

Abstract
Recent studies have shown that the protein CTCF, which plays an important role in insulation and in large-scale organization of chromatin within the eukaryotic nucleus, depends for both activities on recruitment of the cohesin complex. We show here that the interaction of CTCF with the cohesin complex involves direct contacts between the cohesin subunit SA2 and specific regions of the C-terminal tail of CTCF. All other cohesin components are recruited through their interaction with SA2. Expression in vivo of CTCF mutants lacking the C-terminal domain, or with mutations at sites within it required for SA2 binding, disrupts the normal expression profile of the imprinted genes IGF2-H19 and also results in a loss of insulation activity. Taken together, our results demonstrate that specific sites on the C terminus of CTCF are essential for cohesin binding and insulator function. The only direct interaction between CTCF and cohesin involves contact with SA2, which is external to the cohesin ring. This suggests that in recruiting cohesin to CTCF, SA2 could bind first and the ring could assemble subsequently.
INTRODUCTION
CCCTC-binding factor (CTCF) is an evolutionarily conserved zinc finger protein that has been shown to play multiple roles in the regulation of gene expression. (1, 8, 9, 10, 25, 34, 42). Many of its activities probably derive from the ability of CTCF to interact with and stabilize contacts between distant sites within the nucleus (33, 36). The effect of the resulting formation of loop domains in some cases can lead to activation of gene expression by bringing together distant enhancers and promoters, but in its most intensively studied role, a CTCF binding site that lies between an enhancer and a promoter can act as an insulator element to silence a gene (5, 6). The biological significance of this activity first became apparent in studies of the imprinted control region (ICR) of the Igf2-H19 imprinted locus in mouse and human (2, 13, 14, 18), which showed that the ICR had multiple CTCF binding sites that, on the maternal allele, served to prevent a distal enhancer from activating Igf2 expression. On the paternal allele, however, the DNA of the binding sites was methylated, CTCF could not bind, and the enhancer could activate the Igf2 promoter, accounting at least in large part for allele-specific expression.
As with other transcriptional regulatory proteins, CTCF does not act alone. Work over the past several years has revealed a variety of proteins that interact with it, and a number of them appear to be important or essential in assays for insulator activity or stabilization of long-range contacts (43). Among these are the chromatin-remodeling protein CHD8 (16) and suz12, a component of the PRC2 polycomb group complex that is responsible for methylation of lysine 27 on histone H3 and is associated with formation of silencing chromatin structures (24). Why these proteins are essential to CTCF function is not completely understood. However, a more obvious connection to function may be provided by the recent discovery that CTCF recruits the cohesin complex in vivo at the great majority of its binding sites and that depletion of CTCF causes loss of cohesin binding (20, 35, 38, 39, 44, 45). Depletion of cohesin components results in loss of imprinted gene expression at the Igf2-H19 locus, as well as loss of CTCF-dependent long-range contacts within the nucleus, but in most cases, depletion of cohesin components appears not to affect CTCF binding to DNA (12, 29, 31).
The role of cohesin during replication in maintaining the attachment of sister chromatids is reasonably well understood. In yeast, two components of the complex, Smc1 and Smc3, are long antiparallel coiled-coil structures that heterodimerize by interaction at one end (30). The other ends can be bridged and held together by binding a third component, Scc1 (Rad21 in humans), forming a closed circular structure. A fourth component of the complex, Scc3 (SA1/SA2 in humans), interacts with the other components through contact with Scc1 (30). In the most generally accepted model for sister chromatid cohesion, the cohesin complex ring opens to embrace the two chromatids and then closes around them (30). It has been known for some time that some cohesin remains bound to DNA during interphase. The demonstration that this binding is indirect, largely mediated by CTCF, and essential for CTCF function suggests that a related ring-forming mechanism may contribute to the ability of distant CTCF sites to interact. In support of a role for cohesin in long-range interactions, it has been shown recently that cohesin can also be localized at enhancers and core promoters by interactions with Mediator and that this stabilizes loop formation between such enhancers and promoters (17).
To begin to understand the role of cohesin in CTCF function, we asked which subunit components of cohesin, and which protein domains of CTCF, are responsible for the interaction. We found that only Scc3 (SA1/SA2) makes direct contact with CTCF; the ring-forming components of cohesin are recruited through their attachment to SA2. With the use of CTCF mutants, we show that interaction of CTCF with SA2 is confined to the C-terminal tail and within the tail to a specific region. Deletion of this region or mutation of specific amino acids within it interferes with SA2 binding in vitro and recruitment of cohesin components to CTCF sites in vivo. We show that this has striking effects in vivo on the insulator function of CTCF both in reporter constructs and at the Igf2-H19 locus.
MATERIALS AND METHODS
Cell lines and culture.
Plasmids containing various CTCF domains, including the full-length domain, the N terminus plus zinc fingers (Nterm+ZF), and the zinc fingers plus C terminus (ZF+Cterm), were constructed using a poZ vector as described previously (32). All CTCF domains were inserted in the C terminus of a Flag/hemagglutinin (HA) tag. To generate a stably transformed cell line, these constructs were used to transfect a HeLa-S3 cell line. Positive cells coexpressing interleukin 2 receptor (IL-2R) were selected by using an anti-IL-2R magnetic-sorting method, which yields >95% stably expressing cells. HeLa-S3 cells and mouse embryo fibroblasts (MEFs) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 1% penicillin-streptomycin (Invitrogen). K-562 cells were cultured in RPMI 1640 medium supplemented with 2 mM l-glutamine, 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 10 mM HEPES, 1.0 mM sodium pyruvate, 10% fetal bovine serum, and 1% penicillin-streptomycin.
Extraction of nuclei and CTCF purification.
Nuclear extracts were prepared with 6 × 107 stably transformed cells. CTCF-associated proteins were purified using a system described previously with minor modifications (47). Briefly, cells were washed once with 1× phosphate-buffered saline (PBS) solution (pH 7.4) and swollen in hypotonic buffer (10 mM Tris [pH 7.4], 10 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol [DTT] plus protease inhibitors) for 10 min on ice. Subsequently, the cells were homogenized eight times with a “loose” pestle in a Dounce homogenizer. After centrifugation at 2,000 × g for 10 min, the nuclear pellets were resuspended in 0.5 volume of low-salt buffer (20 mM Tris [pH 7.4], 20 mM KCl, 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, protease inhibitors) and homogenized six times with a loose pestle. High-salt buffer (0.5 volume 0.42 M KCl [final]) was then added dropwise to the mixture, which was transferred into a new tube and rotated for 30 min at 4°C. Following centrifugation at 14,000 × g for 15 min, we saved only the pellets for the subsequent experiments, because our initial experiments indicated that CTCF and the cohesin complex components are abundant in the insoluble fraction. The pellet was homogenized 6 times with a Dounce homogenizer in a modified TGM buffer (50 mM Tris [pH 7.9], 25% glycerol, 5 mM MgCl2, 1 mM DTT, protease inhibitors) and digested with 0.1 U/μl micrococcal nuclease for 30 min at room temperature. After centrifugation at 14,000 × g for 15 min, the supernatant was saved. The remaining pellet was homogenized again and further digested with 0.8 U/μl DNase I and 5 μg/ml of RNase A for 30 min at room temperature. Following centrifugation, the soluble solution was combined with the previously saved supernatant and used for CTCF affinity purification.
Immunoprecipitation and Western blotting.
CTCF-associated proteins were affinity purified using the procedure described previously (47). To eliminate protein-protein interactions mediated by DNA, ethidium bromide (final concentration, 250 μg/ml) was added to the incubation mixture. Proteins were separated on an SDS-PAGE gel and detected by Western blot analysis using corresponding antibodies. Goat or rabbit polyclonal antibodies against CTCF, SA1, SA2, SMC1, SMC3, Rad21, and Ku70 were purchased from Abcam Inc. Mouse monoclonal antibodies against Flag and HA were obtained from Sigma-Aldrich. Horseradish peroxidase-conjugated secondary mouse and goat IgG antibodies were purchased from Santa Cruz Biotechnology. Specific antibodies were detected using the ECL+ chemiluminescence kit (Amersham Pharmacia Biotech).
ChIP and quantitative-PCR (qPCR) analysis.
Chromatin immunoprecipitation (ChIP) assays were performed with a ChIP assay kit (Upstate Inc.) following the manufacturer's instructions. In brief, 1 × 106 cells were cross-linked with 1% formaldehyde and sonicated 6 times for 15 s each time using a Sonicator 3000 (Misonix Inc.) with a 24 power setting. The sonicated chromatin was then diluted 10-fold in dilution buffer and precleaned with 50 μl of protein G beads (Amershan Inc.). To perform ChIP, precleaned chromatin (100 mg of protein) and specific antibody (2 μg) were added to a tube that was prefilled with antibody buffer (final volume, 500 μl). The mixture was incubated overnight at 4°C. To precipitate antibody-bound chromatin, 6 μl of protein G beads was added, and the mixture was incubated for 1 h at 4°C. The beads were then washed five times with IP washing buffer and once with 1× Tris-EDTA (TE) buffer (pH 8.0). To release DNA from chromatin, the washed beads were treated with proteinase K and reverse buffer at 65°C overnight. Reversal of cross-links and precipitation of the DNA fragments were conducted by following the manufacturer's instructions.
qPCR was conducted using SYBR green fluorescence and an ABI Prism 7500 (PE Applied Biosytems) detection system.
siRNA.
The small interfering RNA (siRNA) duplexes that specifically target SA2 were purchased from Ambion Inc. and from Dharmacon RNAi Technologies Inc. The siRNA oligonucleotide sequences are available upon request. siRNA was transfected into cells by using an X-tremeGene siRNA transfection reagent from Roche. The transfection efficiency of siRNA was monitored using a green fluorescent protein (GFP)-positive control plasmid and measured by Western blotting analysis.
Reverse transcription (RT)-qPCR analysis.
Total RNA was isolated using an RNeasy mini Kit (Qiagen) and treated with a Turbo DNA Free kit (Ambion). cDNAs were prepared with the AffinityScript QPCR cDNA Synthesis Kit (Stratagene). Quantitative PCR was detected by SYBR green fluorescence. The primers used for quantitative PCR are available upon request. The relative expression levels of IGF2, H19, and CTCF were normalized to that of GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
CTCF cloning and GST pulldown assay.
DNA fragments encoding the different human CTCF domains were amplified with PCR (primer sequences are available upon request). All PCR products were digested with the restriction enzymes XhoI and NotI and cloned into the PGEX-5X1 vector (Amersham Inc.), which carries a glutathione S-transferase (GST) tag. The identities of all plasmids were confirmed by DNA sequencing. GST-fused CTCF proteins were expressed in the Escherichia coli BL21(DE3) host strain (Invitrogen) and purified using glutathione-Sepharose beads according to the manufacturer's instructions (GE Healthcare Inc.). Unlabeled or 35S-labeled SA2 was synthesized using an in vitro transcription-translation T7 Coupled Wheat Germ Extract System (Promega). [35S]methionine was obtained from Amersham Inc. The GST pulldown assay was performed as previously described (15). The GST pulldown SA2 was detected by Western blotting or by autoradiography.
SA2 cloning and MBP pulldown assay.
DNA fragments encoding the different domains of the human SA2 protein were amplified by PCR. To express MBP (maltose-binding protein)-fused SA2 domains, all these PCR products were digested with the restriction enzymes BamHI/EcoRI and SalI and cloned into BamHI/EcoRI- and SalI-digested pMal C2G (New England BioLabs) bacterial expression vector. All the plasmids were confirmed by DNA sequencing. The fusion proteins were expressed in the E. coli BL21 host strain and purified with MBP beads purchased from New England BioLabs. The MBP-purified SA2 domains were incubated with nuclear extracts prepared from K562 cells. The MBP pulldown CTCF was detected by Western blotting.
Insulation assay.
The H19 DMR reporter plasmids (pIHLIE and pIHLME) were previously described (16). The Renilla luciferase control plasmid was purchased from Promega Inc. To perform the luciferase assay, the poZ vectors containing the full-length CTCF or the Nterm+ZF CTCF were cotransfected into HeLa cells together with pIHLIE or pIHLME plus a Renilla plasmid using the Fugene HD transfection system (Roche). Cells were collected and lysed after transfection for 24 h. The activities of both luciferases were measured with a Victor Multilabel Counter (Perkin Elmer) by using the Dual-Luciferase Report Assay System (Promega).
The insulator plasmid pNI-core and its control plasmid pNI were described previously (35, 37). The poZ vectors containing the full-length CTCF or the Nterm+ZF CTCF were cotransfected into K562 cells with pNI or pNI-core plasmid. The expression of the reporter gene, neomycin, was detected by RT-qPCR 24 h after transfection.
RESULTS
CTCF copurifies with the cohesin complex.
In earlier studies, we and others have shown that CTCF interacts with a wide variety of proteins (19, 20, 39, 47). To correlate the recent observations concerning CTCF-cohesin interactions (20, 35, 38, 39, 44, 45) with our earlier studies (47), we extended those studies in which proteins copurifying with Flag-HA-CTCF expressed in HeLa cells were identified by mass spectrometry. Because of technical improvements since our earlier analysis (see Materials and Methods), we were able to identify proteins copurifying with CTCF that had not been detected earlier (49). Among these proteins were five members of the cohesin complex: Smc1, Smc3, Rad21, SA1, and SA2 (data not shown). It had been shown in previous studies that Smc1, Smc3, and Rad21 copurify with CTCF and that SA1 is associated with the CTCF binding site of the human Myc promoter (19, 20, 38, 39). However, copurification of SA2 with CTCF has not been detected previously. We note that SA1 and SA2 share 70% sequence homology and that the divergent sequences of the two proteins are mainly found in the first 75 amino acids of the N-terminal region. Cohesin complexes contain either SA1 or SA2, but the latter is more abundant in somatic cells and does not coexist with SA1 in the same complex (26, 40).
We confirmed the mass spectrometry results by immunoprecipitation of Flag-HA-CTCF, followed by Western blotting with antibodies against cohesin complex components (Fig. 1). Cohesin subunits SA1, SA2, Smc1, and Smc3 were detected; Rad21 was detected only weakly. The CTCF-cohesin interaction appears to be DNA independent. First, the cohesin subunits were still detectable even when micrococcal nuclease (MNase), DNase, and RNase were added to the nuclear extract before immunoprecipitation (data not shown). Second, as a control, we carried out parallel experiments with protein Ku70, which binds to some specific DNA sites adjacent to some CTCF sites (21) and can coprecipitate with CTCF (Fig. 1). In the presence of ethidium bromide, Ku70 association with CTCF was disrupted, whereas the CTCF interactions with cohesin components were not affected (Fig. 1).
Fig. 1.
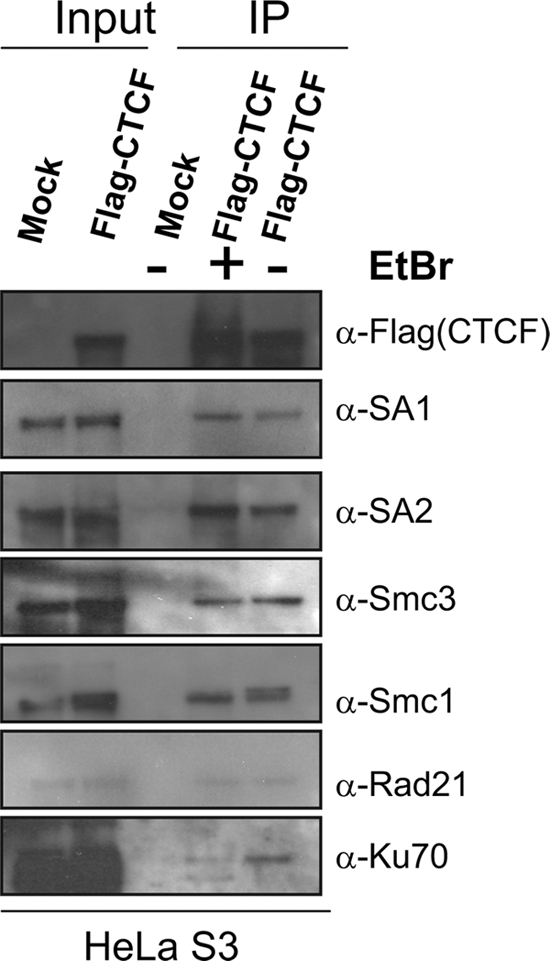
CTCF copurifies with cohesin components. Nuclear extracts were prepared from a HeLa-S3 cell line that stably expresses Flag-HA-CTCF. CTCF-associated proteins were purified with FLAG and HA affinity columns and detected with corresponding antibodies. The Western blots show that components of cohesin coprecipitated with FLAG-HA-CTCF, either with or without treatment with ethidium bromide (EtBr) added to suppress possible DNA-protein interactions.
The above-mentioned experiments did not allow us to distinguish which of the cohesin complex components directly interact with CTCF and which are coprecipitated indirectly. To address this, we carried out GST pulldown assays in which a GST-tagged CTCF peptide (full-length CTCF or truncated forms) was purified and incubated with in vitro-translated SA1, SA2, Smc1, Smc3, or Rad21 (Fig. 2A and B). Western blotting with cohesin subunit antibodies was used to detect interaction; an additional experiment was carried out with 35S-labeled SA2. We found that CTCF binds SA2 and SA1, but not the other cohesin complex components (Fig. 2B, lane 2). This is in contrast to the experiments shown in Fig. 1 with nuclear extracts, where all the components of the cohesin complex were present and could be immunoprecipitated indirectly because of their contact with SA2 or SA1.
Fig. 2.

Identification of interacting domains of CTCF and cohesin components. (A) GST-tagged CTCF constructs used in this and later experiments. (B) Interaction between the constructs in panel A and cohesin components. GST-fused CTCF domains were expressed in E. coli BL21(DE3) and purified with GST beads. Cohesin components were expressed using an in vitro transcription-translation T7-coupled wheat germ extract system in the presence or absence of [35S]cysteine. 35S-labeled cohesin components were detected by autoradiography; those not labeled were detected with specific antibodies. The Coomassie-stained gel shows GST-purified CTCF constructs used in the experiment. (C) Loss of the C terminus of CTCF disrupts CTCF-cohesin interaction in vivo. Various Flag-HA-tagged CTCF domains expressed in HeLa cells were affinity purified, and associated cohesin components were detected by Western blot analysis. (D) MBP-tagged SA2 constructs used to determine the SA2 domain that interacts with CTCF. (E) Interaction of bacterially expressed SA2 with CTCF in a nuclear extract. MBP-fused SA2 peptides were expressed in E. coli BL21(DE3) and purified with MBP beads. MBP-purified SA2 peptides were then incubated with nuclear extracts prepared from the K562 cells. CTCF proteins were detected with antibody. The Coomassie-stained gel shows MBP-purified SA2 constructs used in the experiment. (F) siRNA depletion of SA2 decreases interaction between CTCF and other cohesin complex members. SA2 was transiently knocked down (Input, siRNA) in a HeLa cell line that stably expresses a Flag-HA-tagged CTCF. Flag-CTCF and its associated proteins were affinity purified as for Fig. 1, and the proteins were separated by SDS-PAGE and detected with corresponding antibodies.
To confirm this, we carried out IP experiments in vivo in which siRNA was used to deplete SA2 in HeLa cells expressing Flag-HA-CTCF (Fig. 2F). As expected, the amount of SA2 in the IP fraction was significantly reduced compared to the control. Smc1 and Smc3 abundance was also decreased, consistent with a model in which SA2 interacts directly with CTCF but the other cohesin subunits interact only indirectly, presumably through their contacts with SA2 (see below and Fig. 7D). We note that CTCF interaction with SA1 was not affected. This result is in agreement with previous reports that SA1 and SA2 exist exclusively in separate cohesin complexes (27) and indicates that SA1, like SA2, directly interacts with CTCF. In the following experiments, we focused our attention on the direct interaction between CTCF and SA2. We did not study the equivalent complex with SA1, which in any case is present in much smaller amounts in somatic cells (26, 40), but we expect the interactions to be similar.
Fig. 7.

Binding of Flag-CTCFs and SA2 at the mouse H19 ICR insulator in HeLa cells transfected with pIHLIE or pIHLME. (A) Schematic diagrams of the reporter constructs pIHLIE and pIHLME, showing regions used for PCR (short lines under Ins regions). The two regions are not distinguished by these primers. (B) Flag-tagged CTCF occupancy at the insulator region measured by ChIP using an anti-FLAG antibody. HeLa cells were cotransfected with poZ vector carrying the full-length CTCF, N-term+ZF CTCF, or the poZ vector only combined with either pIHLIE or pIHLME plus a Renilla luciferase control plasmid. The error bars indicate standard deviations. (C) SA2 occupancy measured by ChIP, using an antibody against SA2. (D) Model depicting the interaction between CTCF and the cohesin components.
The C terminus of CTCF is essential for CTCF-cohesin interaction.
To identify the CTCF domain that interacts with SA2, we constructed and expressed in bacteria additional GST-fused CTCF proteins (see Materials and Methods) containing or missing the N-terminal, C-terminal, or zinc finger domain (Fig. 2A). GST pulldown assays were used to test the interaction between CTCF domains and in vitro-translated 35S-labeled SA2 or unlabeled in vitro-translated SA2 (Fig. 2B, lanes 3 to 7). We found that the C terminus of CTCF is essential for CTCF-SA2 interaction: the C terminus alone, similarly to the full-length protein, showed high affinity for SA2, whereas the N terminus or the zinc fingers alone, as well as the Nterm+ZF construct, failed to interact with in vitro-translated SA2. In parallel, we performed immunoprecipitation analyses using HeLa cells that stably expressed various CTCF domains. Western blotting indicated that the CTCF construct ZF+Cterm (amino acids 265 to 727) coprecipitated with SA2 almost as efficiently as full-length CTCF, whereas the CTCF construct Nterm+ZF (amino acids 1 to 579) lost interaction with SA2 (Fig. 2C).
We carried out reciprocal experiments to identify the region of SA2 that interacts with CTCF. MBP fusion constructs carrying various SA2 domains (Fig. 2D) were used to immunoprecipitate CTCF from nuclear extracts; CTCF was detected by Western blotting (Fig. 2E). The interaction with CTCF requires at a minimum the region of SA2 from amino acids 162 to 290; interaction appears to be stronger when a more extended sequence (162 to 993) is used.
The C terminus of CTCF contains several evolutionarily conserved motifs (23, 34). These have been described as the “nucleoside triphosphate binding motif” (NTP) (amino acids 580 to 590), the phosphorylation site (P site) (amino acids 604 to 611), and the nuclear localization signal (NLS) (amino acids 591 to 601). To determine which motifs contributed to the CTCF-SA2 interaction, we first analyzed the SA2 binding capacity in a series of CTCF truncations in which the C terminus was increasingly deleted either from the N-terminal end or from the C-terminal end. As shown in Fig. 2, the GST-tagged CTCF fragments were expressed bacterially and incubated with in vivo-translated SA2. Deletions from the N-terminal end showed that a peptide extending from amino acid 575 to the C terminus could bind in vitro-translated SA2 (Fig. 3B, lane 8). Truncations of CTCF from both the N terminus and C terminus showed that a shorter peptide containing amino acids 575 to 611 was still capable of binding to SA2 (Fig. 3B, lane 4). Further truncations from the N terminus revealed that peptide 591-727 also bound SA2 (Fig. 3C, lane 6), whereas peptide 1-591 did not, but both 1-601 and 1-611 did (Fig. 3D, lanes 6, 7, and 9). These results indicate that at least when carrying out large-scale deletions, neither the P site nor the NTP region appears to be important for the CTCF-SA2 interaction. In contrast, deletions of the NLS region failed to pull down SA2. To explore these interactions in greater detail, we constructed full-length GST-CTCF site mutants with amino acid substitutions in the NTP, NLS, or P-site domain (Fig. 4). In experiments similar to those in Fig. 2 and 3, GST pulldown assays were used to test interactions with in vitro-translated SA2. We found that, with full-length CTCF constructs, mutations in either the NTP, NLS, or P site were sufficient to interfere with interaction between SA2 and CTCF. This result differs from that obtained with some truncated CTCF constructs described above, in which the regions containing amino acids 1 to 597, 1 to 613, 602 to 727, or 612 to 727 were deleted (Fig. 3C, lanes 7 and 8, and D, lanes 7 and 9), and reveals that in the full-length protein the NTP site, the NLS region, and the P site each play a role in stabilizing interactions between SA2 and CTCF, which is supported by experiments described below.
Fig. 3.

Determination of CTCF domains that interact with SA2 in vitro. (A) Diagram showing subdomains of the CTCF C terminus as designated by Klenova et al. (23). Numbering of amino acids is in the sequence, and the corresponding numbers in the nucleotide sequence are under the arrows above. (B, C, and D) (Top) Western blots in which various bacterially expressed GST-CTCF peptides were purified with GST beads and incubated with SA2 expressed in vitro using the wheat germ extract system. The interaction was detected with SA2 antibody. The initial and final residue numbers for each peptide are shown above the lanes. (Bottom) Coomassie-stained gels of the purified CTCF constructs.
Fig. 4.

Mutations in the CTCF C terminus disrupt the interaction between CTCF and SA2. (A) Diagram showing each mutated amino acid within the NTP binding site, NLS region, and P site on the CTCF C terminus, where PDGVEGENGGET was replaced with PDGVAGANGGET, KKSKR with RRGE, and SKKEDSSD with AKKEDAAD. (B) Full-length CTCF, C-terminal peptide of CTCF, and full-length CTCF carrying mutations were expressed in BL21(DE3) and purified with GST beads. SA2 was expressed in a wheat germ extract system as shown in Fig. 2 and 3. The Coomassie-stained gel shows the purified CTCF constructs. The interaction was detected with SA2 antibody.
The C terminus of CTCF is required for insulator activity at the Igf2/H19 imprinted region.
We next asked whether the cohesin binding properties of the CTCF mutants described above were reflected in CTCF insulator function. The cell lines stably expressing full-length CTCF or CTCF mutants and used for the CTCF-cohesin interaction measurements described above were not suitable for the insulator studies, because overexpression of these CTCFs affects DNA methylation genome-wide (11). We found that stable overexpression of the full-length CTCF or CTCF mutants had varying effects on DNA methylation patterns at CTCF binding sites in the Igf2-H19 ICR, which could alter CTCF binding independently of any effects of lack of recruitment of cohesin to those sites (data not shown). For that reason, we used transient-transfection methods to introduce CTCF constructs into cells. The Ifg2-H19 locus is imprinted, and in untransfected cells, the binding of CTCF to the maternal imprinted control region results in suppression of Igf2 expression and elevation of H19 expression from the maternal allele (2, 13, 18).
To measure the effect of transient expression of CTCF C-terminally truncated mutants on expression of Igf2 and H19 in MEFs, we first examined the DNA methylation status at the ICR. No change in DNA methylation levels was observed between the control cells and the transiently transfected cells (data not shown). Transient expression of full-length CTCF had no significant effect on Igf2 or H19 mRNA levels compared to mock-transfected cells, but expression of the C-terminally truncated mutant (Nterm+ZF) resulted in an increase in Igf2 expression and a decrease in H19 expression (Fig. 5A, B, and C). This is consistent with a model in which the truncated mutant acts as a competitive inhibitor of full-length CTCF, resulting in a loss of insulating activity at the ICR correlated with loss of SA2 binding (Fig. 5D and E; see Fig. 7). We note that the increase in Igf2 expression on introduction of the mutant CTCF is larger than the expected 2-fold, suggesting that the mutant may have direct effects on the Igf2 gene independent of insulating action. These results parallel those obtained by Wendt et al. (45) when Scc1 (Rad21) was depleted and confirm both that recruitment of cohesin is essential for CTCF insulator function at the Igf2-H19 locus and that the C terminus of CTCF mediates that recruitment.
Fig. 5.

Igf2 and H19 expression in a MEF line (A to C) or HeLa cells (D and E). MEFs were transiently transfected with a poZ vector carrying the human full-length CTCF or a peptide containing the N terminus and zinc finger domain (Nterm+ZF) or with the poZ vector only. mRNA extracted from each cell line was used to generate cDNA. RT-qPCR was used to measure IGF2 and H19 expression. The relative expression levels of IGF2, H19, and CTCF were normalized to that of GAPDH. (A) IGF2 expression. (B) H19 expression. (C) Expression of Flag-CTCF constructs. (D and E) Igf2 and H19 expression in HeLa cells with SA2 depleted using siRNA. mRNA extracted from each cell line was used to generate cDNA. RT-qPCR was used to measure IGF2 and H19 expression. (D) IGF2 expression. (E) H19 expression. The error bars indicate standard deviations.
To explore these effects further, we carried out experiments with reporter constructs pIHLIE and pIHLME, designed to measure CTCF-dependent insulator function (Fig. 6A) (16). HeLa cells were transiently transfected with one of these reporter constructs, together with a plasmid expressing either Flag-tagged full-length CTCF, a CTCF mutant lacking the C terminus, or an empty-vector control plus a plasmid expressing a cytomegalovirus (CMV)-Renilla plasmid as a standard for transfection efficiency. As has been observed in earlier studies, introduction of the full-length CTCF resulted in lower expression of luciferase in pIHLIE, where access of the enhancer to the promoter is impeded, than in pIHLME, where the downstream CTCF site is mutated and CTCF binding is reduced (Fig. 7B). In contrast, the CTCF mutant with a C-terminal deletion failed to function as an insulator: luciferase expression levels were similar in pIHLIE and pIHLME (Fig. 6B). Experiments with CTCF carrying the NTP site mutations gave similar results (Fig. 6B).
Fig. 6.

Deletion of the C terminus of CTCF results in loss of CTCF-mediated insulation function. (A) Schematic diagram of the reporter constructs pIHLIE and pIHLME that were used in the insulation assay (45). In the pIHLIE reporter plasmid, expression of luciferase is controlled by a wild-type mouse ICR insulator (Ins), which allows CTCF to bind and blocks the interaction between the enhancer (Enh) and the luciferase promoter, leading to low expression of luciferase. In the pIHLME control plasmid, the mouse ICR insulator is replaced with mutated sequences (Mut Ins) that do not bind CTCF, resulting in normal expression of luciferase. (B) HeLa cells were cotransfected with poZ vector carrying the full-length CTCF, Nterm+ZF CTCF, CTCF NTP mutant, or the poZ vector only combined with either pIHLIE or pIHLME plus a Renilla luciferase control plasmid. Cell lysates were prepared 24 h after transfection with plasmids and used to measure luciferase activities. The relative luciferase activity for each cell lysate was normalized to that of Renilla luciferase. The error bars indicate standard deviations. (C) Schematic diagrams of the reporter constructs pNI (control) and pNI-5′HS4 core insulator plasmid that were also used in insulation assays. The pNI-core plasmid contains a neomycin reporter gene, activation of which is blocked by two copies of the core insulator element, containing a CTCF site, from the 5′ HS4 site upstream of the chicken β-globin locus. (D) K562 cells were cotransfected with poZ vector carrying the full-length CTCF, Nterm+ZF CTCF, CTCF NTP mutant, or the poZ vector only combined with either pNI or pNI-core plasmid. cDNA was prepared from cells 24 h after transfection with plasmids to measure expression of the Neomycin reporter. For each transfection, the relative expression level of Neomycin was normalized against GAPDH expression. (E and F) SA2 is required for CTCF-mediated insulation function. K562 cells were depleted of SA2 by treatment with siRNA for 48 h. The SA2 knockdown efficiency was detected by Western blotting. These cells were transfected with either pNI or pNI-core plasmid and analyzed for Neomycin expression as in panel D.
To test whether the loss of insulation function is due to reduced SA2 recruitment, we first performed ChIP analysis using an antibody against Flag that detects the Flag-tagged CTCFs. We found that both the Flag-tagged full-length CTCF and the CTCF mutant lacking the C terminus bound to the mouse H19 ICR insulator element within pIHLIE (Fig. 7B). However, SA2 binding to this element was much reduced when the C terminus of CTCF was deleted (Fig. 7C). The primers used should detect binding of CTCF at either of the insulator (Ins) sites, and strong binding of both WT and mutant CTCF species is detected on pIHLIE. Note, however, that binding is reduced by much more than half on pIHLME, where the Ins element adjacent to the enhancer is mutated. This suggests that in this construct cooperative interactions between the two sites may help stabilize CTCF binding. This, of course, makes pIHLME an even more effective control. To confirm that introduction of exogenous CTCF did not affect DNA methylation on the test plasmids, we carried out HpaII/MspI digestion of the ICR site on pIHLIE/pIHLME. During the period following transfection over which the preceding experiments were conducted, the site remained unmethylated (data not shown).
In addition, we tested the effect of overexpressing CTCF on insulator function with another pair of reporter plasmids: the pNI and pNI-core plasmids (35, 37) (Fig. 6C). We transiently transfected the pNI or pNI-core plasmid into human K562 erythroid cells. Neomycin expression analysis confirmed the results shown in Fig. 6A: expression of CTCF lacking the C terminus, or of the NTP mutant, resulted in loss of insulator activity (Fig. 6D). In parallel experiments, we transfected the pNI or pNI-core plasmid into SA2-depleted K562 cells. In agreement with previous reports that cohesin is required for CTCF-mediated insulation activity (45), depletion of SA2 also led to loss of insulation activity (Fig. 6E and F). Collectively, these results indicate that the CTCF C terminus is essential for its insulator function and that mutations at sites within the C terminus that affect SA2 binding impair this function.
DISCUSSION
The observations, reported by several laboratories, that CTCF recruits the cohesin complex to at least the majority of CTCF sites and that the ability of CTCF to function as an enhancer-blocking insulator depends on this recruitment raise obvious questions about the nature of the interaction between CTCF and cohesin. Although the earlier studies revealed that members of the cohesin complex—Smc1, Smc3, and Rad 21—could all be immunoprecipitated with CTCF, it had not been known which of these components interacts with CTCF directly or with what sites on CTCF. We have addressed these questions in this study. Our IP experiments show that, although CTCF coprecipitates all members of the cohesin complex—Smc1, Smc3, Rad21, and SA2 (Fig. 1)—only the interaction with SA2 is direct. The other cohesin proteins are recruited through their contact with SA2 (Fig. 2A, B, and C).
Recently, it has been reported that depletion of cohesin in HeLa cells results in an increase in IGF2 transcript levels and a decrease in H19 levels, consistent with the idea that cohesin binding to the Igf2-H19 ICR is necessary to block activation by downstream enhancers, the established function of the CTCF-mediated enhancer-blocking insulator activity (45). We therefore asked what the effect would be of introducing into mouse embryo fibroblasts or HeLa cells CTCF mutants that could not recruit cohesin. As shown in Fig. 5, expression of the truncated CTCF (Nterm+ZF), lacking the C terminus, resulted in a marked increase in Igf2 expression and a decrease in H19, reflecting interference in recruitment of SA2 to the ICR. We confirmed these effects by using two additional assays based on plasmid reporter constructs designed to detect insulation activity associated with CTCF binding (Fig. 6).
In these experiments, it was important to determine that expression of CTCF mutants did not affect levels of DNA methylation at the CTCF binding sites, since that could lead to changed binding properties at the sites. For that reason, we made use of short-duration transient transfection of both CTCF mutants and reporter plasmids. Studies involving long-term changes in CTCF expression levels may give rise to changes in DNA methylation that complicate the interpretation of the results.
Conclusions.
The well-established ability of the cohesin complex to link sister chromatids during S phase has naturally led to the suggestion that it has a related function when bound to CTCF: the ability to stabilize interactions between distant chromatin sites within the nucleus, leading to formation of loop domains. In this model, the details of the disposition of the cohesin complex remain unclear. Presumably, as in models of the role of cohesin during mitosis, the closed ring composed of Smc1, Smc3, and Rad21 bound to a CTCF site would open and encircle some target on a distant CTCF site. It may be relevant, therefore, that the only cohesin subunit interacting directly with CTCF is SA2 (or in some cases SA1), which is not integral to the closed cohesin circle and could act as a bridge between CTCF and the cohesin circle (Fig. 7D). This does not exclude the possibility of indirect binding mechanisms in which secondary proteins bind both to CTCF and to other cohesin components and help stabilize the CTCF-cohesin interaction (46). It has been reported that during cohesin assembly in meiosis in Caenorhabditis elegans (3) and in grasshoppers (41) the SMC proteins can assemble first, and Scc3(SA2) associates with the complex independently. This suggests that, in the course of recruiting the cohesin complex to CTCF, SA2 could bind first and the remainder of the complex could assemble subsequently on that site (Fig. 7D).
Much of the CTCF molecule is also free to engage in other interactions, and evidence from other laboratories shows that it does make contact with a variety of regulatory factors both in vivo and in vitro. Among those factors for which the regions of interaction on CTCF have been identified, CHD8, Sin3, and YB-1 interact with sites in the zinc finger domain (16, 22, 28). We have found that the DEAD box protein p68 also interacts with the zinc finger domain of CTCF (46). The regulatory factor Kaiso is reported to interact with the C terminus of CTCF, but the region of critical interaction does not overlap with the cohesin binding domain. Kaiso binds specifically to DNA and suppresses insulation when its binding site is adjacent to the CTCF site (7). The large subunit of RNA polymerase II is reported to be recruited to CTCF binding sites through an interaction with the C terminus of CTCF (4, 7). We do not know whether the contact points in this case overlap with or are distinct from the SA2 site or whether more than one factor can be attached to the C terminus at a given moment, though of course they may “time share” occupancy. The C terminus of CTCF contains many amino acids that could undergo covalent modification. In particular, the four serine residues that are targets of phosphorylation have been shown to be important for c-myc expression: replacement of the serines results in enhanced repression of c-myc, though it does not affect binding of CTCF to the nearby sites on DNA (23). Given that replacement of these serines also affects SA2 binding to CTCF (Fig. 4), it is possible that at least one effect of phosphorylation is to interfere with cohesin recruitment and insulator-related (i.e., loop-forming) functions. Quite recent work has shown that cohesin can also be recruited to regulatory sites on chromatin by Mediator. In addition to identifying the components of the Mediator complex that are involved in this interaction, it will be important to determine whether SA2 plays the same role at these sites in binding of cohesin.
ACKNOWLEDGMENTS
We thank José Luis Barbero for SA2 plasmids, Masahiko Negishi for the Smc1 plasmid, and Marisa S. Bartolomei for the MEF cell line. We are grateful to Matthias Merkenschlager and Suzana Hadjur for initially interesting us in this problem and for critical advice. We also thank members of the Felsenfeld laboratory for helpful discussions and critical reading of the manuscript.
This work was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH.
Footnotes
Published ahead of print on 28 March 2011.
REFERENCES
- 1. Baniahmad A., Steiner C., Kohne A. C., Renkawitz R. 1990. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell 61:505–514 [DOI] [PubMed] [Google Scholar]
- 2. Bell A. C., Felsenfeld G. 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405:482–485 [DOI] [PubMed] [Google Scholar]
- 3. Chan R. C., et al. 2003. Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature 423:1002–1009 [DOI] [PubMed] [Google Scholar]
- 4. Chernukhin I., et al. 2007. CTCF interacts with and recruits the largest subunit of RNA polymerase II to CTCF target sites genome-wide. Mol. Cell. Biol. 27:1631–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chung J. H., Bell A. C., Felsenfeld G. 1997. Characterization of the chicken beta-globin insulator. Proc. Natl. Acad. Sci. U. S. A. 94:575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung J. H., Whiteley M., Felsenfeld G. 1993. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74:505–514 [DOI] [PubMed] [Google Scholar]
- 7. Defossez P. A., et al. 2005. The human enhancer blocker CTC-binding factor interacts with the transcription factor Kaiso. J. Biol. Chem. 280:43017–43023 [DOI] [PubMed] [Google Scholar]
- 8. De La Rosa-Velázquez I. A., Rincon-Arano H., Benitez-Bribiesca L., Recillas-Targa F. 2007. Epigenetic regulation of the human retinoblastoma tumor suppressor gene promoter by CTCF. Cancer Res. 67:2577–2585 [DOI] [PubMed] [Google Scholar]
- 9. Filippova G. N., et al. 2002. Tumor-associated zinc finger mutations in the CTCF transcription factor selectively alter tts DNA-binding specificity. Cancer Res. 62:48–52 [PubMed] [Google Scholar]
- 10. Gaszner M., Felsenfeld G. 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 7:703–713 [DOI] [PubMed] [Google Scholar]
- 11. Guastafierro T., et al. 2008. CCCTC-binding factor activates PARP-1 affecting DNA methylation machinery. J. Biol. Chem. 283:21873–21880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hadjur S., et al. 2009. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 460:410–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hark A. T., et al. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486–489 [DOI] [PubMed] [Google Scholar]
- 14. Holmgren C., et al. 2001. CpG methylation regulates the Igf2/H19 insulator. Curr. Biol. 11:1128–1130 [DOI] [PubMed] [Google Scholar]
- 15. Huang S., Qiu Y., Stein R. W., Brandt S. J. 1999. p300 functions as a transcriptional coactivator for the TAL1/SCL oncoprotein. Oncogene 18:4958–4967 [DOI] [PubMed] [Google Scholar]
- 16. Ishihara K., Oshimura M., Nakao M. 2006. CTCF-dependent chromatin insulator is linked to epigenetic remodeling. Mol. Cell 23:733–742 [DOI] [PubMed] [Google Scholar]
- 17. Kagey M. H., et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanduri C., et al. 2000. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 10:853–856 [DOI] [PubMed] [Google Scholar]
- 19. Kang H., Lieberman P. M. 2009. Cell cycle control of Kaposi's sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J. Virol. 83:6199–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang H., Lieberman P. M. 2009. Cell cycle control of KSHV latency transcription by CTCF-cohesin interactions. J. Virol. 83:6199–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katz D. J., Beer M. A., Levorse J. M., Tilghman S. M. 2005. Functional characterization of a novel Ku70/80 pause site at the H19/Igf2 imprinting control region. Mol. Cell. Biol. 25:3855–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klenova E., et al. 2004. YB-1 and CTCF differentially regulate the 5-HTT polymorphic intron 2 enhancer which predisposes to a variety of neurological disorders. J. Neurosci. 24:5966–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klenova E. M., et al. 2001. Functional phosphorylation sites in the C-terminal region of the multivalent multifunctional transcriptional factor CTCF. Mol. Cell. Biol. 21:2221–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li T., et al. 2008. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol. Cell. Biol. 28:6473–6482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lobanenkov V. V., et al. 1990. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5:1743–1753 [PubMed] [Google Scholar]
- 26. Losada A. 2007. Cohesin regulation: fashionable ways to wear a ring. Chromosoma 116:321–329 [DOI] [PubMed] [Google Scholar]
- 27. Losada A., Yokochi T., Kobayashi R., Hirano T. 2000. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol. 150:405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lutz M., Baniahmad A., Renkawitz R. 2000. Modulation of thyroid hormone receptor silencing function by co-repressors and a synergizing transcription factor. Biochem. Soc. Trans. 28:386–389 [PubMed] [Google Scholar]
- 29. Mishiro T., et al. 2009. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 28:1234–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nasmyth K., Haering C. H. 2009. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 43:525–558 [DOI] [PubMed] [Google Scholar]
- 31. Nativio R., et al. 2009. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 5:e1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogawa H., Ishiguro K., Gaubatz S., Livingston D. M., Nakatani Y. 2002. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296:1132–1136 [DOI] [PubMed] [Google Scholar]
- 33. Ohlsson R., Lobanenkov V., Klenova E. 2010. Does CTCF mediate between nuclear organization and gene expression? Bioessays 32:37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohlsson R., Renkawitz R., Lobanenkov V. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 17:520–527 [DOI] [PubMed] [Google Scholar]
- 35. Parelho V., et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132:422–433 [DOI] [PubMed] [Google Scholar]
- 36. Phillips J. E., Corces V. G. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Recillas-Targa F., Bell A. C., Felsenfeld G. 1999. Positional enhancer-blocking activity of the chicken beta-globin insulator in transiently transfected cells. Proc. Natl. Acad. Sci. U. S. A. 96:14354–14359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rubio E. D., et al. 2008. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U. S. A. 105:8309–8314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stedman W., et al. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 27:654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sumara I., Vorlaufer E., Gieffers C., Peters B. H., Peters J. M. 2000. Characterization of vertebrate cohesin complexes and their regulation in prophase. J. Cell Biol. 151:749–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valdeolmillos A. M., et al. 2007. Sequential loading of cohesin subunits during the first meiotic prophase of grasshoppers. PLoS Genet. 3:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vostrov A. A., Quitschke W. W. 1997. The zinc finger protein CTCF binds to the APBbeta domain of the amyloid beta-protein precursor promoter. Evidence for a role in transcriptional activation. J. Biol. Chem. 272:33353–33359 [DOI] [PubMed] [Google Scholar]
- 43. Wallace J. A., Felsenfeld G. 2007. We gather together: insulators and genome organization. Curr. Opin. Genet. Dev. 17:400–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wendt K. S., Peters J. M. 2009. How cohesin and CTCF cooperate in regulating gene expression. Chromosome Res. 17:201–214 [DOI] [PubMed] [Google Scholar]
- 45. Wendt K. S., et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801 [DOI] [PubMed] [Google Scholar]
- 46. Yao H., et al. 2010. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev. 24:2543–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yusufzai T. M., Tagami H., Nakatani Y., Felsenfeld G. 2004. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell 13:291–298 [DOI] [PubMed] [Google Scholar]