

Abstract
Analysis of sequenced bacterial genomes revealed that the genomes encode more than 30% hypothetical and conserved hypothetical proteins of unknown function. Among proteins of unknown function that are conserved in anaerobes, some might be determinants of the anaerobic way of life. This study focuses on two divergent clusters specifically found in anaerobic microorganisms and mainly composed of genes encoding conserved hypothetical proteins. We show that the two gene clusters DVU2103-DVU2104-DVU2105 (orp2) and DVU2107-DVU2108-DVU2109 (orp1) form two divergent operons transcribed by the σ54-RNA polymerase. We further demonstrate that the σ54-dependent transcriptional regulator DVU2106, located between orp1 and orp2, collaborates with σ54-RNA polymerase to orchestrate the simultaneous expression of the divergent orp operons. DVU2106, whose structural gene is transcribed by the σ70-RNA polymerase, negatively retrocontrols its own expression. By using an endogenous pulldown strategy, we identify a physiological complex composed of DVU2103, DVU2104, DVU2105, DVU2108, and DVU2109. Interestingly, inactivation of DVU2106, which is required for orp operon transcription, induces morphological defects that are likely linked to the absence of the ORP complex. A putative role of the ORP proteins in positioning the septum during cell division is discussed.
INTRODUCTION
Sulfate-reducing microorganisms (SRM) are anaerobic microorganisms that exhibit a mode of growth based on the reduction of sulfate as a terminal electron acceptor. They are ubiquitous in anoxic habitats, where they play an important role in both sulfur and carbon cycles (26). The broad range of compounds that SRM are able to use as substrates for growth (26) illustrates their great metabolic versatility. In addition to their ecological value in the sulfur cycle, SRM are also important in biotechnology, since they are used in heavy metal bioremediation and sulfur compound removal from waste compounds and flue gas (40). On the other hand, they also cause serious problems in industry because of the production of sulfide, which is highly reactive, corrosive, and toxic (3). The recent emergence of genomic techniques has brought new insights into the way of life of SRM. So far, more than 20 complete genomes of SRM are available. The analysis of the genome of the deltaproteobacterium Desulfovibrio vulgaris Hildenborough revealed that 34% of it encodes hypothetical and conserved hypothetical proteins of unknown function (19, 27). Among them, several are conserved in anaerobes, suggesting that they serve critical cellular functions in the anaerobic way of life of D. vulgaris Hildenborough and other SRM.
A comparative analysis of genomes from anaerobic and aerobic microorganisms using the cluster of groups of orthologous proteins (COG) database (53) has led to the identification of 33 COGs that are specific to the anaerobic way of life (18). Twenty-eight were correlated with known functions, such as protection against oxidative conditions in anaerobic organisms, e.g., superoxide scavenging (COG2033) or oxygen reduction (COG1773). Five COGs contain uncharacterized conserved proteins that were assumed to play a crucial role in anaerobiosis, including COG1433. Very little information is available regarding COG1433, except for the nuclear magnetic resonance (NMR) structures of three proteins: Methanobacterium MTH1175 (Protein Data Bank [PDB] 1eo1) and Thermotoga maritima TM1816 (PDB 1t3v) and TM1290 (PDB 1rdu) (12, 13, 20). Although these three proteins share only 30% sequence identity, their three-dimensional structures are highly similar, with all the secondary structures superimposable. Their three-dimensional structures resemble that of RNase H (PDB 1cxq) but differ from it in key aspects that make unlikely a nuclease activity for these proteins (13). Despite the availability of the three-dimensional structures, no physiological function has been yet assigned to this class of proteins.
In D. vulgaris Hildenborough, three protein locus tags, DVU2107, DVU2108, and DVU2109, belong to COG1433. DVU2107 and DVU2108 are two putative proteins of 183 and 119 residues, respectively, with a full-length COG1433 domain. Interestingly, DVU2108 exhibits 48% sequence identity with the orange protein (ORP) from Desulfovibrio gigas. ORP is a monomeric soluble protein that contains a novel metal sulfide (S2MoS2CuS2MoS2) cluster noncovalently bound to the polypeptide chain (8, 24, 42). DVU2109 is a 487-residue protein composed of two domains: the N-terminal domain (residues 1 to 297) belongs to COG0489 (ATPases involved in chromosome partitioning), and the C-terminal domain (residues 375 to 480) belongs to COG1433. Phylogenetic analysis shows that homologues of DVU2108 genes are present in most archaeal genomes and several bacterial genomes and tend to be systematically clustered with DVU2103 and DVU2104 homologues (50). The DVU2103 and DVU2104 proteins share 41% similarity to each other and belong to COG1149. This COG comprises MinD superfamily P-loop ATPases with an additional ferredoxin domain (53). Additionally, the N-terminal domain of DVU2103 exhibits significant homology with proteins belonging to COG2894, which comprises septum formation inhibitor-activating ATPases. Although the function of COG1149 proteins is yet to be discovered, it is noteworthy that the COG is distributed only in anaerobic microorganisms.
The consistent clustering pattern of DVU2103, DVU2104, and DVU2108 genes across taxonomically diverse microorganisms led the authors to propose that the gene cluster shares similar evolutionary histories and is involved in common cellular functions (50). Accordingly, transcriptome studies suggest that the gene set is involved in the lifestyle change of D. vulgaris Hildenborough from syntrophy to sulfate reduction (50).
In D. vulgaris Hildenborough, DVU2108 is located in a cluster divergent from genes DVU2103 and DVU2104. Indeed, the DVU2103, DVU2104, and DVU2105 genes form a cluster with a transcriptional direction opposite that of a second cluster composed of the DVU2107, DVU2108, and DVU2109 genes (see Fig. S1 in the supplemental material). These divergent gene clusters are separated by DVU2106, annotated as a σ54-dependent transcription factor (27). Altogether, these genes compose a large cluster, named here the orp gene cluster.
σ54-dependent transcription factors, also called enhancer-binding activator proteins (EBPs), bound to regulatory conserved sequences usually located upstream of σ54-dependent promoters. EBP-catalyzed ATP hydrolysis is required for opening the σ54-RNA polymerase promoter complexes to initiate transcription (44, 47, 51, 59). Most EBPs participate in signal transduction circuits that respond to environmental cues (9, 16, 23). Homologues of the EBP-encoding gene, DVU2106, are found colocated with homologues of DVU2103, DVU2104, and DVU2108 in most Deltaproteobacteria but are missing in the Archaea, Firmicutes, and Thermotogaceae (see Fig. S1 in the supplemental material). This finding suggests a link between DVU2106 and the transcription of DVU2103, DVU2104, and DVU2108 genes, but to our knowledge, no data have been reported so far for σ54-dependent regulation in Desulfovibrio species. The presence of a single copy of a gene annotated as RNA polymerase σ54 factor and the identification of 70 potential σ54 promoters by computational prediction (10) support the idea that the σ54-RNA polymerase might govern the expression of numerous genes in D. vulgaris Hildenborough.
In this paper, we characterize the transcriptional regulation of the orp gene cluster. We identify two divergent operons, named here orp2 and orp1, which are regulated by the product of the DVU2106 gene and the σ54-bound RNA polymerase. We further show that DVU2106 downregulates its own expression. Finally, we show that this gene cluster encodes a protein complex in vivo, the absence of which induced an aberrant cellular morphology.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and growth conditions.
The strains of Escherichia coli and D. vulgaris Hildenborough and the plasmids used in this study are listed in Table S1 in the supplemental material. The primers used in the study are listed in Table S2 in the supplemental material. The E. coli strains DH5α and MW3064 were grown at 37°C in Luria-Bertani (LB) medium supplemented with the appropriate antibiotic when required (0.27 mM for ampicillin and 0.15 mM for chloramphenicol). E. coli MW3064 was grown with 0.3 mM 2,6-diaminopimelic acid. Cultures (up to 100 ml) of D. vulgaris Hildenborough were prepared in medium C (45) at 32°C in an anaerobic chamber (COY Laboratory Products) filled with a 10% H2-90% N2 mixed-gas atmosphere, and large-scale cultures (20 liters) were carried out in 300-liter fermentors. D. vulgaris Hildenborough was grown until the optical density at 600 nm (OD600) reached 0.4 for RNA preparation and 0.8 for purification of Strep-tag II (strep)-tagged proteins. The medium was supplemented with the appropriate antibiotic concentrated at 0.17 mM for kanamycin and 0.15 mM for thiamphenicol when specified.
Methanosarcina barkeri (DSM 804) was purchased from the Deutsche Sammlung von Mikroorganismen and Zellkulturen (Braunschweig, Germany).
Methanosarcina monocultures were grown in CCM medium (58) lacking lactate but amended with 5 mM acetate, and the headspace was filled with 80% H2-20% CO2.
Cocultures of D. vulgaris Hildenborough and M. barkeri were grown in the dark at 37°C in CCM medium amended with 30 mM lactate with the headspace filled with 80% H2-20% CO2 in Hungate tubes equipped with rubber stoppers and screw tops with shaking at 200 rpm. Cocultures were established by inoculating 10% (vol/vol) of an exponentially growing culture of D. vulgaris Hildenborough and 10% (vol/vol) of an exponentially growing culture of M. barkeri.
DNA manipulations.
Standard protocols were used for cloning and transformations. All restriction endonucleases and DNA modification enzymes were purchased from New England Biolabs. PCRs were performed with Expand High Fidelity from Roche Diagnostics Corp. Chromosomal DNA was purified using the Wizard Genomic DNA purification kit from Promega. DNA fragments and plasmids were excised or purified using MiniElute kits from Qiagen.
Construction of E. coli mutant strains.
The W3110 lacZ derivative was constructed by a one-step inactivation procedure using λ red technology (14). Briefly, the kanamycin cassette from plasmid pKD4 was PCR amplified using two oligonucleotides carrying 5′ extensions complementary to the sequence upstream of the lacZ start codon and to sequence downstream of the lacZ stop codon. The PCR product was then electroporated into the W3110 strain carrying plasmid pKD46. lacZ::kan strains were selected on kanamycin-LB plates supplemented with isopropyl-β-d-galactopyranoside (IPTG) and bromo-chloro-indolyl-β-d-galactopyranoside (X-Gal). The absence of the lacZ gene in kanamycin-resistant and white colonies was verified by PCR. The mutation was then transferred to W3110 by P1 transduction. For construction of W3110 lacZ rpoN, the rpoN::kan cassette from W3110 rpoN::kan (4) was transferred into W3110 lacZ by P1 transduction.
Construction of a DVU2106 gene disruptant.
For construction of the DVU2106 gene disruptant, DNA corresponding to the Per-Arnt-Sim (PAS) domain of DVU2106 (residues 1 to 128) was amplified by PCR and subcloned into the XhoI and SpeI sites of the plasmid pNot19Cm-Mob-XS (see Table S1 in the supplemental material) to obtain p2106PAS. p2106PAS was transferred into E. coli MW3064 and subsequently transferred by conjugational gene transfer into D. vulgaris Hildenborough. Cells that had recombined the target disruptant gene into the chromosome were selected for their resistance to thiamphenicol, and the construction was verified by PCR.
Gene tagging on the D. vulgaris Hildenborough chromosome.
For the construction of genes fused to the C-terminal, two strategies were used depending on the position of the gene to be tagged in the operon. For DVU2103 and DVU2109 (the last gene of each operon), 500 bp upstream of the insertion site, i.e., the stop codon of the target gene, was amplified from D. vulgaris Hildenborough genomic DNA. For DVU2108, located in the middle of the operon, both the DVU2107 and DVU2108 genes were amplified by PCR. DNA fragments were amplified by using the specific forward primers shown in Table S2 in the supplemental material (Nter2103SpeI for DVU2103-strep, DVU2107 for DVU2108-strep, and 2109strep_comp for DVU2109-strep) containing, respectively, a restriction site for SpeI, for EcoRI, and for XhoI. For amplification of the 3′ region flanking the Strep-tag II insertion site, a specific reverse primer containing the Strep-tag II-specific sequence encoding the 8 amino acids (WSHPQFEK) (49) and restriction sites for SpeI (DVU2103-strep and DVU2109-strep) and BamHI (DVU2108-strep) was used. The three PCR products were digested with the appropriate enzymes and purified. The 900-bp SpeI fragment encoding DVU2103-strep, the 1,232-bp EcoRI-BamHI fragment encoding DVU2108-strep, and the 1,500-bp XhoI-SpeI fragment encoding DVU2109-strep were subcloned into pNot19Cm-Mob-XS (see Table S1 in the supplemental material) previously digested with the appropriate enzymes to obtain p2103-strep, p2108-strep, and p2109-strep, respectively. The resulting plasmids were transferred into E. coli MW3064 and subsequently transferred into D. vulgaris Hildenborough by conjugational gene transfer with E. coli as described previously (57). Cells that had recombined the tagged genes into the chromosome were selected by their resistance to thiamphenicol, and each construction was verified by PCR (using a primer containing the Strep-tag II sequence and specific primers containing nucleotides homologous to a gene present on the chromosome before the conjugational event).
Purification of strep-tagged proteins from D. vulgaris Hildenborough.
D. vulgaris Hildenborough cells in the late exponential growth phase (20 liters; OD600 = 0.8 to 0.9) were harvested and resuspended in buffer W (100 mM Tris-HCl [pH 8], 150 mM NaCl) and Complete protease inhibitor EDTA free (from Roche Applied Science) and disrupted twice with a French press. The mixture was then centrifuged for 1 h at 27,000 × g at 4°C, and the supernatant after filtration was loaded (0.5 ml/min) onto a Strep-Tactin Superflow high-capacity cartridge H-Pr (IBA; BioTAGnology) on an ÄKTAprime Plus chromatography workstation system (GE Healthcare) previously equilibrated with buffer W. The column was washed (2 ml/min) with 50 ml of the same buffer, and the strep-tagged fractions were eluted (1 ml/min) using buffer E (100 mM Tris-HCl [pH 8], 150 mM NaCl, 2.5 mM desthibiotin). The 3-ml strep-tagged protein complex fractions were concentrated 10 times using Amicon Ultra centrifugal filter devices (cutoff, 5 kDa) from Millipore and stored at −80°C for further analysis by SDS-PAGE.
SDS-PAGE.
Forty micrograms of each strep-tagged protein complex fraction was separated on a 12.5% SDS-polyacrylamide gel and stained with Bio-Safe Coomassie (Bio-Rad). Protein bands were cut out, digested with trypsin, and analyzed by mass spectrometry.
Mass spectrometry.
Tryptic digestion of excised gel plugs, ion trap, and matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) protein identification were performed as previously described (25).
Cloning and purification of DVU2106c.
The sequence encoding a truncated version of the DVU2016 protein (amino acids 127 to 458; DVU2106c) was amplified from genomic DNA and cloned into the pET19b plasmid (Novagen). The DVU2106c protein fragment was purified from 500 ml of E. coli BL21(DE3) culture induced for 16 h at 22°C with 200 μM IPTG. Under these conditions, recombinant DVU2106c was essentially found in inclusion bodies. The cell pellet was resuspended in 10 ml of 20 mM Tris-HCl (pH 8.0), 100 mM NaCl (TN buffer) supplemented with urea (8 M) and protease inhibitors (Complete EDTA free; Roche), and the cells were disrupted with a French press. The insoluble material was removed by centrifugation for 45 min at 90,000 × g. Histidine-tagged proteins were immobilized on an ion metal affinity chromatography resin (Cobalt Talon resin; Clontech) preequilibrated in TN buffer supplemented with urea. Proteins were eluted in an imidazole gradient, and the fractions containing concentrated and pure proteins were pooled and dialyzed stepwise against TN buffer containing 6 M, 4 M, 2 M, and no urea. At this step, a large portion of recombinant DVU2106c precipitated and was removed by centrifugation for 15 min at 20,000 × g. The concentration of the protein preparation was determined by the absorbance at 280 nm using the theoretical molar extinction coefficient calculated on the ExPASy website (http://www.expasy.ch/tools/protparam.html).
Purification of the E. coli σ54 factor.
Cloning of the E. coli rpoN gene into the pET19b vector and purification of σ54 were carried out as previously described (4). The Eσ54 holoenzyme was reconstituted by mixing purified σ54 with RNA polymerase (RNAP) core enzyme (Epicentre Biotechnologies) at a 2:1 ratio.
Electrophoretic mobility shift assay (EMSA).
Gel shift experiments were carried out with Eσ70 and Eσ54 and with the DVU2106 enhancer-binding protein fragment (DVU2106c) essentially as previously described (4). Briefly, for Eσ54, 32P-labeled probes were mixed with Eσ54 (premixed as an RNAP core enzyme at a σ54 ratio of 1:2) in STA buffer (25 mM Tris-acetate [pH 8.0], 8 mM magnesium acetate, 10 mM KCl, 1 mM dithiothreitol [DTT], and 3.5% polyethylene glycol [PEG] 6000) in the presence of sonicated salmon sperm DNA (10 μg/ml) and bovine serum albumin (BSA) (50 μg/ml). Controls included incubation with RNAP core enzyme or with double-stranded DNA competitors (consensus σ54- and Fur-binding boxes obtained by annealing of two complementary oligonucleotides). Binding of Eσ70 was tested similarly using commercial RNAP holoenzyme saturated in σ70 (Epicentre Biotechnologies). For DVU2016c, the protein was mixed with 32P-labeled promoter probes in a binding reaction mixture containing sonicated salmon sperm DNA (10 μg/ml), BSA (100 μg/ml), 10 mM MgCl2, 25 mM KCl, 5% glycerol, 1 mM DTT, 10 mM acetyl-phosphate in 25 mM Tris-HCl (pH 8). After binding for 20 min, samples were resolved on a prerun 9% acrylamide gel in Tris-borate buffer. The gels were fixed in 10% trichloroacetic acid for 10 min and exposed to Kodak BioMax MR films. RNAP core and (σ70-saturated) holoenzymes were purchased from Epicentre Biotechnology.
Promoter cloning in E. coli and β-galactosidase assay.
Transcriptional fusion reporter plasmids (listed in Table S1 in the supplemental material) were constructed by a double-PCR technique, allowing amplification of the gene or promoter of interest flanked by extensions annealing to the target vector (2, 55). The product of the first PCR was used as oligonucleotides for a second PCR with the target vector as a template (custom oligonucleotides are listed in Table S2 in the supplemental material). For lacZ transcriptional fusions, the promoter sequences were PCR amplified from D. vulgaris Hildenborough genomic DNA and cloned downstream of the T7 promoter into the pT7.5 vector (52). The lacZ gene, encoding beta-galactosidase, was cloned downstream of each promoter. For production plasmids, the DVU2106 gene fused to an N-terminal FLAG epitope (DYKDDDDK) was PCR amplified from genomic DNA and cloned into the pOK12 vector (56) downstream of the lac promoter. All constructs were verified by restriction analyses and DNA sequencing.
The activity of the β-galactosidase reporter was measured from mid-exponential growth phase cells (OD600, 0.8) as previously described (36). Each enzymatic assay was performed in triplicate, starting from each biological triplicate (from independent plasmid transformations).
Promoter cloning in D. vulgaris Hildenborough and β-glucuronidase assay.
For the construction of uidA translational fusions, DNA fragments corresponding to the promoters of interest (DVU2105 and DVU2107) were amplified by PCR and subcloned into the XhoI and SpeI sites of the plasmid pNot19Cm-Mob-XS (see Table S1 in the supplemental material). Before amplifying the DVU2105 promoter, a mutagenesis step by overlapping PCR was performed to remove an NdeI site in the promoter sequence (custom oligonucleotides are listed in Table S2 in the supplemental material). The uidA gene, encoding β-glucuronidase, was amplified by PCR from E. coli K-12 and cloned downstream of each promoter to obtain pNot19p2105::uidA and pNot19p2107::uidA. These plasmids were transferred into E. coli MW3064 and subsequently transferred by conjugational gene transfer into D. vulgaris Hildenborough. Cells that had recombined the translational fusions into the chromosome instead of the wild-type (WT) gene were selected by their resistance to thiamphenicol, and the construction on the chromosome was verified by PCR.
β-Glucuronidase assays were performed in a buffer consisting of 50 mM sodium phosphate (pH 7), 10 mM 2-mercaptoethanol, 0.1% Triton X-100, and 1 mM p-nitrophenyl β-d-glucuronide. Reactions occurred at 37°C, and p-nitrophenol absorbance was measured at 415 nm (29).
RNA preparation.
Total RNA was isolated using the following procedure. Three independent cultures of D. vulgaris Hildenborough (80 ml) were grown to exponential growth phase (OD595, 0.4). The cells were harvested and resuspended in 200 μl of 10 mM Tris, pH 8. RNA was prepared using the High Pure RNA kit from Roche Diagnostics according to the manufacturer's instructions, with an extra DNase I digestion step in order to eliminate contaminating DNA.
Real-time quantitative PCR (qRT-PCR).
Ten micrograms of total extracted RNA was reverse transcribed by using random hexamers. Real-time PCR was performed on a LightCyclerFastStart DNA MasterPlus SYBR green I Kit (Roche Diagnostics). cDNA was mixed with 0.25 μM each primer and 2 μl of master mix in a 10-μl final volume. The primer pairs used to quantify the selected gene expression levels are shown in Table S2 in the supplemental material. PCR was carried out with one cycle at 95°C for 8 min, followed by up to 45 cycles at 95°C for 12 s, 60°C for 6 s, and 72°C for 20 s. The specificities of the accumulated products were verified by melting-curve analysis. The fluorescence derived from the incorporation of SYBR green I into the double-stranded PCR products was measured at the end of each cycle to determine the amplification kinetics of each product. The fit point method described by the manufacturer was then applied to the results. Briefly, a horizontal noise band was determined, as well as a log line fitting the exponential portion of the amplification curve. The intersections of these log lines with the horizontal noise lines identified the crossing points. The crossing points were determined for each gene under three conditions. The Relative Expression Software Tool (REST) was used to calculate the relative expression of each gene under each condition (43) using the 16S RNA gene as a reference for normalization.
Determination of transcriptional start sites.
Total RNAs extracted from thhe strain D. vulgaris Hildenborough were hybridized with a primer complementary to positions +32 to +30 relative to the putative ATG of orp1. Primer-rev was 32P labeled by using [γ32P]ATP and T4 polynucleotide kinase (Biolabs). A total of 5 μg of RNAs and 4 ng of labeled primer were incubated together with 200 units of Superscript III reverse transcriptase (Invitrogen) for 50 min at 55°C, followed by 10 min at 70°C to inactivate the enzyme. The sequencing ladder was PCR amplified with the same labeled primer and a 5′ primer hybridizing to positions 143 to 208 relative to the ATG of orp2. The sequencing reaction was performed using the Thermo Sequenase Cycle Sequencing Kit (USB Corporation). Extension and sequencing products were separated onto a 6 M urea-8% acrylamide (19:1) gel.
Microscopic imaging.
For phase-contrast imaging, 5 μl of a stationary-phase culture cell was placed on a microscope slide and covered with a coverslip. Images were taken on an Olympus confocal IX81. For cell length measurement, stationary-phase cells were examined on freshly prepared poly-l-lysine-treated slides. The images were viewed using a Zeiss Phomi3, and cell length was measured using Olympus Image-Pro Plus. The average cell length was determined from two independent cultures (>1,000 cells were counted in each experiment).
Miscellaneous.
Protein concentrations from cell extracts were determined with the Bradford (Bio-Rad) protein assay reagent.
RESULTS
The orp gene cluster is organized in three distinct transcriptional units.
The transcription organization of the orp gene cluster was investigated by reverse transcription from total RNAs prepared from D. vulgaris Hildenborough cells. PCR analyses of the corresponding cDNAs were carried out to test the amplification of intergenic regions (primer pairs are listed in Table S2 in the supplemental material). Amplicons were obtained for intergenic regions between DVU2103 and DVU2104 (Fig. 1A, a) and between DVU2104 and DVU2105 (Fig. 1A, b), while no amplification product was obtained between DVU2105 and DVU2106 (Fig. 1A, c). These data indicate that the DVU2103-DVU2104-DVU2105 genes are organized as an operon (named here orp2) (Fig. 1B). Intergenic regions were amplified between DVU2107 and DVU2108 (Fig. 1A, e) and between DVU2108 and DVU2109 (Fig. 1A, f), while no amplicon was observed between DVU2106 and DVU2107 (Fig. 1A, d), indicating that DVU2107-DVU2108-DVU2109 belong to the same transcriptional unit (named here orp1) (Fig. 1). Thus, DVU2106 appeared as a monocistronic unit, transcribed independently of the two polycistronic transcripts, as no amplification was observed between DVU2105 and DVU2106 (Fig. 1A, c) or between DVU2106 and DVU2107 (Fig. 1A, d). As controls, no amplification product was obtained, whatever the primer pairs, when RNAs were used as templates in the PCR, indicating that the RNA preparations were not contaminated by genomic DNA. Altogether, these data show that the orp gene cluster is organized in three different transcriptional units, the orp1 and orp2 divergent operons separated by the monocistronic transcriptional unit, DVU2106 (Fig. 1B).
Fig. 1.

orp gene cluster organization in D. vulgaris Hildenborough. (A) PCR products of intergenic regions between DVU2103-DVU2104 (a), DVU2104-DVU2105 (b), DVU2105-DVU2106 (c), DVU2106-DVU2107 (d), DVU2107-DVU2108 (e), DVU2108-DVU2109 (f), and DVU2109-DVU2110 (g) from RNA (lanes 1), cDNA (lanes 2), and genomic DNA (lanes 3). Molecular size markers are indicated on the left and right (in bp). (B) Structural organization of the orp gene cluster. The arrows indicate the direction of transcription. (C) Relative quantification of orp gene expression by real-time PCR of DVU2104, DVU2106, and DVU2108 transcripts. The averages of three independent biological samples are shown, with standard deviations calculated from replicates. The amount of DVU2108 transcripts was taken as a reference (arbitrary units).
The transcriptional status of the two divergent operons and of DVU2106 on lactate/sulfate medium was determined by quantitative real-time PCR using primer pairs designed from the coding sequences of DVU2104, DVU2108, and DVU2106, respectively. Figure 1C shows that the expression levels of the two operons were similar, while expression of DVU2106 was about 300 times lower. These data show that the divergent orp1 and orp2 operons are transcribed at similar levels when D. vulgaris Hildenborough is cultured under anaerobic lactate/sulfate conditions.
To characterize the cis elements required for transcription of the orp1 and orp2 operons, in silico analyses were carried out using the Softberry (BProm) and PromScan algorithms. These analyses predicted a putative σ54 promoter with conserved −12 and −24 elements (GTGGAACGGAACGTGCTC) upstream of the orp2 operon, at 43 and 54 nucleotides (nt) upstream of the ATG initiation codon of DVU2105, respectively. On the other hand, no promoter was clearly identified upstream of the translational start point of DVU2107 according to the genome annotation (27).
We then used a primer extension assay to map the transcription start sites of both operons. Two extension products were obtained by primer extension from orp2, with only one base difference, which located the transcriptional start point of orp2 (nt 2200221) 10 bp downstream of the −12 element of the predicted σ54-dependent promoter (Fig. 2A and B). Surprisingly, one extension product was obtained for DVU2107, locating the transcriptional start point of orp1 in the annotated coding region of DVU2107 (nt 2202069) (Fig. 2A and B). In silico analysis of the experimentally determined DVU2107 transcriptional start point revealed the presence of a putative σ54 promoter (CCGGCATCATTCCTGCTT), with the −12 element located 13 bp upstream of the DVU2107 transcriptional start site (Fig. 2B). Accordingly, a new translational initiation codon for DVU2107 could be identified 22 bp downstream of the transcriptional start site, together with a putative Shine-Dalgarno sequence located 9 bp upstream of the new start codon (Fig. 2B). These data strongly suggest that DVU2107 has been misannotated in the genome and that it encodes a 136-residue protein (instead of 183 residues as previously reported [27]).
Fig. 2.

Identification of signal transcription of orp1, orp2, and DVU2106. (A) Primer extension analysis using RNA extracted from D. vulgaris Hildenborough. The arrowheads indicate the primer extension products generated using primers DVU2105_rev(+1) (annealing within the DVU2105 gene) (left) and DVU2107_rev (annealing within the DVU2107 gene) (right). Lanes C, T, A, and G correspond to the sequence reads generated using the same primers. Sequences around the transcriptional +1 position are indicated, and the sequence patterns are shown on the right. (B) Sequences of the DVU2105-DVU2106 and DVU2106-DVU2107 intergenic regions with the positions of the transcriptional start sites. The nucleotide numbers correspond to the locations in the genome sequence (27). The predicted σ54 promoters of orp2 and orp1 are boxed. The predicted Shine-Dalgarno (SD) consensuses are shown in boldface, and the initiation codons of DVU2105 and DVU2107 are underlined.
The E. coli σ54-RNA polymerase specifically binds to the promoter regions of orp1 and orp2.
Consensus sequences for σ54 factor binding were detected 35 bp and 43 bp upstream of the translational start sites of the orp1 and orp2 operons, respectively, suggesting that transcription of both operons was dependent on σ54-RNA polymerase. We then tested the binding of the E. coli σ54-bound RNA polymerase (Eσ54) with the orp1 and orp2 promoters by EMSA. Eσ54 bound to the orp1 and orp2 promoter fragments (see Fig. 4A and B). Further competition experiments showed that a consensus Fur box duplex did not interfere with Eσ54 binding to the orp1 and orp2 promoters, whereas a consensus σ54 box duplex did compete. To gain further insights into the orp2 promoter organization, two fragments of 89 bp and 103 bp were generated, located at the 5′ and 3′ ends, respectively (Fig. 3). As predicted, due to the location of the putative σ54 sequence, Eσ54 bound to the 3′ fragment (Fig. 4D) but did not shift the 5′ fragment (Fig. 4C). Hence, the promoter regions of the orp1 and orp2 operons specifically recruit the σ54 factor in vitro.
Fig. 4.
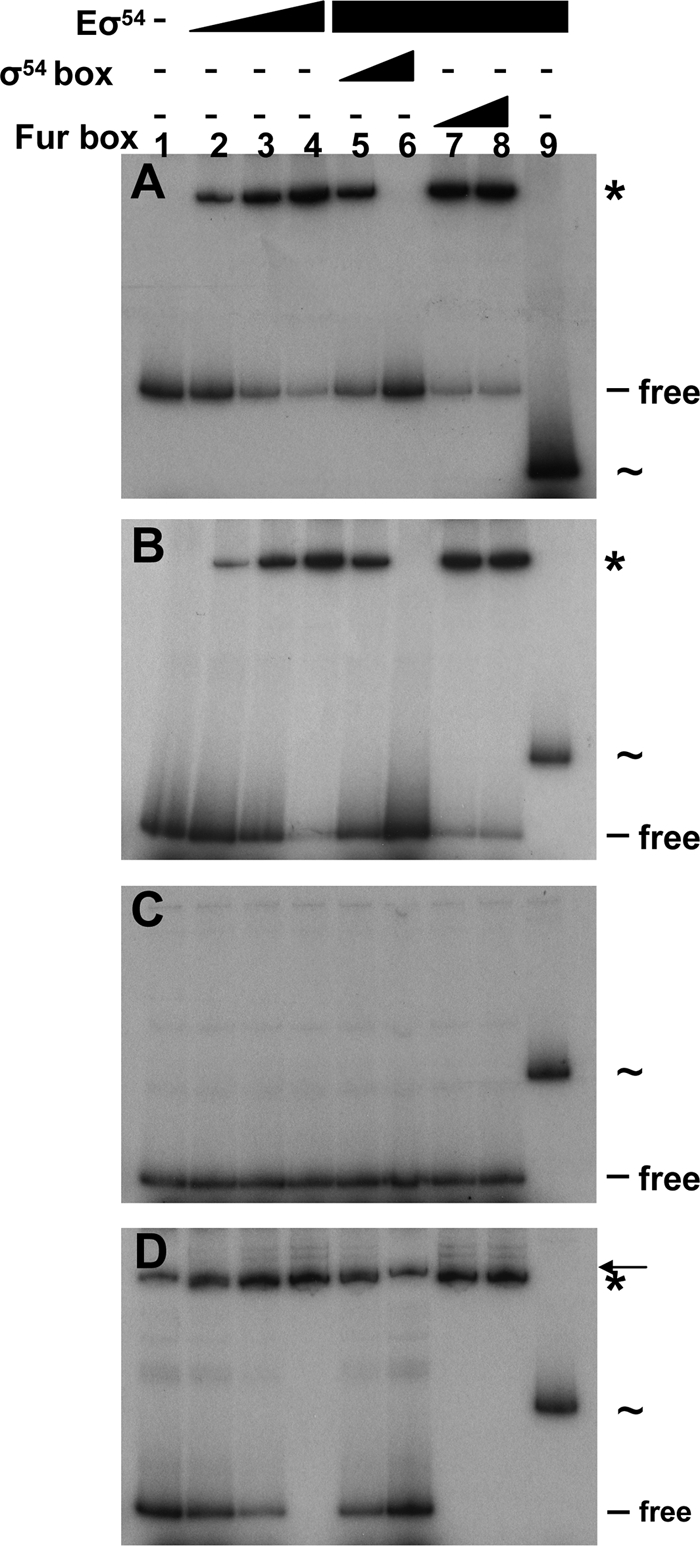
The σ54-RNA polymerase interacts with the promoter regions of orp1 and orp2. Shown is EMSA of the promoter regions of orp1 (A), orp2 (B), and the 5′ (C) and 3′ (D) fragments of the orp2 promoter with a reconstituted σ54-RNA polymerase complex (Eσ54) (lane 1, no protein; lane 2, 25 nM; lane 3, 100 nM; lane 4, 250 nM). Competition experiments using a double-stranded consensus σ54-binding box (lane 5, molecular ratio, probe-competitor, 1:5; lane 6, molecular ratio, 1:25) or consensus Fur-binding box (lane 7, molecular ratio, probe-competitor, 1:5; lane 8, molecular ratio, 1:25). Retarded probe-Eσ54 complexes are indicated by asterisks. As a control, a 260-bp fragment of the σ70-dependent E. coli sci-1 promoter (indicated by ∼) was incubated in the presence of Eσ54 (250 nM; lane 9). The arrow in panel D indicates an unrelated amplicon obtained by PCR when the 5′ fragment of the orp2 promoter was amplified.
Fig. 3.

Sequence analyses of the orp1 and orp2 promoters. The −12/−24 elements of the orp1 and orp2 σ54 promoters are indicated by solid-line boxes, and the putative −10/−35 elements of the DVU2106 σ70 promoter are indicated by dashed boxes. The drawing emphasizes the positions of the two putative palindromic DVU2106-binding sequences. The positions and lengths of the probes used for EMSA are indicated by arrows.
The σ54-dependent transcriptional regulator DVU2106 binds specifically to imperfect palindrome sites within the promoter regions of both orp1 and orp2.
The protein encoded by DVU2106 consists of 458 amino acid residues (50.6 kDa) related to the σ54-dependent transcriptional factor family that contains PAS, AAA-type ATPase, and DNA-binding domains. Because σ54-dependent transcription requires the activity of a cognate σ54-dependent transcriptional factor, we tested whether the product of the DVU2106 gene was able to form a complex with the orp1 and orp2 promoter regions. Pilot experiments showed that the full-length DVU2106 protein fused to an N-terminal histidine tag was unstable and was rapidly degraded into a stable C-terminal degradation product of 36 kDa. We next generated a truncated form of DVU2106 with the N-terminal PAS domain deleted (called here DVU2016c). Despite the stability of DVU2016c, it aggregated as inclusion bodies. DVU2106c was purified to homogeneity from urea-solubilized inclusions and used for EMSA after urea removal. Figure 5 shows that DVU2106c bound to the 360-bp fragment of the orp1 promoter region (Fig. 5A), as well as to the 174-bp fragment corresponding to the orp2 promoter (Fig. 5B). The use of the two shorter orp2 fragments showed that DVU2106c clearly bound to the 5′ extremity of the orp2 promoter (Fig. 5C), whereas it was unable to shift the 3′ fragment (Fig. 5D).
Fig. 5.
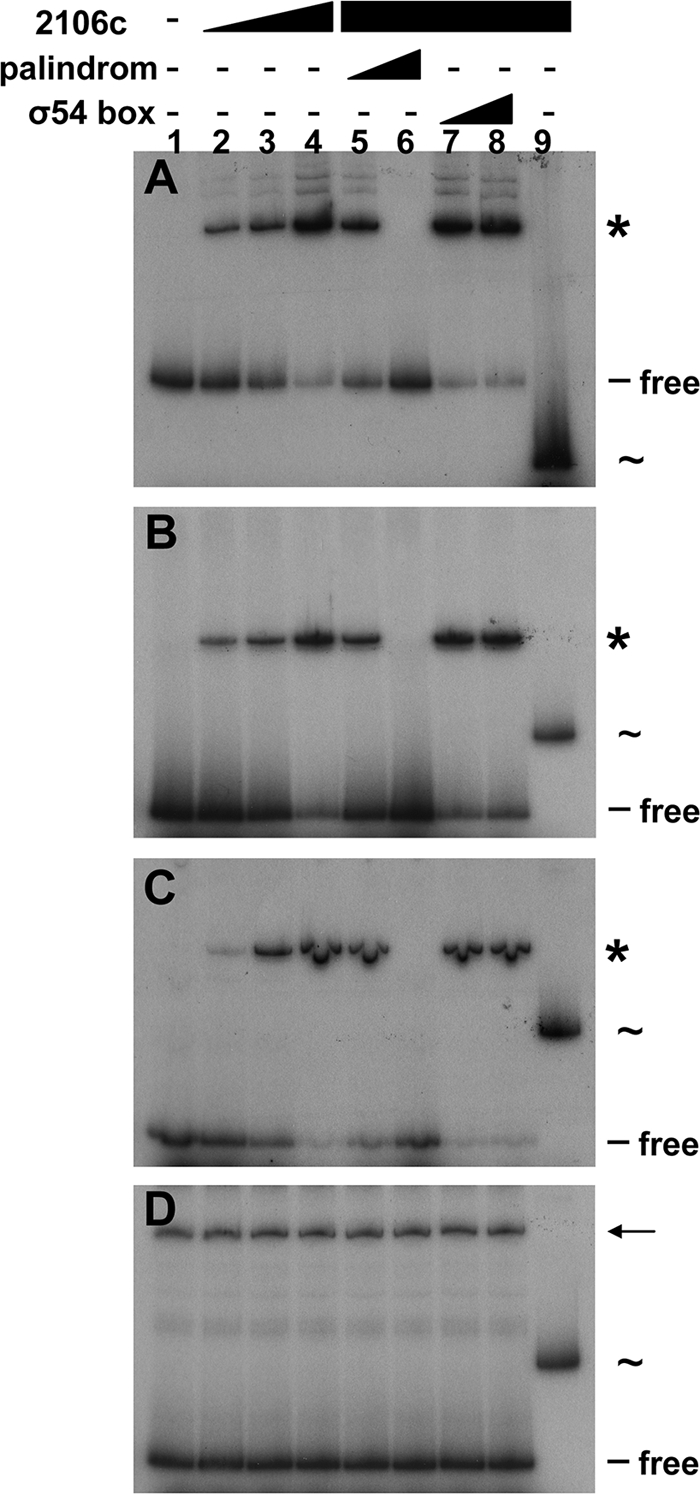
The σ54-dependent transcriptional regulator DVU2106 interacts with the intergenic region of orp1 and orp2. Shown is EMSA of the promoter regions of orp1 (A), orp2 (B), and the 5′ (C) and 3′ (D) fragments of the orp2 promoter using purified DVU2106c (lane 1, no protein; lane 2, 100 nM; lane 3, 200 nM; lane 4, 500 nM). Competition experiments using a double-stranded putative palindromic DVU2106-binding sequence (lane 5, molecular ratio, probe-competitor, 1:5; lane 6, molecular ratio, 1:25) or a consensus σ54-binding box (lane 7, molecular ratio, probe-competitor, 1:5; lane 8, molecular ratio, 1:25). Retarded probe-DVU2106c complexes are indicated by asterisks. As a control, a 260-bp fragment of the σ70-dependent E. coli sci-1 promoter (indicated by ∼) was incubated in the presence of DVU2106c (500 nM; lane 9). The arrow in panel D indicates an unrelated amplicon obtained by PCR when the 5′ fragment of the orp2 promoter was amplified.
Sequence alignment of the intergenic regions upstream of orp1 and orp2 revealed the presence of two imperfect palindrome sequences (palindrome sequences separated by a nonpalindrome spacer) of 17 bp (nt 2201931 to 2201948 and nt 200325 to 2200308) exhibiting 88% identity (GGGCGYRTTTTGCGCCC). These palindromes were located upstream of the σ54-binding sequence at positions −131 and −88, respectively, from the transcriptional start sites of orp1 and orp2 (Fig. 3). Because it has been proposed that imperfect palindrome sequences are involved in the fixation of transcriptional regulators (33), the ability of DVU2106c to bind these imperfect palindrome sequences was assayed in EMSA competition experiments. Cold duplex DNA corresponding to these sequences, or to the σ54-binding box as a control, was added to the EMSA mixture in a 1:5 or 1:25 ratio (promoter/competitor) (Fig. 5). Although the σ54-binding box binding sequence duplexes did not interfere with DVU2106c-DNA complex formation with the orp1 and orp2 promoter regions, molecular excesses of 17-bp GGGCGYRTTTTGCGCCC sequence abolished DVU2106c binding to both promoters (Fig. 5). The results shown in Fig. 5 lead the conclusion that the DVU2106 σ54-dependent transcriptional factor binds to specific imperfect palindrome sequences present in both the orp1 and orp2 promoter regions.
DVU2106 recruits the σ54-RNA polymerase to activate the transcription of both orp1 and orp2.
We have shown that both E. coli σ54-RNA polymerase and the transcriptional regulator DVU2106 bind specifically to the intergenic region of the orp1 and orp2 operons. To fully assess the influence of σ54-RNA polymerase and the putative transcriptional regulator DVU2106 on the expression of the two divergent operons and of DVU2106 itself, gene fusions with lacZ were constructed and introduced into the heterologous host E. coli. β-Galactosidase activities were measured in both σ54-proficient (wild-type strain) and σ54-deficient (ΔrpoN strain) backgrounds. For the orp1 and orp2 promoter fusions, we detected a very low activity, and no difference was observed in the presence or absence of σ54 (Fig. 6A and C). We therefore expressed DVU2106 from a compatible multicopy plasmid. Figure 6A and C show that the activity of the fusions increased 4- to 6-fold in the WT background in the presence of DVU2106, while no increase was detected when DVU2106 was produced in a ΔrpoN strain. These data show that the simultaneous presence of σ54-RNA polymerase and DVU2106 is required for the transcription of both orp1 and orp2. In contrast, experiments performed with a DVU2106 promoter fusion showed that the expression of DVU2106 was independent of σ54 (Fig. 6B). However, overproduction of DVU2106 led to a reproducible, significant 2-fold decrease of DVU2106-lacZ fusion activity (Fig. 6B). This suggests that DVU2106 negatively retrocontrols its own expression.
Fig. 6.

Activities of the reporter fusions in the heterologous host E. coli. The lacZ reporter fusions with the promoter regions of orp1 (A), DVU2106 (B), and orp2 (C) are represented on the left. The transcript start sites are indicated by bent arrows. The positions of the −10 and −35 consensus sequences of the σ70 promoter and of the −12 and −24 consensus sequences of the σ54 promoters are indicated by black rectangles. The β-galactosidase activities of these reporter fusions in various backgrounds are shown on the right: the E. coli wild-type strain and its isogenic rpoN derivative in the absence (dark-gray and white bars, respectively) or presence (light-gray and black bars, respectively) of DVU2106 production. The activity is the average of three independent measurements (the error bars show the standard deviations).
The heterologous experiments described above, using E. coli as the host, showed that DVU2106 regulates the expression of orp1 and orp2, as well as its own expression. To investigate its role in D. vulgaris Hildenborough, we constructed an isogenic strain in which the DVU2106 gene was replaced on the chromosome by a truncated version that produced a protein composed of only the PAS domain, i.e., devoid of both the ATPase central and C-terminal DNA-binding domains (DvH p2106::2106PAS). We then compared the expression levels of the orp1 and orp2 operons in the WT and DvH(p2106::2106PAS) strains by qRT-PCR. The expression ratio of orp1 [log2(absolute gene regulation) = −5.02 ± 0.3] and orp2 [log2(absolute gene regulation) = −5.2 ± 0.09] showed that the transcript levels were approximately 40 times lower in the DvH(p2106::2106PAS) strain than in the WT strain. The expression ratio of DVU2106 [log2(absolute gene regulation) = 3.14 ± 0.8] revealed that its expression was 12 times higher in the mutant strain. These results are consistent with the experiments performed in the E. coli reporter strains and demonstrate that, in D. vulgaris Hildenborough, DVU2106 activates the transcription of the two divergent operons orp1 and orp2 and negatively retrocontrols its own expression.
DVU2106 displaces Eσ70 at the DVU2106 promoter.
Our data showed that the expression of DVU2106 was σ54-RNA polymerase independent. In silico analysis of the DVU2106 promoter indicated the presence of putative σ70-binding elements. Putative Eσ70 −10 and −35 promoter elements are present 37 bp and 53 bp, respectively, upstream of the initiation codon of DVU2106 (Fig. 7A). The 5′ 89-bp fragment of the orp2 promoter encompassing the promoter region of DVU2106 was therefore used for EMSA with Eσ70. Figure 7B shows that the Eσ70 holoenzyme is able to bind to the DVU2106 promoter region, while no shift was detected when Eσ70 was mixed with the 3′ orp2 promoter fragment (data not shown). Interestingly, the putative σ70 −10 element overlaps with the imperfect palindrome DVU2106-binding sequence (Fig. 7A). The β-galactosidase data and the in silico predictions therefore suggest that DVU2106 might displace the σ70-RNA polymerase holoenzyme from the DVU2106 promoter, thus acting as a negative regulator of its own expression. To experimentally test this hypothesis, we tested whether Eσ70 and DVU2106c could simultaneously interact with the DVU2106 promoter using EMSA. Figure 7B shows that when DVU2106c was added to the DNA-Eσ70 mixture, the Eσ70-promoter complex was displaced in favor of a DVU2106c-promoter complex. As a control, competition experiments were carried out with NtrC, an E. coli EBP. As shown in Fig. 7B, the presence of a large amount of NtrC did not shift the DVU2106 promoter and did not affect Eσ70 binding to the DVU2106 promoter. These data show that DVU2106c binding specifically displaces Eσ70 from the DVU2106 promoter, likely by direct competition at overlapping binding sequences (Fig. 7A).
Fig. 7.

DVU2106 represses its own transcription. (A) The promoter of DVU2106. The putative −10 and −35 elements of the σ70 DVU2106 promoter are underlined and in boldface. The proposed Shine-Dalgarno consensus is shown in boldface, and the initiation codon is underlined. The DVU2106-binding site is indicated by a box showing its overlap with the putative −10 element. (B) EMSA of the 5′ fragment of the orp2 promoter using the σ70-RNA polymerase complex (Eσ70) (lane 1, no protein; lane 2, 25 nM; lane 3, 100 nM; lane 4, 250 nM), the core enzyme (E) (lane 8, RNAP devoid of the sigma factor, 250 nM), or the purified 2106 protein (lane 9, 200 nM; lane 10, 500 nM). Competition experiments between Eσ70 and 2106c (lane 5, 250 nM Eσ70 plus 200 nM 2106c; lane 6, 250 nM Eσ70 plus 500 nM 2106c; lane 7, 250 nM Eσ70 plus 500 nM unrelated E. coli NtrC EBP).
The DVU2103, DVU2104, DVU2105, DVU2108, and DVU2109 proteins form a physiological complex in D. vulgaris Hildenborough cells.
The coregulation of the orp1 and orp2 operons suggests that the corresponding gene products collaborate in the same pathway. We therefore defined the interaction network among these proteins by using an endogenous in vivo pulldown method in D. vulgaris Hildenborough. We constructed pNot19Cm-Mob-XS, a vector containing a chloramphenicol resistance cassette as a selection marker and a mobilization cassette allowing conjugational transfer from E. coli to D. vulgaris Hildenborough. This plasmid was designed so that it could be used as a template for the insertion of any tagged gene in its original genetic locus on the chromosome of D. vulgaris Hildenborough. By using this strategy, DVU2103, DVU2108, and DVU2109 were strep tagged and used as bait in endogenous pulldown experiments. The tagged proteins and their physiological partners were immobilized by affinity purification on a Strep-Tactin resin, and proteins were identified by MALDI-TOF or electrospray ionization ion trap (ESI-IT) mass spectrometry on SDS-PAGE (Fig. 8A). Nonspecific interaction proteins were determined by loading wild-type D. vulgaris Hildenborough extract on the Strep-Tactin column (see Table S4 in the supplemental material). The nonspecific interacting proteins included several classical abundant proteins, like ribosomal proteins, elongation factors, or chaperones, as well as some highly abundant proteins in D. vulgaris Hildenborough, such as adenylsulfate reductase and pyruvate carboxylase.
Fig. 8.

Endogenous pulldown experiments using C-terminally strep-tagged 2103, 2108, and 2109 as bait. (A) SDS-PAGE of the affinity purification fractions. The strep-tagged (St) protein used as bait is indicated at the top of each lane, and the orp gene clusters identified by mass spectrometry are indicated by arrows. The degradation products of DVU2109 are indicated by plus signs. The molecular mass markers are indicated in kDa. (B) Summary of the ORP interaction network deduced from panel A (DVU2108 partners, solid line; DVU2109 partners, dashed line, DVU2103 partners, dotted line).
Strep-tagged DVU2103 and strep-tagged DVU2108 were both identified on the polyacrylamide gel with the expected molecular weight for the corresponding protein fusion. In the case of strep-tagged DVU2109, in addition to the band corresponding to the expected molecular weight, several degradation products of the tagged proteins were detected and identified on the polyacrylamide gel (Fig. 8A). The complexities of the interaction networks were variable, depending on the bait. Relatively few interacting proteins were detected for DVU2108 and DVU2109 compared to DVU2103 (Fig. 8A). DVU2104, DVU2105, DVU2108, and DVU2109 were identified as prey when strep-tagged DVU2103 was used as bait (Fig. 8A; see Table S3 in the supplemental material). Reciprocally, DVU2103, DVU2104, and DVU2105 coeluted with strep-tagged DVU2108 and DVU2109 (Fig. 8A; see Table S3 in the supplemental material). Those data provide the first evidence that DVU2103, DVU2104, DVU2105, DVU2108, and DVU2109 physically interact in vivo and form a physiological multiprotein complex (Fig. 8B).
orp1 and orp2 transcriptional analysis of syntrophic coculture between D. vulgaris Hildenborough and a methanogen.
It has been shown by differential transcriptome analyses that DVU2103, DVU2104, and DVU2108 of D. vulgaris Hildenborough were 2- to 4-fold upregulated following the lifestyle shift from syntrophy to sulfate reduction (50). Mutualist interaction between D. vulgaris Hildenborough and archaeon Methanococcus species, when acetate is the sole carbon source, has been reported and showed that the two organisms collaborate to survive (28). In this case, D. vulgaris Hildenborough ferments lactate to produce acetate, carbon dioxide, and hydrogen and relies on hydrogen and acetate scavenging by Methanococcus to convert these compounds to methane (6). To test whether these genes were involved in the lifestyle shift change or in the different metabolisms driven by the culture conditions, D. vulgaris Hildenborough was grown either in cococulture with M. barkeri (syntrophy conditions) or in monoculture in lactate/sulfate medium (sulfate reduction conditions). The transcription levels of orp1 and orp2 were monitored with chromosomal β-glucuronidase gene fusions under both conditions. We constructed two strains [DvH(p2105::uidA) and DvH(p2107::uidA)] in which translational fusions at the start codon of DVU2105 and DVU2107 were inserted at their respective loci in the chromosome. Expressions of orp2 and orp1 were 2-fold higher in monocultures (461 ± 185 units for orp1 and 1,017 ± 379 units for orp2) than in cocultures (277 ± 14 units for orp1 and 514 ± 20 units for orp2). As a control, no significant β-glucuronidase activity could be detected in the WT strain.
These results show that the two operons were downregulated during syntrophic growth compared to sulfate respiration.
Inactivation of DVU2106 affects cell morphology.
Our data showed that in the DvH(p2106::2106PAS) strain, the production of a truncated form of the σ54-dependent transcriptional regulator, DVU2106, induced a drastic downregulation of both the orp1 and orp2 operons. Interestingly, this strain grew more slowly in lactate/sulfate medium than the WT D. vulgaris Hildenborough strain (see Fig. S2 in the supplemental material). This growth defect is coupled to a morphological defect: phase-contrast microscopy analysis indicated that the DvH(p2106::2106PAS) cells exhibited heterogeneous cell morphology with emergence of irregular spiral cells (Fig. 9B) compared to the classical slightly curved rods of the wild-type strain (Fig. 9A). This morphology defect was quantified. Cell distribution plots showed that 80% of the WT cells exhibited a length between 1 and 1.6 μm (Fig. 9C). The distribution was broadened with mutant cells, which exhibited only 54% with a 1- to 1.6-μm cell length, with 7% elongated cells (>2.2 μm) and 28% shorter cells (<1 μm) (2% and 9%, respectively, in the WT) (Fig. 9C).
Fig. 9.

Effect of DVU2106 inactivation on cell morphology. (A and B) Phase-contrast micrographs of cells of wild-type D. vulgaris Hildenborough (A) and DvH(p2106::2106PAS) (B). (C) Histograms plotting cell length frequency distributions of wild-type D. vulgaris Hildenborough and DvH(p2106::2106PAS).
DISCUSSION
σ54-RNA polymerase and a cognate EBP control the expression of two divergent operons, orp1 and orp2.
In this work, we have reported the transcriptional regulation analysis of two gene clusters from the anaerobe D. vulgaris Hildenborough that encode proteins of unknown function and are conserved in 28 genomes of anaerobic microorganisms. Our results showed that DVU2107, DVU2108, and DVU2109 form the first operon (orp1), while DVU2103, DVU2104, and DVU2105 belong to a second, divergent transcriptional unit (orp2). The determination of the transcriptional start sites allowed us to identify two σ54-dependent −24/−12 elements upstream of orp1 and orp2, respectively. Both sequences are similar to the published conservative regions of the bacterial σ54-dependent promoter (5, 7, 35). In addition, we experimentally demonstrated that the expression of orp1 and orp2 was dependent on the σ54-RNA polymerase. It should be noted that the σ54 promoter located in the intergenic region of orp2 was previously identified by computational prediction and was suggested to regulate DVU2106 (10). However, we showed here that this functional σ54 promoter controls the expression of orp2 (DVU2105-DVU2103) and not that of DVU2106.
Transcription at σ54 promoters requires, in addition to the RNA polymerase associated with σ54, a specialized transcription factor, called EBP (7). The EBPs usually bind to regulatory DNA sequences upstream of the promoter, and DNA bending allows the EBP to interact with σ54-RNA polymerase. EBP-catalyzed ATP hydrolysis is required to open the σ54-RNA polymerase promoter complex and to initiate transcription (44, 47, 51, 59). In the D. vulgaris Hildenborough genome, 37 genes are annotated as encoding EBPs, and 70 σ54 promoters have been identified previously by computational predictions (10, 34). In contrast, only a single σ54-RNA polymerase factor-encoding gene has been annotated. To our knowledge, Myxococcus xanthus is the sole organism having a higher number of EBPs (30, 52) in its genome. Like D. vulgaris Hildenborough, M. xanthus is a deltaproteobacterium. Extensive studies have been performed on this microorganism to understand the function of these numerous EBPs. Examination of the gene functions under the control of σ54 does not suggest a unifying area of cellular functions in which σ54 operates. A number of these EBPs are involved in development, heat shock response, and motility, suggesting that the σ54 factor is at the nexus of a large regulatory network (31). Recently, depletion of rpoN in M. xanthus led to aberrant dividing cells, suggesting that some of the σ54-regulated genes may be important for cell division (22).
The high abundance of putative σ54-dependent genes and σ54-associated EBPs supports the idea that the σ54-RNA polymerase governs the expression of numerous genes in D. vulgaris Hildenborough. No study has been performed, to our knowledge, on σ54 regulation in Desulfovibrio species, and this work thus represents the first study of genes transcribed by σ54-RNA polymerase in the organism. In the orp gene cluster, the two divergent operons orp1 and orp2 are separated by a monocistronic gene, DVU2106, encoding an EBP homologue. We showed that this EBP binds to the promoter regions of orp1 and orp2 and positively controls the expression of the two operons and negatively modulates its own expression. Negative retrocontrol by DVU2106 of its own expression could be explained by a colocation of the σ70-RNA polymerase promoter (−10 element) and the DVU2106-binding site. We also demonstrated competition between DVU2106 and the σ70-RNA polymerase at the DVU2106 promoter. One may hypothesize that direct competition and RNAP displacement occur due to overlapping binding sequences; however, due to the poor consensus −10 and −35 elements, it remains possible that the short half-life of the RNAP-promoter complex leaves the site transiently unoccupied, allowing DVU2106 binding. It has already been reported for the regulation of other system, like the l-arabinose operon (48) or the torCAD operon (1) in E. coli, that transcriptional regulators can act either as activator or repressor. Looking for potential DVU2106-binding sites by BLAST searching both intergenic regions indicated the existence of a conserved 17-bp imperfect palindrome motif (GGGCGYRTTTTGCGCCC). Genome scanning failed to identify any other potential DVU2106-binding site in D. vulgaris Hildenborough, suggesting that DVU2106 specifically regulates the orp1 and orp2 operons. It should be noted that DVU2106 colocates with DVU2103, DVU2104, and DVU2108 genes in most Deltaproteobacteria but is missing in the Archaea, Firmicutes, and Thermotogaceae (see Fig. S1 in the supplemental material).
Taken together, these results demonstrate that the regulation of gene expression in Desulfovibrio species can be studied in E. coli by providing the cognate transcription factors.
Most σ54-associated EBPs share a domain structure that includes three domains (39): the C-terminal DNA-binding domain, the central module carrying the ATPase activity and responsible for interaction with the σ54-RNA polymerase, and the N-terminal regulatory domain. Activation of σ54-dependent transcription is controlled by environmental cues through the regulatory module, which is the most variable portion in EBP structures. In silico analyses suggest that the N-terminal regulatory domain of DVU2106 is part of the PAS domain family (37). PAS domains occur in all kingdoms of life (21) and regulate processes as diverse as nitrogen fixation in rhizobia (15), phototrophism in plants (11), circadian behavior in insects (41), and gating of ion channels in vertebrates (38).
After having delineated the regulatory mechanism underlying orp gene expression, it is important to identify the signal sensed by DVU2106 and to understand the physiological advantage conferred by this gene cluster. As a start for future prospects, it is noteworthy that PAS domains have been suggested to be involved in oxygen, light, or redox potential sensing (54). Given the specific association of orp genes with the anaerobic lifestyle, it will be interesting to understand the link between the PAS domain and the Desulfovibrio lifestyle.
The physiological function of the orp gene cluster.
The clustering pattern across taxonomically remotely related species, the congruent patterns in their phylogenetic trees, and their synchronized expression suggest that DVU2103, DVU2104, and DVU2108 are functionally related. By using endogenous pulldown experiments, we provided evidence that DVU2103, DVU2104, DVU2105, DVU2108, and DVU2109 physically interact to form a physiological multiprotein complex called here the ORP complex. As mentioned previously, results from transcriptome analyses led to the hypothesis that DVU2103, DVU2104, and DVU2108 may play roles related to the lifestyle change of D. vulgaris from syntrophy to sulfate reduction (50). Comparison of orp1 and orp2 expression in monoculture and coculture with a methanogen allowed us to propose that the orp gene cluster is involved in a specific metabolism during sulfate reduction rather than in the lifestyle change from syntrophy to sulfate reduction.
A surprising result emerges from the morphological phenotype of cells producing a truncated version of DVU2106. The truncated DVU2106 fragment is unable to interact with the intergenic regions of both operons and thus cannot activate orp1 and orp2 expression. Compared to the wild-type strain, the mutant strain exhibits morphological defects that may be attributed to the absence of transcription of genes dependent on DVU2106. As discussed above, the DVU2106-binding site appears to be restricted to the orp1 and orp2 operons, suggesting that the absence of the ORP complex is responsible for the morphological phenotype of the mutant strain. It should be noted that inactivation of DVU2106 is not deleterious for D. vulgaris Hildenborough, but the morphological changes observed suggest a defect in cell division or the cell growth control processes.
Such morphological defects exhibiting heterogeneous cell morphology have been described in the min mutant strains of E. coli (32, 46). The min locus in E. coli is an operon containing three loci, minC, minD, and minE (17). The minC, minD, and minE products work in concert to prevent septation at potential division sites located near the cell pole (32, 46). D. vulgaris Hildenborough genome scanning failed to identify MinC and MinE homologues. In contrast, DVU2103 and DVU2104 are identified as MinD-like proteins belonging to COG1149, which comprises MinD superfamily P-loop ATPases with an atypical additional ferredoxin domain. The function of the COG1149 proteins, distributed only in anaerobic microorganisms, has yet to be discovered.
The phenotype observed in the absence of the ORP complex, the absence of minCDE genes in the D. vulgaris Hildenborough genome, and the homology of DVU2103 and DVU2104 with the MinD P-loop ATPase family led us to propose that these gene clusters could be involved in the proper location of the Z ring at the midcell in Desulfovibrio and more generally in anaerobes. These microorganisms may use a specific mechanism involving the ORP complex for positioning the septum, constituting an original mechanism of cell division under specific environmental conditions.
Concluding remarks.
Taken together, these results led to a model of the regulation of the orp operons, shown in Fig. 10. In this model, the σ54 transcriptional regulator, DVU2106, represses its own expression and collaborates with σ54 to activate and synchronize the transcription of the two divergent orp operons. Both the orp1 and orp2 operons encode proteins that form the physiological ORP complex. Inactivation of DVU2106 induced morphological defects that are probably linked to the absence of the ORP complex. The putative role of the ORP complex in the proper location of the Z ring at the cell midpoint needs further study to be accurately determined.
Fig. 10.

Schematic representation of the regulatory mechanisms of the orp gene cluster in D. vulgaris Hildenborough. The positions of the σ54 and σ70 promoters are indicated by bent arrows, and the DVU2106-binding sequences are indicated by hatched rectangles. The DVU2106 transcriptional regulator plays a positive role in the expression of the σ54-dependent orp1 and orp2 operons and exerts a negative retrocontrol on its own σ70-dependent expression.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of Marielle Bauzan to growing bacteria; Sabrina Lignon for mass spectroscopy analysis; Yann Denis for transcriptomic facilities; Alain Bernadac for useful advice on microscopy; and Marie-Claire Durand, Elise Courvoisier-Dezord, and Marthe Gavioli for technical assistance. We thank Laetitia Pieulle, Amel Latifi, and Vincent Méjean for helpful comments and discussion.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
Published ahead of print on 29 April 2011.
REFERENCES
- 1. Ansaldi M., Simon G., Lepelletier M., Méjean V. 2000. The TorR high-affinity binding site plays a key role in both torR autoregulation and torCAD operon expression in Escherichia coli. J. Bacteriol. 182:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aschtgen M. S., Gavioli M., Dessen A., Lloubès R., Cascales E. 2010. The SciZ protein anchors the enteroaggregative Escherichia coli type VI secretion system to the cell wall. Mol. Microbiol. 75:886–899 [DOI] [PubMed] [Google Scholar]
- 3. Barton L., Fauque G. 2009. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv. Appl. Microbiol. 68:41–98 [DOI] [PubMed] [Google Scholar]
- 4. Bernard C. S., Brunet Y. R., Gavioli M., Lloubès R., Cascales E. 2011. Regulation of type VI secretion gene clusters by sigma54 and cognate enhancer binding proteins. J. Bacteriol. 193:2158–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bose D., et al. 2008. Organization of an activator-bound RNA polymerase holoenzyme. Mol. Cell 32:337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brockhurst M. A., Colegrave N., Rozen D. E. 2011. Next-generation sequencing as a tool to study microbial evolution. Mol. Ecol. 20:972–980 [DOI] [PubMed] [Google Scholar]
- 7. Buck M., Gallegos M. T., Studholme D. J., Guo Y., Gralla J. D. 2000. The bacterial enhancer-dependent sigma(54) (sigma(N)) transcription factor. J. Bacteriol. 182:4129–4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bursakov S. A., et al. 2004. Antagonists Mo and Cu in a heterometallic cluster present on a novel protein (orange protein) isolated from Desulfovibrio gigas. J. Inorg. Biochem. 98:833–840 [DOI] [PubMed] [Google Scholar]
- 9. Calogero S., et al. 1994. RocR, a novel regulatory protein controlling arginine utilization in Bacillus subtilis, belongs to the NtrC/NifA family of transcriptional activators. J. Bacteriol. 176:1234–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chhabra S., et al. 2006. Global analysis of heat shock response in Desulfovibrio vulgaris Hildenborough. J. Bacteriol. 188:1817–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christie J. M., et al. 1998. Arabidopsis NPH1: a flavoprotein with the properties of a photoreceptor for phototropism. Science 282:1698–1701 [DOI] [PubMed] [Google Scholar]
- 12. Columbus L., Peti W., Etezady-Esfarjani T., Herrmann T., Wüthrich K. 2005. NMR structure determination of the conserved hypothetical protein TM1816 from Thermotoga maritima. Proteins 60:552–557 [DOI] [PubMed] [Google Scholar]
- 13. Cort J., Yee A., Edwards A., Arrowsmith C., Kennedy M. 2000. NMR structure determination and structure-based functional characterization of conserved hypothetical protein MTH1175 from Methanobacterium thermoautotrophicum. J. Struct. Funct. Genomics 1:15–25 [DOI] [PubMed] [Google Scholar]
- 14. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. David M., et al. 1988. Cascade regulation of nif gene expression in Rhizobium meliloti. Cell 54:671–683 [DOI] [PubMed] [Google Scholar]
- 16. Debarbouille M., Gardan R., Arnaud M., Rapoport G. 1999. Role of bkdR, a transcriptional activator of the sigL-dependent isoleucine and valine degradation pathway in Bacillus subtilis. J. Bacteriol. 181:2059–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Boer P. A., Crossley R. E., Rothfield L. I. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641–649 [DOI] [PubMed] [Google Scholar]
- 18. Dermoun Z., et al. 2010. TM0486 from the hyperthermophilic anaerobe Thermotoga maritima is a thiamin-binding protein involved in response of the cell to oxidative conditions. J. Mol. Biol. 400:463–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elias D., et al. 2009. Expression profiling of hypothetical genes in Desulfovibrio vulgaris leads to improved functional annotation. Nucleic Acids Res. 37:2926–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Etezady-Esfarjani T., et al. 2004. NMR structure determination of the hypothetical protein TM1290 from Thermotoga maritima using automated NOESY analysis. J. Biomol. NMR 29:403–406 [DOI] [PubMed] [Google Scholar]
- 21. Finn R. D., et al. 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34:D247–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. García-Moreno D., et al. 2009. A vitamin B12-based system for conditional expression reveals dksA to be an essential gene in Myxococcus xanthus. J. Bacteriol. 191:3108–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gardan R., Rapoport G., Débarbouillé M. 1997. Role of the transcriptional activator RocR in the arginine-degradation pathway of Bacillus subtilis. Mol. Microbiol. 24:825–837 [DOI] [PubMed] [Google Scholar]
- 24. George G. N., et al. 2000. A novel protein-bound copper-molybdenum cluster. J. Am. Chem. Soc. 122:8321–8832 [Google Scholar]
- 25. Guiral M., et al. 2009. New insights into the respiratory chains of the chemolithoautotrophic and hyperthermophilic bacterium Aquifex aeolicus. J. Proteome Res. 8:1717–1730 [DOI] [PubMed] [Google Scholar]
- 26. Hansen T. 1994. Metabolism of sulfate-reducing prokaryotes. Antonie Van Leeuwenhoek 66:165–185 [DOI] [PubMed] [Google Scholar]
- 27. Heidelberg J., et al. 2004. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22:554–559 [DOI] [PubMed] [Google Scholar]
- 28. Hillesland K. L., Stahl D. A. 2010. Rapid evolution of stability and productivity at the origin of a microbial mutualism. Proc. Natl. Acad. Sci. U. S. A. 107:2124–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jefferson R. A., Burgess S. M., Hirsh D. 1986. Beta-glucuronidase from Escherichia coli as a gene-fusion marker. Proc. Natl. Acad. Sci. U. S. A. 83:8447–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jelsbak L., Givskov M., Kaiser D. 2005. Enhancer-binding proteins with a forkhead-associated domain and the sigma54 regulon in Myxococcus xanthus fruiting body development. Proc. Natl. Acad. Sci. U. S. A. 102:3010–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kroos L. 2005. Eukaryotic-like signaling and gene regulation in a prokaryote that undergoes multicellular development. Proc. Natl. Acad. Sci. U. S. A. 102:2681–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lutkenhaus J. 2007. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu. Rev. Biochem. 76:539–562 [DOI] [PubMed] [Google Scholar]
- 33. Martínez M., Colombo M. V., Palacios J. M., Imperial J., Ruiz-Argüeso T. 2008. Novel arrangement of enhancer sequences for NifA-dependent activation of the hydrogenase gene promoter in Rhizobium leguminosarum bv. viciae. J. Bacteriol. 190:3185–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meier T., Schickor P., Wedel A., Cellai L., Heumann H. 1995. In vitro transcription close to the melting point of DNA: analysis of Thermotoga maritima RNA polymerase-promoter complexes at 75 degrees C using chemical probes. Nucleic Acids Res. 23:988–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merrick M. J. 1993. In a class of its own—the RNA polymerase sigma factor sigma 54 (sigma N). Mol. Microbiol. 10:903–909 [DOI] [PubMed] [Google Scholar]
- 36. Miller J. H. 1975. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 37. Möglich A., Ayers R. A., Moffat K. 2009. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 17:1282–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morais Cabral J. H., et al. 1998. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95:649–655 [DOI] [PubMed] [Google Scholar]
- 39. Morett E., Segovia L. 1993. The sigma 54 bacterial enhancer-binding protein family: mechanism of action and phylogenetic relationship of their functional domains. J. Bacteriol. 175:6067–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muyzer G., Stams A. J. 2008. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6:441–454 [DOI] [PubMed] [Google Scholar]
- 41. Nambu J. R., Lewis J. O., Wharton K. A., Crews S. T. 1991. The Drosophila single-minded gene encodes a helix-loop-helix protein that acts as a master regulator of CNS midline development. Cell 67:1157–1167 [DOI] [PubMed] [Google Scholar]
- 42. Pauleta S., et al. 2007. NMR assignment of the apo-form of a Desulfovibrio gigas protein containing a novel Mo-Cu cluster. Biomol. NMR Assign. 1:81–83 [DOI] [PubMed] [Google Scholar]
- 43. Pfaffl M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Popham D. L., Szeto D., Keener J., Kutsu S. 1989. Function of a bacterial activator protein that binds to transcriptional enhancers. Science 243:629–635 [DOI] [PubMed] [Google Scholar]
- 45. Postgate J. 1966. Media for sulphur bacteria. Lab. Pract. 15:1239–1244 [PubMed] [Google Scholar]
- 46. Rothfield L., Taghbalout A., Shih Y. L. 2005. Spatial control of bacterial division-site placement. Nat. Rev. Microbiol. 3:959–968 [DOI] [PubMed] [Google Scholar]
- 47. Sasse-Dwight S., Gralla J. D. 1990. Role of eukaryotic-type functional domains found in the prokaryotic enhancer receptor factor sigma 54. Cell 62:945–954 [DOI] [PubMed] [Google Scholar]
- 48. Schleif R. 2000. Regulation of the L-arabinose operon of Escherichia coli. Trends Genet. 16:559–565 [DOI] [PubMed] [Google Scholar]
- 49. Schmidt T. G., Skerra A. 2007. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2:1528–1535 [DOI] [PubMed] [Google Scholar]
- 50. Scholten J., Culley D., Brockman F., Wu G., Zhang W. 2007. Evolution of the syntrophic interaction between Desulfovibrio vulgaris and Methanosarcina barkeri: involvement of an ancient horizontal gene transfer. Biochem. Biophys. Res. Commun. 352:48–54 [DOI] [PubMed] [Google Scholar]
- 51. Studholme D. J., Dixon R. 2003. Domain architectures of sigma54-dependent transcriptional activators. J. Bacteriol. 185:1757–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tabor S., Richardson C. C. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U. S. A. 82:1074–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tatusov R. L., Koonin E. V., Lipman D. J. 1997. A genomic perspective on protein families. Science 278:631–637 [DOI] [PubMed] [Google Scholar]
- 54. Taylor B. L., Zhulin I. B. 1999. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 63:479–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van den Ent F., Löwe J. 2006. RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67:67–74 [DOI] [PubMed] [Google Scholar]
- 56. Vieira J., Messing J. 1991. New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene 100:189–194 [DOI] [PubMed] [Google Scholar]
- 57. Voordouw G., et al. 1990. Functional expression of Desulfovibrio vulgaris Hildenborough cytochrome c3 in Desulfovibrio desulfuricans G200 after conjugational gene transfer from Escherichia coli. J. Bacteriol. 172:6122–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walker C., et al. 2009. The electron transfer system of syntrophically grown Desulfovibrio vulgaris. J. Bacteriol. 191:5793–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wedel A., Kutsu S. 1995. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes Dev. 9:2042–2052 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.