were taken for Western blotting during the experiment performed in Fig. 2C. We observed that Rad53 was hyperphosphorylated in wild-type cells at both 25°C and 35°C and in rmi1-1 cells at 25°C (Fig. 2D). Moreover, Rad53 dephosphorylation occurred by 2 h after removal of the MMS. In contrast to rmi1Δ mutants (19), Rad53 was also hyperphosphorylated in rmi1-1 cells at 35°C and remained phosphorylated even after 4 h of recovery from the MMS treatment. Therefore, DNA damage checkpoint activation is proficient in rmi1-1 cells, and the persistent activation of this checkpoint probably accounts for the G2/M delay observed in rmi1-1 cells following MMS treatment.
Replication-associated branched DNA structures persist in rmi1-1 cells after a perturbed S phase.
The specific roles of Rmi1 during S phase are poorly defined. Therefore, we used pulsed-field gel electrophoresis (PFGE) analysis to monitor the kinetics of completion of DNA replication in wild-type and rmi1-1 cells after MMS treatment. In this system, intact chromosomes are resolved into distinct bands, which can be visualized by ethidium bromide staining. However, replicating chromosomes are unable to enter the gel because branched-DNA replication/repair intermediates impair their electrophoretic migration (27, 40). To investigate DNA replication kinetics following a transient exposure to MMS, samples were taken for PFGE analysis during the experiment in Fig. 2C. We observed that, in contrast to the G1-arrested samples, the majority of chromosomes were retained in the wells in the samples taken after a 1.5 h treatment with 0.0167% MMS (Fig. 3 A). Following the removal of MMS, the majority of chromosomes re-entered the gel within 2 h in wild-type and rmi1-1 cells at 25°C and in wild-type cells at 35°C. However, chromosomal integrity was clearly perturbed in rmi1-1 cells at 35°C, with reduced band intensities still evident after 4 h of recovery from the MMS treatment (Fig. 3A). To quantify this effect, the DNA was transferred to a membrane by Southern blotting and then hybridized with a probe that binds to ARS305 on chromosome III (Fig. 3B). The relative ratio of chromosome III in the gel to chromosome III retained in the wells was then quantified (Fig. 3C). We observed that the majority of chromosome III was retained in the wells in the presence of MMS in all strains examined. Following the removal of MMS, chromosome III had largely migrated into the gel by 2 h in wild-type cells at 25°C and 35°C and in rmi1-1 cells at 25°C. However, 56% of chromosome III DNA remained in the wells in rmi1-1 cells at 35°C after 4 h of recovery from the MMS treatment, compared to only 21% in rmi1-1 cells at 25°C. This suggests that more branched DNA structures persist in rmi1-1 cells at 35°C after MMS treatment than in either wild-type cells (at both 35°C and 25°C) or rmi1-1 cells at 25°C (Fig. 3C). We propose that these structures consist of unprocessed, postreplicative HRR intermediates, late-stage DNA replication intermediates, or a combination of the two.
Fig. 3.

Branched DNA structures persist in rmi1-1 cells after a perturbed S phase. (A) Chromosome integrity is impaired following MMS treatment in rmi1-1 cells. Samples were taken from strains in Fig. 2C, and analyzed at the indicated times by pulsed-field gel electrophoresis (PFGE). C, yeast chromosomal DNA marker; α, α-factor-arrested sample; + M, sample taken after 1.5 h exposure to 0.0167% MMS; − M, sample taken after 50 min in the absence of MMS. 1, 2, 3 and 4 refer to the number of hours after the MMS was removed from the cultures. (B) Branched structures impair chromosome III electrophoretic mobility following MMS treatment in rmi1-1 cells. The DNA was transferred by Southern blotting and then hybridized with a probe that binds to ARS305 on chromosome III. (C) Quantification of chromosome III migration. The intensity of chromosome III in the wells versus the gel in panel B was quantified for each time point. The data are from three independent experiments. Error bars show standard errors.
X-shaped DNA molecules that are sensitive to the E. coli RusA HJ resolvase persist when Rmi1 is impaired.
Previous studies have shown that RAD51-dependent, MMS-induced, X-shaped DNA molecules accumulate in cells in which Sgs1, Top3, or Rmi1 is impaired (48, 55, 58). Therefore, we used neutral-neutral 2D gel electrophoresis technique to investigate whether similar types of DNA molecules also accumulate in rmi1-1, mus81Δ, mms4Δ, slx1Δ, slx4Δ, or yen1Δ cells (12, 51). Strains were released from G1 arrest into medium containing 0.0167% MMS for 1.5 h. Origin firing at ARS305 was detectable in wild-type, rmi1-1, and rmi1-1 rad51Δ cells after incubation in MMS for 15 min, as revealed by the presence of DNA replication bubbles, Y-shaped structures, and origin-associated X-shaped DNA molecules (Fig. 4 B). All strains examined progressed through S phase with similar kinetics, apart from rmi1-1 rad51Δ cells, which exhibited impaired S-phase progression (Fig. 4C). This is consistent with the observation that mutation of RAD51 causes impaired S-phase progression in the presence of MMS, an effect that is largely due to hyperactive checkpoint activation in these cells (55). We observed that MMS-induced, X-shaped DNA molecules accumulated in rmi1-1 cells but not in rad51 rmi1-1Δ cells (Fig. 4C). This suggests that, unlike the origin-associated X-shaped DNA molecules observed after incubation in MMS for 15 min (Fig. 4B), these intermediates are RAD51 dependent and are, therefore, unprocessed HRR intermediates (51). The disappearance of the majority of DNA replication intermediates from ARS305 in wild-type and rmi1-1 rad51Δ cells after 1.5 h exposure to MMS can be explained by replication forks having progressed beyond the boundaries of the genomic region being studied by this time. These results are in agreement with published findings for rmi1Δ mutants (58) and further verify that our rmi1-1 strain phenocopies an rmi1Δ mutant at 35°C. Importantly, MMS-induced, X-shaped DNA molecules were not observed in mus81Δ, slx1Δ, or yen1Δ strains (Fig. 4C). In addition, X-shaped DNA molecules were not observed in mms4Δ or slx4Δ cells (data not shown). Since X-shaped DNA molecules arise only in the absence of a functional RTR complex, and not in the absence of any single HJ resolvase, this suggests that the processing of these structures is most likely performed primarily by the RTR complex.
Fig. 4.

Expression of the E. coli RusA HJ resolvase diminishes MMS-induced X-shaped DNA molecules in rmi1-1 cells. (A) DNA structures detectable using the 2D electrophoresis technique. Bubble (light gray), Y-shaped (dark gray), and X-shaped DNA molecules (black) are indicated. (B) Origin firing at ARS305 is unaffected in rmi1-1 cells. Wild-type, rmi1-1, and rmi1-1 rad51Δ strains were released from G1 arrest at 35°C into medium containing 0.0167% MMS. After 15 min, DNA replication bubbles, Y-shaped structures, and origin-associated, X-shaped DNA structures were detectable by 2D gel electrophoresis at the early-firing ARS305 origin on chromosome III. (C) RAD51-dependent, MMS-induced, X-shaped DNA molecules persist in rmi1-1 cells but not in strains lacking putative HJ resolvases. The indicated strains were released from G1 arrest at 35°C into medium containing 0.0167% MMS for 1.5 h. DNA replication intermediates were detected at ARS305 by 2D gel electrophoresis. RAD51-dependent X-shaped DNA molecules are indicated by the arrow. The lower panels indicate that wild-type, rmi1-1, mus81Δ, slx1Δ, and yen1Δ cells progress through S phase with similar kinetics, as measured by flow cytometry. (D) Overexpression of the RusA HJ resolvase diminishes MMS-induced X-shaped DNA molecules in rmi1-1 cells. rmi1-1-GFP, rmi1-1-RusA, and rmi1-1-RusAD70N strains were released from G1 arrest into medium containing 0.033% MMS. Protein expression was induced during the G1 arrest, and throughout the subsequent incubation, by the addition of 2% galactose. Cultures were released from G1 arrest at 35°C and were then incubated at 37°C after 1 h to promote robust HJ resolvase activity. Samples were taken for 2D gel electrophoresis after 7 h exposure to 0.033% MMS.
Recent findings from our laboratory indicate that the RAD51-dependent, X-shaped DNA molecules forming in cells lacking either functional Sgs1 or Top3 can be resolved in vivo by the E. coli HJ resolvase, RusA (53). This suggests that these X-shaped DNA molecules contain HJs. To investigate whether the X-shaped DNA molecules that accumulate in rmi1-1 cells also contain HJs, pYES2 plasmids that express NLS-GFP (control), NLS-RusA-GFP, or the nuclease-defective allele NLS-RusAD70N (10, 39) were transformed into rmi1-1 cells. These proteins are referred to here as GFP, RusA, and RusAD70N, respectively. Cells harboring these plasmids were grown to log phase at 25°C and were then arrested in G1 at 35°C. Protein expression was induced by the addition of galactose during the G1 arrest and was maintained by the presence of galactose in the growth medium throughout the experiment. The cells were released from the G1 arrest into medium containing 0.033% MMS for 7 h (and switched to 37°C after 1 h to promote HJ resolvase activity). The more prolonged, and higher dose of, MMS treatment used causes a strong X-shaped DNA molecule signal in rmi1-1 cells and also allows a large window of time in which RusA can act upon X-shaped DNA structures. At this time, persistent X-shaped DNA molecules were detectable at ARS305 in rmi1-1 cells expressing GFP or RusAD70N but not in rmi1-1 cells expressing RusA (Fig. 4D). We note, however, that RusA expression exacerbated, rather than suppressed, the poor growth of rmi1-1 strains (data not shown). We conclude that RusA is able to cleave the RAD51-dependent, X-shaped DNA molecules arising in rmi1-1 cells. We propose, therefore, that the X-shaped DNA molecules in rmi1-1 cells contain HJs.
Mus81-Mms4 can process the X-shaped DNA molecules that persist in cells impaired for Top3 or Rmi1.
To further investigate the genetic interactions between RMI1 and putative HJ resolvase genes, rmi1-1 mus81Δ, rmi1-1 mms4Δ, rmi1-1 slx1Δ, rmi1-1 slx4Δ, and rmi1-1 yen1Δ strains were grown to log phase at 25°C and then arrested in G1 at 35°C (Fig. 5 A). Strains were then released from G1 arrest into medium containing 0.0167% MMS. After a 1.5-h exposure to MMS, similar levels of X-shaped DNA molecules were detectable in all of the strains analyzed (Fig. 5A). This suggests that loss of putative HJ resolvases does not noticeably cause increased formation of the X-shaped DNA molecules in rmi1-1 cells. To examine if Mus81-Mms4, Slx1-Slx4, or Yen1 has a role in resolution of the X-shaped DNA molecules, the fate of these intermediates was examined following the removal of MMS, as described for Fig. 2C. Although all of the strains analyzed were able to traverse S phase with similar kinetics, the flow cytometry traces for the rmi1-1 mus81Δ, rmi1-1 mms4Δ, rmi1-1 slx1Δ, and rmi1-1 slx4Δ cells revealed a broadened peak of DNA content by 4 h after the MMS treatment. This is indicative of cell segregation defects during an aberrant mitosis (56) and may explain the synthetic lethal genetic interactions observed when these genes are mutated in cells with impaired Rmi1 (Fig. 1F). As observed above (Fig. 2C), rmi1-1 cells exhibited a G2/M delay relative to wild-type cells. After 4 h of recovery from the MMS treatment, the X-shaped DNA molecules were reduced considerably in intensity in rmi1-1, rmi1-1 slx1Δ, rmi1-1 slx4Δ, and rmi1-1 yen1Δ cells (Fig. 5A). In contrast, X-shaped DNA molecules were still detectable in rmi1-1 mus81Δ and rmi1-1 mms4Δ cells, and furthermore, these did not decrease in intensity by this time (Fig. 5A). Taken together, these data reveal that rmi1-1 mus81Δ and rmi1-1 mms4Δ cells attempt to undergo mitosis with unprocessed HRR intermediates following MMS treatment. Most significantly, these data reveal an important role for the Mus81-Mms4 endonuclease in the resolution of DNA damage-induced HRR intermediates when Rmi1 is impaired.
Fig. 5.

Inactivation of Top3 or Rmi1 in the absence of Mus81-Mms4 impairs the resolution of MMS-induced X-shaped DNA molecules. (A) Resolution of MMS-induced X-shaped DNA molecules is impaired in rmi1-1 cells lacking MUS81 or MMS4. The indicated strains were released from G1 arrest at 35°C into medium containing 0.0167% MMS for 1.5 h and then resuspended in drug-free medium as described for Fig. 2C. DNA replication intermediates arising at ARS305 were examined by 2D gel electrophoresis after a 1.5-h exposure to 0.0167% MMS and after 4 h of recovery following the removal of MMS. (B) Resolution of MMS-induced, X-shaped DNA molecules is impaired in mus81Δ cells overexpressing TOP3Y356F. The indicated strains were released from G1 arrest into medium containing 0.0167% MMS for 1.5 h and then resuspended in drug-free medium to allow recovery as described for Fig. 2C. DNA replication intermediates arising at ARS305 were examined by 2D gel electrophoresis after 4 h of recovery following the removal of MMS.
TOP3Y356F is a dominant negative allele of TOP3 lacking the catalytic (decatenation) activity of Top3, which causes persistence of MMS-induced, X-shaped DNA molecules when overexpressed (55). We utilized this to investigate whether Mus81-Mms4 is also required to resolve MMS-induced X-shaped DNA molecules when Top3 is impaired. The experiment was conducted as described for Fig. 5A, using T344 wild-type, mus81Δ, slx1Δ, and yen1Δ strains overexpressing TOP3Y356F. All of these strains progressed through S phase with similar kinetics, as revealed by flow cytometry (Fig. 5B). After 4 h of recovery from MMS, the X-shaped DNA molecules had largely disappeared in wild-type, yen1Δ, and slx1Δ cells overexpressing TOP3Y356F. However, the X-shaped DNA molecules were still detectable in mus81Δ cells overexpressing TOP3Y356F (Fig. 5B). With these data, we reveal an important role for Mus81-Mms4 in the resolution of damage-induced HRR intermediates when Top3 or Rmi1 is impaired.
Branched DNA structures persist throughout the genome when Mus81 and Rmi1 are both impaired.
Given that Mus81-Mms4 has an important role in the resolution of MMS-induced, X-shaped DNA molecules at ARS305, we next investigated the consequences of inactivating Rmi1 and Mus81 on a genome-wide level using PFGE. Wild-type, rmi1-1, mus81Δ, and rmi1-1 mus81Δ cells were grown to log phase at 25°C, arrested in G1 at 35°C, and then released from G1 arrest into medium containing 0.0167% MMS. After incubation for 1.5 h, the MMS was washed off and the cells were allowed to recover in drug-free medium for 4 h (as described for Fig. 2C). In each of the strains, the chromosomes migrated into the gel in the G1-arrested samples but not in the MMS-treated samples (Fig. 6). Following the removal of MMS, the majority of chromosomes re-entered the gel within 2 h in wild-type and mus81Δ strains. As observed previously (Fig. 3C), branched DNA structures that impede the electrophoretic mobility of the chromosomes persist in rmi1-1 cells (Fig. 6). Interestingly, this defect was markedly exacerbated in rmi1-1 mus81 cells and affected all chromosomes, indicating that branched DNA structures persist throughout the genome in this double mutant (Fig. 6). These data reveal an important genome-wide role for Mus81 in the resolution of replication-associated, branched DNA structures when Rmi1 is impaired.
Fig. 6.
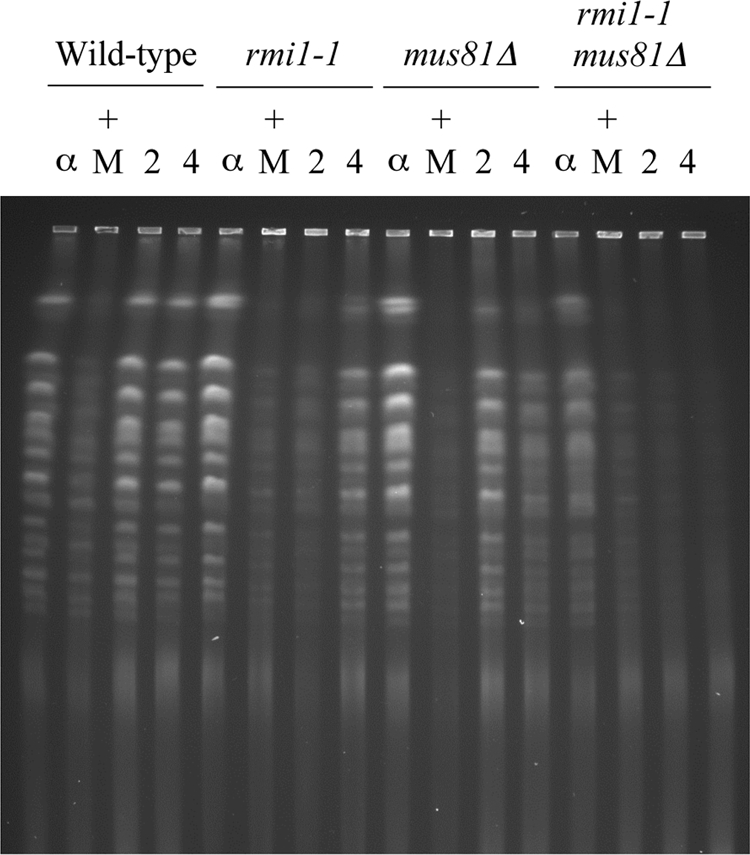
Deletion of MUS81 exacerbates defects in rmi1-1 mutants. The indicated strains were released from G1 arrest at 35°C into medium containing 0.0167% MMS for 1.5 h and then resuspended in drug-free medium as described for Fig. 2C. Samples were taken following α-factor arrest (α), after 1.5 h exposure to 0.0167% MMS (+ M), after 2 h of recovery from MMS treatment (2), and after 4 h of recovery (4). The samples were analyzed by PFGE.
DISCUSSION
To examine phenotypes caused by loss of Rmi1 function, we isolated a temperature-sensitive mutant (rmi1-1 mutant). The rmi1-1 strain exhibits wild-type growth at 25°C but severe growth inhibition at 35°C. Similar to an rmi1Δ mutant, rmi1-1 cells are highly sensitive to MMS at 35°C, and this sensitivity is suppressed by mutation of RAD51 or SGS1 (19, 62). Furthermore, it was demonstrated previously using 2D gel electrophoresis that RAD51-dependent, MMS-induced, X-shaped DNA molecules accumulate in rmi1Δ mutants (58), and here we demonstrate that they also accumulate in rmi1-1 cells. Collectively, these data indicate that the rmi1-1 temperature-sensitive mutant is a novel inducible system in which to analyze phenotypes caused by acute Rmi1 impairment.
The DNA damage checkpoint effector kinase, Rad53, becomes hyperphosphorylated in response to MMS in wild-type cells (66). However, this is partially defective in rmi1Δ or top3Δ cells, suggesting that the RTR complex can influence the DNA damage checkpoint (18, 19). In addition, Top3 genetically interacts with the checkpoint protein, Crb2, in S. pombe (16). To examine DNA damage checkpoint activation in rmi1-1 strains, we exposed rmi1-1 cells to MMS and observed that, in contrast to results reported for an rmi1Δ mutant (19), Rad53 becomes hyperphosphorylated to wild-type levels. We propose that the Rmi1 protein is required for DNA damage checkpoint activation and, although the function of Rmi1 is clearly impaired in rmi1-1 cells at 35°C, the presence of even defective Rmi1 is sufficient for normal DNA damage checkpoint activation. This proposal is supported by the finding that Rad53 is hyperphosphorylated after MMS treatment in wild-type cells in which the catalytically inactive TOP3Y356F allele is overexpressed (55). As hRMI1 and hRMI2 are required for formation of BLM foci at the site of DNA damage or replication blockage, it is possible that S. cerevisiae Rmi1 also has a role in the recruitment of Sgs1 and Top3 to replicons damaged by MMS (73, 89). Hence, the ability of Rmi1 to influence checkpoint activation may depend on its ability to stabilize the RTR complex.
Deletion of MUS81, MMS4, SLX1, or SLX4 in an sgs1Δ, top3Δ, or rmi1Δ background causes synthetic lethality, precluding detailed analysis of these genetic interactions (3, 4, 19, 31, 61, 62). To investigate these genetic interactions further, we deleted MUS81, MMS4, SLX1, or SLX4 in the rmi1-1 strain. The resulting strains are viable at 25°C but exhibit synthetic lethality and cell cycle defects at 35°C. Following a transient exposure to MMS, the RAD51-dependent, X-shaped DNA molecules that accumulate in rmi1-1 cells are eventually resolved, revealing a redundant resolution activity when Rmi1 is impaired. The kinetics of X-molecule disappearance is unaffected in cells lacking SLX1, SLX4, or YEN1. In contrast, loss of MUS81 or MMS4 in rmi1-1 cells impairs resolution of these X-shaped intermediates, suggesting that Mus81-Mms4 is the predominant activity responsible for their resolution. These results were recapitulated when Top3 was impaired in mus81Δ, slx1Δ, and yen1Δ strains. We conclude that the resolution of HRR intermediates arising in mitotic cells in the absence of a functional RTR complex primarily requires Mus81-Mms4. Although this mechanism may be conserved in higher eukaryotes, it is also possible that alternative mechanisms are invoked to process HRR intermediates in these organisms.
Previously, it was proposed that the MMS-induced, RAD51-dependent, X-shaped DNA molecules that arise in the absence of RTR complex components are HJs, sister-chromatid junctions, or hemicatenanes (11, 48, 54, 56). However, for the following reasons, we propose that the unprocessed X-shaped DNA molecules that accumulate when Top3/Rmi1 and Mus81-Mms4 are both impaired consist of intact and/or nicked HJs. First, the X-shaped DNA molecules persisting in cells with impaired Rmi1 can be resolved by E. coli RusA in vivo. Second, Mus81 can resolve HJ-containing substrates both in vivo and in vitro (25, 79, 80). Third, the RTR complex catalyzes DHJ dissolution in vitro (5, 17, 49, 85), and the turnover of DHJs that form during DSB repair in diploid cells is defective in sgs1 mutants (15). Moreover, our data are consistent with the RTR complex, rather than HJ resolvases, being the primary pathway for the processing of HJ-containing DNA molecules that arise following exposure to MMS, because these structures persist only in cells impaired for components of the RTR complex, not in mus81Δ, mms4Δ, slx1Δ, slx4Δ, or yen1Δ mutants (48, 55, 58). Our analyses did not reveal a major role for Yen1 in processing MMS-induced X-shaped DNA molecules when Rmi1 or Top3 are impaired. This is perhaps not surprising because yen1Δ sgs1Δ double mutants show a level of MMS sensitivity similar to that of the sgs1Δ single mutant (8). However, our findings do not exclude a role for Yen1 in the resolution of HRR intermediates. For example, Mus81 and Yen1 have redundant roles in the resolution of an HJ-containing plasmid-based molecule in vivo, and mus81Δ yen1Δ cells are more sensitive to MMS and HU than is either of the single mutants (8, 79). It is likely that, while each HJ processing enzyme plays a defined role in wild-type cells, there may be some functional redundancy between these proteins, which is revealed only when one or more of the enzymes are inactivated. Nevertheless, if Yen1 does play a role in the cleavage of X-shaped DNA molecules in mus81 rmi1-1 cells, it must be quite inefficient, because these structures are not resolved in mus81 rmi1-1 cells even 4 h after the removal of MMS. One possibility is that Yen1 functions primarily in a particular phase of the cell cycle and may not, therefore, promote the resolution of X-shaped DNA molecules in mus81 rmi1-1 cells in late-S/G2 phase. In our system at least, the role of Slx1-Slx4 in processing MMS-induced, X-shaped DNA molecules also appears to be a minor one in comparison with that of the RTR complex or Mus81-Mms4. Consistent with this, the synthetic lethality caused by deletion of MUS81 or MMS4 in sgs1Δ, top3Δ, and rmi1Δ strains is suppressed by deletion of early HRR genes such as RAD51, whereas the synthetic lethality caused by deletion of SLX1 or SLX4 is not (3, 19, 31, 62). This indicates that the synthetic lethality probably relates to functions of Slx1-Slx4 in processes other than HRR. Interestingly, loss of Slx1-Slx4 or Mus81 causes defects in the faithful replication of rDNA repeats in the absence of Sgs1 (43, 45), suggesting that chromosome XII integrity impacts the viability of sgs1 slx4 and sgs1 mus81 mutants. However, defining the specific problems occurring at this locus, and throughout other regions of the genome, will require further investigation, as discussed below.
Using the rmi1-1 temperature-sensitive mutant, we demonstrated that loss of Rmi1 function during S phase leads to a G2/M delay and mitotic DNA segregation defects. A transient exposure to MMS leads to an exacerbation of this G2/M delay and causes a high level of branched DNA structures to arise. These structures arise on a genome-wide level, not just at specific loci, as revealed by PFGE. We propose that these branched structures contain unresolved, postreplicative HRR intermediates, as observed at ARS305. Although at least some of the branched DNA structures could also contain late-stage replication intermediates, such as those arising at sites of converging replication forks, it has recently been shown that programmed replication termination at TER sites is mediated by Top2 and is Top3 independent (32). However, it is still possible that the RTR complex may have a role in replication termination at non-TER sites, or in resolving topological constraints at converging forks as they approach the zone in which replication termination occurs.
Our findings using the rmi1-1 temperature-sensitive mutant are consistent with a role for Rmi1 in the processing of HJ-containing HRR intermediates that arise during S phase. It is our proposal that the MMS-induced X-shaped DNA structures consist largely of DHJs, and the most plausible source of these DHJs is during the HRR-mediated filling of postreplicative ssDNA gaps. Furthermore, we propose that the DHJ dissolution pathway is dominant over Mus81-Mms4, or any other HJ resolution pathways, in resolving DHJs. Indeed, it could be advantageous for cells to favor DHJ dissolution by the RTR complex over resolution by HJ resolvases, because DHJ dissolution produces exclusively noncrossover products, whereas HJ resolution would produce a mixture of noncrossover and crossover products (77, 86). Preferential production of noncrossovers as an endpoint for HRR would minimize potentially deleterious effects of crossing-over during HRR, such as chromosomal rearrangement, missegregation, and loss of heterozygosity. It is also possible that DHJ dissolution is the primary pathway because conventional HJ resolution is either error prone or inefficient. Given the greater ability of Mus81-Mms4 to resolve nicked rather than intact HJs in vitro (21, 23, 24, 35), it is possible that the X-shaped DNA molecules that are resolved by Mus81-Mms4 in cells impaired for Rmi1 or Top3 comprise either nicked HJs or intact HJs that are converted to nicked junctions. Further work will be required to fully understand the different physiological situations in which the RTR complex, Mus81-Mms4, Slx1-Slx4, and Yen1 are required.
ACKNOWLEDGMENTS
The anti-Rad53 antibody was kindly provided by Marco Foiani. We thank the members of the Genome Integrity Group and the DNA Damage and Repair Group for their helpful comments.
This work was supported by Cancer Research UK and the Bloom's Syndrome Foundation.
Footnotes
Published ahead of print on 22 February 2011.
REFERENCES
- 1. Amberg D., Burke D., Strathern J. 2005. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Ashton T. M., Hickson I. D. 2010. Yeast as a model system to study RecQ helicase function. DNA Repair (Amsterdam) 9:303–314 [DOI] [PubMed] [Google Scholar]
- 3. Bastin-Shanower S. A., Fricke W. M., Mullen J. R., Brill S. J. 2003. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol. Cell. Biol. 23:3487–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bellaoui M., et al. 2003. Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J. 22:4304–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bennett R. J., Keck J. L., Wang J. C. 1999. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J. Mol. Biol. 289:235–248 [DOI] [PubMed] [Google Scholar]
- 6. Bennett R. J., Noirot-Gros M. F., Wang J. C. 2000. Interaction between yeast sgs1 helicase and DNA topoisomerase III. J. Biol. Chem. 275:26898–26905 [DOI] [PubMed] [Google Scholar]
- 7. Bernstein K. A., Rothstein R. 2009. At loose ends: resecting a double-strand break. Cell 137:807–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blanco M. G., Matos J., Rass U., Ip S. C., West S. C. 2010. Functional overlap between the structure-specific nucleases Yen1 and Mus81-Mms4 for DNA-damage repair in S. cerevisiae. DNA Repair (Amsterdam) 9:394–402 [DOI] [PubMed] [Google Scholar]
- 9. Bloom D. 1954. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs—probably a syndrome entity. Ama Am. J. Dis. Child. 88:754–758 [PubMed] [Google Scholar]
- 10. Bolt E. L., Sharples G. J., Lloyd R. G. 1999. Identification of three aspartic acid residues essential for catalysis by the RusA Holliday junction resolvase. J. Mol. Biol. 286:403–415 [DOI] [PubMed] [Google Scholar]
- 11. Branzei D., et al. 2006. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127:509–522 [DOI] [PubMed] [Google Scholar]
- 12. Brewer B. J., Fangman W. L. 1987. The localization of replication origins on ARS plasmids in Saccharomyces cerevisiae. Cell 51:463–471 [DOI] [PubMed] [Google Scholar]
- 13. Broberg K., et al. 2007. Genetic variant of the human homologous recombination-associated gene RMI1 (S455N) impacts the risk of AML/MDS and malignant melanoma. Cancer Lett. 258:38–44 [DOI] [PubMed] [Google Scholar]
- 14. Broberg K., et al. 2009. Association between polymorphisms in RMI1, TOP3A, and BLM and risk of cancer, a case-control study. BMC Cancer 9:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bzymek M., Thayer N. H., Oh S. D., Kleckner N., Hunter N. 2010. Double Holliday junctions are intermediates of DNA break repair. Nature 464:937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caspari T., Murray J. M., Carr A. M. 2002. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 16:1195–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cejka P., Plank J., Bachrati C., Hickson I., Kowalczykowski S. 2010. Dissolution of double Holliday junctions via migration and decatenation by Sgs1-Top3-Rmi1. Nat. Struct. Mol. Biol. 17:1377–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chakraverty R. K., et al. 2001. Topoisomerase III acts upstream of Rad53p in the S-phase DNA damage checkpoint. Mol. Cell. Biol. 21:7150–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang M., et al. 2005. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J. 24:2024–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen C. F., Brill S. J. 2007. Binding and activation of DNA topoisomerase III by the Rmi1 subunit. J. Biol. Chem. 282:28971–28979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X. B., et al. 2001. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 8:1117–1127 [DOI] [PubMed] [Google Scholar]
- 22. Chu W. K., Hickson I. D. 2009. RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9:644–654 [DOI] [PubMed] [Google Scholar]
- 23. Ciccia A., McDonald N., West S. C. 2008. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 77:259–287 [DOI] [PubMed] [Google Scholar]
- 24. Constantinou A., Chen X. B., McGowan C. H., West S. C. 2002. Holliday junction resolution in human cells: two junction endonucleases with distinct substrate specificities. EMBO J. 21:5577–5585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cote A. G., Lewis S. M. 2008. Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol. Cell 31:800–812 [DOI] [PubMed] [Google Scholar]
- 26. Deans A. J., West S. C. 2009. FANCM connects the genome instability disorders Bloom's syndrome and Fanconi anemia. Mol. Cell 36:943–953 [DOI] [PubMed] [Google Scholar]
- 27. Desany B. A., Alcasabas A. A., Bachant J. B., Elledge S. J. 1998. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 12:2956–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dunderdale H. J., et al. 1991. Formation and resolution of recombination intermediates by E. coli RecA and RuvC proteins. Nature 354:506–510 [DOI] [PubMed] [Google Scholar]
- 29. Ellis N. A., German J. 1996. Molecular genetics of Bloom's syndrome. Hum. Mol. Genet. 5:1457–1463 [DOI] [PubMed] [Google Scholar]
- 30. Erdeniz N., Mortensen U. H., Rothstein R. 1997. Cloning-free PCR-based allele replacement methods. Genome Res. 7:1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fabre F., Chan A., Heyer W. D., Gangloff S. 2002. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. U. S. A. 99:16887–16892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fachinetti D., et al. 2010. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 39:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fekairi S., et al. 2009. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138:78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frei C., Gasser S. M. 2000. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 14:81–96 [PMC free article] [PubMed] [Google Scholar]
- 35. Fricke W. M., Bastin-Shanower S. A., Brill S. J. 2005. Substrate specificity of the Saccharomyces cerevisiae Mus81-Mms4 endonuclease. DNA Repair (Amsterdam). 4:243–251 [DOI] [PubMed] [Google Scholar]
- 36. Fricke W. M., Brill S. J. 2003. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 17:1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fricke W. M., Kaliraman V., Brill S. J. 2001. Mapping the DNA topoisomerase III binding domain of the Sgs1 DNA helicase. J. Biol. Chem. 276:8848–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gangloff S., McDonald J. P., Bendixen C., Arthur L., Rothstein R. 1994. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol. 14:8391–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giraud-Panis M. J., Lilley D. M. 1998. Structural recognition and distortion by the DNA junction-resolving enzyme RusA. J. Mol. Biol. 278:117–133 [DOI] [PubMed] [Google Scholar]
- 40. Hennessy K. M., Lee A., Chen E., Botstein D. 1991. A group of interacting yeast DNA replication genes. Genes Dev. 5:958–969 [DOI] [PubMed] [Google Scholar]
- 41. Hoadley K. A., et al. 2010. Structure and Cellular Roles of the RMI core complex from the Bloom syndrome dissolvasome. Structure 18:1149–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hovland P., Flick J., Johnston M., Sclafani R. A. 1989. Galactose as a gratuitous inducer of GAL gene expression in yeasts growing on glucose. Gene 83:57–64 [DOI] [PubMed] [Google Scholar]
- 43. Ii M., Ii T., Brill S. J. 2007. Mus81 functions in the quality control of replication forks at the rDNA and is involved in the maintenance of rDNA repeat number in Saccharomyces cerevisiae. Mutat. Res. 625:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ip S. C., et al. 2008. Identification of Holliday junction resolvases from humans and yeast. Nature 456:357–361 [DOI] [PubMed] [Google Scholar]
- 45. Kaliraman V., Brill S. J. 2002. Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr. Genet. 41:389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kitao S., et al. 1999. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 22:82–84 [DOI] [PubMed] [Google Scholar]
- 47. Li X., Heyer W. D. 2008. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 18:99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liberi G., et al. 2005. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 19:339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lo Y. C., et al. 2006. Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity. Mol. Cell. Biol. 26:4086–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Longtine M. S., et al. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961 [DOI] [PubMed] [Google Scholar]
- 51. Lopes M., Cotta-Ramusino C., Liberi G., Foiani M. 2003. Branch migrating sister chromatid junctions form at replication origins through Rad51/Rad52-independent mechanisms. Mol. Cell 12:1499–1510 [DOI] [PubMed] [Google Scholar]
- 52. Lundin C., et al. 2005. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 33:3799–3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mankouri H. W., Ashton T. M., Hickson I. The Sgs1 complex directly processes Holliday junctions formed during homologous recombination repair in S. cerevisiae. Proc. Natl. Acad. Sci. U. S. A., in press [Google Scholar]
- 54. Mankouri H. W., Hickson I. D. 2007. The RecQ helicase-topoisomerase III-Rmi1 complex: a DNA structure-specific ‘dissolvasome’? Trends Biochem. Sci. 32:538–546 [DOI] [PubMed] [Google Scholar]
- 55. Mankouri H. W., Hickson I. D. 2006. Top3 processes recombination intermediates and modulates checkpoint activity after DNA damage. Mol. Biol. Cell 17:4473–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mankouri H. W., Ngo H. P., Hickson I. D. 2009. Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 20:1683–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mankouri H. W., Ngo H. P., Hickson I. D. 2007. Investigating the role(s) of the Sgs1-Top3-Rmi1 complex in homologous recombination repair in S. cerevisiae. Mutagenesis 22:448 [Google Scholar]
- 58. Mankouri H. W., Ngo H. P., Hickson I. D. 2007. Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol. Biol. Cell 18:4062–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moynahan M. E., Jasin M. 2010. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11:196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mullen J. R., Kaliraman V., Brill S. J. 2000. Bipartite structure of the SGS1 DNA helicase in Saccharomyces cerevisiae. Genetics 154:1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mullen J. R., Kaliraman V., Ibrahim S. S., Brill S. J. 2001. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157:103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mullen J. R., Nallaseth F. S., Lan Y. Q., Slagle C. E., Brill S. J. 2005. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol. Cell. Biol. 25:4476–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Munoz I. M., et al. 2009. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35:116–127 [DOI] [PubMed] [Google Scholar]
- 64. Nishino T., Ariyoshi M., Iwasaki H., Shinagawa H., Morikawa K. 1998. Functional analyses of the domain structure in the Holliday junction binding protein RuvA. Structure 6:11–21 [DOI] [PubMed] [Google Scholar]
- 65. Oakley T. J., Goodwin A., Chakravety R. K., Hickson I. D. 2002. Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair 1:463–482 [DOI] [PubMed] [Google Scholar]
- 66. Paulovich A. G., Hartwell L. H. 1995. A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82:841–847 [DOI] [PubMed] [Google Scholar]
- 67. Payne M., Hickson I. D. 2009. Genomic instability and cancer: lessons from analysis of Bloom's syndrome. Biochem. Soc. Trans. 37:553–559 [DOI] [PubMed] [Google Scholar]
- 68. Prabu J. R., et al. 2006. Structure of Mycobacterium tuberculosis RuvA, a protein involved in recombination. Acta Crystallogr. Sect. F Struct. Biol. Cryst Commun. 62:731–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Raynard S., Bussen W., Sung P. 2006. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIα, and BLAP75. J. Biol. Chem. 281:13861–13864 [DOI] [PubMed] [Google Scholar]
- 70. Raynard S., et al. 2008. Functional role of BLAP75 in BLM-topoisomerase IIIα-dependent Holliday junction processing. J. Biol. Chem. 283:15701–15708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Santocanale C., Diffley J. F. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395:615–618 [DOI] [PubMed] [Google Scholar]
- 72. Siitonen H. A., et al. 2003. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum. Mol. Genet. 12:2837–2844 [DOI] [PubMed] [Google Scholar]
- 73. Singh T. R., et al. 2008. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 22:2856–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. So S., Adachi N., Lieber M. R., Koyama H. 2004. Genetic interactions between BLM and DNA ligase IV in human cells. J. Biol. Chem. 279:55433–55442 [DOI] [PubMed] [Google Scholar]
- 75. Southern E. M. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503–517 [DOI] [PubMed] [Google Scholar]
- 76. Sun H., Treco D., Szostak J. W. 1991. Extensive 3′-overhanging, single-stranded DNA associated with the meiosis-specific double-strand breaks at the ARG4 recombination initiation site. Cell 64:1155–1161 [DOI] [PubMed] [Google Scholar]
- 77. Svendsen J. M., Harper J. W. 2010. GEN1/Yen1 and the SLX4 complex: Solutions to the problem of Holliday junction resolution. Genes Dev. 24:521–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Svendsen J. M., et al. 2009. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138:63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tay Y. D., Wu L. 2010. Overlapping roles for Yen1 and Mus81 in cellular Holliday junction processing. J. Biol. Chem. 285:11427–11432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taylor E. R., McGowan C. H. 2008. Cleavage mechanism of human Mus81-Eme1 acting on Holliday-junction structures. Proc. Natl. Acad. Sci. U. S. A. 105:3757–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Van Maldergem L., et al. 2006. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 43:148–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vindigni A., Marino F., Gileadi O. 2010. Probing the structural basis of RecQ helicase function. Biophys. Chem. 149:67–77 [DOI] [PubMed] [Google Scholar]
- 83. Wach A., Brachat A., Pohlmann R., Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793–1808 [DOI] [PubMed] [Google Scholar]
- 84. Wang F., et al. 2010. Crystal structures of RMI1 and RMI2, two OB-fold regulatory subunits of the BLM complex. Structure 18:1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wu L., et al. 2006. BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. U. S. A. 103:4068–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu L., Hickson I. D. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426:870–874 [DOI] [PubMed] [Google Scholar]
- 87. Xu D., et al. 2008. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 22:2843–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yang J., Bachrati C. Z., Ou J., Hickson I. D., Brown G. W. 2010. Human topoisomerase IIIα is a single-stranded DNA decatenase that is stimulated by BLM and RMI1. J. Biol. Chem. 285:21426–21436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yin J., et al. 2005. BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J. 24:1465–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yu C. E., et al. 1996. Positional cloning of the Werner's syndrome gene. Science 272:258–262 [DOI] [PubMed] [Google Scholar]