

Abstract
Background
Many RNA viruses do not have a single, representative genome but instead form a set of related variants that has been called a quasispecies. The sequence variability of such viruses presents a significant bioinformatics challenge. In order for the sequence information to be understood, the complete mutational spectrum needs to be distilled to a biologically relevant and analyzable representation.
Results
Here, we develop a "selection mapping" algorithm--QUASI--that identifies the positively selected variants of viral proteins. The key to the selection mapping algorithm is the identification of particular replacement mutations that are overabundant relative to silent mutations at each codon (e.g., threonine at hemagglutinin position 262). Selection mapping identifies such replacement mutations as positively selected. Conversely, selection mapping recognizes negatively selected variants as mutational "noise" (e.g., serine at hemagglutinin position 262).
Conclusion
Selection mapping is a fundamental improvement over earlier methods (e.g., dN/dS) that identify positive selection at codons but do not identify which amino acids at these codons confer selective advantage. Using QUASI's selection maps, we characterize the selected mutational landscapes of influenza A H3 hemagglutinin, HIV-1 reverse transcriptase, and HIV-1 gp120.
Background
Antigenic drift and the generation of viral quasispecies
Some RNA viruses form a quasispecies--a set of related viral variants that coexist in field populations and even within single infected individuals (reviewed in [1,2,3,4,5]). The emergence of immunologically distinct members of a viral quasispecies through mutation and subsequent immune selection is called "antigenic drift." Antigenic drift is thought to be important in human immunodeficiency virus (HIV) infection and the continuing seasonal influenza epidemics because immunity generated against one viral quasispecies member selects for escape variants. Attributed in part to antigenic drift are the moderately high failure rate and the short-lived efficacy of influenza vaccines [6], the failure of synthetic foot-and-mouth disease virus vaccines [7], and the inability of recombinant HIV vaccines to provide complete protection against field strains of the virus [8].
The hemagglutinin (HA) envelope surface glycoprotein--the major neutralizing determinant of influenza A--is a classic example of an antigenically drifting protein [9]. Walter Gerhard and colleagues demonstrated that the immune pressure exerted by monoclonal antibodies (Abs) selects for HA escape mutants in model systems [10,11]. Later, Dimmock and colleagues showed that polyclonal anti-sera also select for escape mutants [12,13]. Similarly, much of the observed variability of glycoprotein 120 (gp120), the principal surface antigen of HIV, is thought to reflect antigenic drift [14,15,16,17]. The correlation of intra-patient viral diversity with immune response strength has been cited as evidence that the immune response is a selective factor in HIV antigenic drift [18,19,20,21,22].
Phylogenetic analyses describe divergence within a viral population, and these methods have been used to infer the selective advantages of viral variation [18,19,20,21,22,23,24,25,26,27,28,29,30]. A more direct indication of the selective advantage gained through variation is an observed overabundance of replacement mutations relative to silent mutations in viral proteins [31]. Such analyses of gp120 and its V regions indicate that replacement mutations are generally over-represented in this protein and thus appear to confer selective advantage to HIV-1 [22,24,25,26,27,28,29,30,32,33,34,35]. In more detailed analyses, several groups tested individual codons for replacement mutations that are, as an aggregate, overabundant [23,36,37,38]. However, none of these methods determine which replacement mutations are actually positively selected. Also, when replacement mutations of varying fitness are lumped together, positively selected mutations may remain undetected among negatively selected mutations.
To overcome these limitations, we have developed a "selection mapping" algorithm. The cornerstone of selection mapping is the testing of each observed replacement mutation at each codon to identify those particular replacement mutations that are overabundant relative to silent mutations at that codon. Such replacement mutations are determined to be positively selected. Negatively selected variants are recognized as "noise" and are thereafter ignored. Here, we use the selection mapping method to identify the positively selected variants of influenza A HA (H3 serotype), HIV-1 reverse transcriptase (RT), and HIV-1 gp120.
Results and Discussion
Selection map of influenza A H3 hemagglutinin
QUASI identifies 25 HA codons where one or more replacement mutations are positively selected in the influenza A H3 virus (Fig. 1). [From our neutral drift testing of QUASI, we expect a maximum of about 2-3 false positives (see Materials and Methods)]. The distribution of these positively selected codons is of particular interest. Without exception, the codons where variants are positively selected are on the HA surface (Fig. 2, left). The parsimonious explanation of this result is that HA variants are primarily selected for escape from B-cell immunity. If T-cell immunity is instead the primary selective force affecting HA variation, then either all T-cell immunity escape variants are coincidentally solvent-exposed, or HA T-cell epitopes are determined by Ab protection [39]. These 25 codons include 13 outside those identified as positively selected by Walter Fitch and colleagues [40]. We attribute our new findings primarily to sites where many variants are negatively selected but where at least one variant is positively selected. The Fitch group also identifies 6 additional codons as positively selected. We believe that these are false positives caused by the Fitch group's assumption that HA is, on average, neutrally drifting; these six codons may be less negatively selected than average, but they nevertheless appear to be negatively selected. Additionally, our data, while largely the same as those analyzed by the Fitch group, do have some differences.
Figure 1.

Selection Map of influenza A HA (H3 serotype). Positively selected variants are at those codons where one or more mutations from the consensus confer selective advantage on the virus (capitalized and listed below the consensus). Lowercase letters indicate mutations where the neutral drift hypothesis is not ruled out. Negatively selected variants are not shown. The antigenic sites [41] are colored: A, red; B, yellow; C, green; D, blue. The selective advantage of the mutations at sites 156, 186, and 276 may have been conferred during viral passage in culture, so these mutations may not be positively selected in the field [58, 59].
Figure 2.
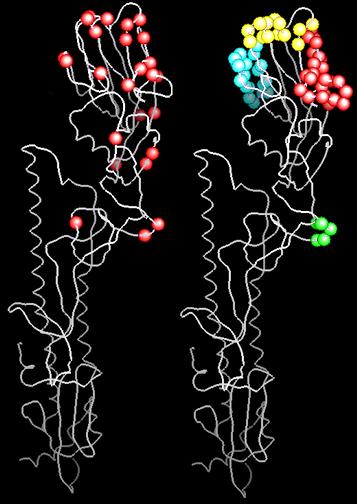
Positively Selected Sites vs. Antigenic Sites. Spatial relationships of the influenza HA positively selected variants plotted on the 3Å resolution HA crystal structure [41]. The white line is the backbone worm. Relevant Cα atoms are colored. Left: Advantageous Variants. Codons where variants are positively selected are colored red. Right: Antigenic Sites. The antigenic sites [41] are colored: A, red; B, yellow; C, green; D, blue. Figure 2 was generated using GRASP [60].
Wiley et al. proposed four antigenic sites where field and laboratory mutations could be grouped on the HA surface [41]. These putative antigenic sites are indicated in Figure 1 and the right side of Figure 2. Positively selected variants are correlated (Fisher's exact test) with antigenic site A (p = 5.27 × 10-3), antigenic site B (p = 1.12 × 10-3), and antigenic site C (p = 1.0 × 10-5). In contrast, antigenic site D is not particularly correlated with positively selected variants. We believe the lack of positively selected variants spanning antigenic site D may explain a decades-old puzzle. In the fully assembled HA protein, site D is buried in the trimer interface and therefore is not generally accessible to Ab [41,42]. At the time that the antigenic sites were proposed, residues in and around the trimer interface were variable and found on the monomer surface, so they were grouped and labeled as antigenic site D even though it was unclear how site D was recognized by Ab [41]. In fact, the only two positively selected variants QUASI identifies in site D are solvent-exposed at the extreme edge of the HA trimer (Fig. 2). Based on QUASI's selection map of HA, we now conclude that those mutations found in the trimer-buried portion of HA do not confer significant advantage on influenza A. That is, while the trimer-buried portion of antigenic site D includes the sound and fury of variability, it signifies nothing.
While QUASI finds that antigenic sites A-C are significantly associated with positively selected variation, this association may be a simple consequence of the sites' surface exposure. As can be seen in the left-hand side of Figure 2, positively selected variants are scattered across the entire exposed surface of HA. There are positively selected variants outside the antigenic sites, and there are subregions of the antigenic sites where variation is negatively selected. Thus, it may be more appropriate to view antigenic sites as more positional than functional. Indeed, more recent demarcations of antigenic sites enlarge antigenic sites A-D and add an additional antigenic site, site E, and these sites are now considered primarily positional rather than functional [6,43].
Selection map of HIV-1 gp120
At 123 codons, QUASI's selection map of gp120 indicates that one or more non-consensus amino acids are positively selected in HIV-1 (Fig. 3). Most positively selected variants appear to be on the gp120 surface (not shown), but in contrast to HA, gp120 includes several monomer-buried positively selected variants (at sites I225, V270, N295, H333, I345, T387, I424, and L453). Additionally, some of the positively selected variants that appear to be solvent-exposed may normally be buried (some loops are absent from the core protein crystal structure [44]). The burial of positively selected variants in the gp120 monomer confirms that gp120 quasispeciation is not selected solely for escape from B-cell immunity.
Figure 3.

Selection Map of HIV-1 gp120. Positively selected variants are at those codons where one or more mutations from the consensus confer selective advantage on the virus (capitalized and listed below the consensus). Lowercase letters indicate mutations where the neutral drift hypothesis is not ruled out. Negatively selected variants are not shown. Numbering is according to the HXBc2 sequence, where residue one is the amino-terminal methionine of the signal sequence. The asterisk indicates a 310Q-311R insert in HXBc2 that is not common enough to include in the profile (the alignment gap is the consensus sequence, and 2H+σ < 1.5). The hyphens in the numbering indicate codons absent from HXBc2. Many of the positively selected gp120 variants lie outside the variable regions, V1-V5, and are instead found in the constant regions proposed by Starcich et al. [61]. This is not troubling in that the V1-V5 designations were based on an analysis of only five gp120 sequences, so the "variable" and "constant" designations are not truly indicative of variability (B. Foley, Los Alamos, pers. commun.; this paper). For more appropriate positional references, we indicate secondary structure motifs [44]. The four numbered regions are repetitive and gap-filled and for which we could not generate a reasonable alignment; because we are unable to align these subregions reliably and because the alignment is a prerequisite of our analysis, we cannot reasonably comment further on these regions. The CD4BS epitopes are yellow; CD4i epitopes are blue; positions that belong to both CD4BS and CD4i are green [45].
Two competition-group epitopes have been identified for broadly neutralizing anti-gp120 Abs: the CD4-binding site (CD4BS) and the CD4-induced (CD4i) epitopes (references in [45]). Each epitope includes only a single non-consensus positively selected variant (Fig. 3). Thus, broadly neutralizing Abs appear to be those that engage few protein positions where variation is positively selected. Based on this observation, we propose that the neutralizing spectra of Abs may be predicted if the epitopes are known. For example, we would predict that the anti-CD4BS Abs will have particular trouble recognizing gp120 molecules carrying the positively selected variant of the CD4BS epitope (D→N at codon 474). This prediction is fulfilled in that the 15e anti-CD4BS monoclonal Ab fails to react to gp120 from HIV strain RF, a strain that carries this D→N mutation [46]. We also predict that gp120 molecules carrying the positively selected I→F mutation at codon 423 will be poorly recognized by the anti-CD4i Abs.
It is worth commenting on the GPGRAF motif (gp120 residues 312-317) that is sometimes (though increasingly rarely) referred to as "highly conserved." Because QUASI identifies non-consensus variants at two codons of this motif as positively selected (Fig. 3), it may be inappropriate to refer to the GPGRAF motif as "highly conserved." Instead, five positively selected variants appear to exist at this region in addition to GPGRAF: GPGKAF, GPGRTF, GPGKTF, GPGRVF, and GPGKVF.
Kwong et al. roughly divide the gp120 three-dimensional structure into outer (β9-β19 and β22-β24) and inner (N-α1, β4-β8, and α5-C) domains joined by a bridging sheet (β2, β3, β20, and β21) [44]. As indicated by Kwong et al., all three domains include variable regions; as QUASI shows, diversity in all three regions is positively selected (Fig. 3). The selective advantage we find rendered by diversity in some of these regions (e.g., the "silent face") has been attributed to neutral drift [44,45], thus QUASI's results run counter to previous interpretations. That is, QUASI finds that diversity in regions proposed to be inaccessible to the immune system nevertheless confers selective advantage on HIV. QUASI's findings may be consistent with the existence of gaps in the carbohydrate groups thought to mask the silent face from gp120 from immune surveillance. Interestingly, in carbohydrate-building models of gp120, these gaps correspond to codons where we find variation is positively selected (P. Kwong, pers. commun.).
A "non-neutralizing" face has been identified where binding Abs generally do not neutralize HIV when gp120 is oligomerized [45,47]; these data were interpreted as indicating that the non-neutralizing face is occluded in the trimer and that binding Abs are raised against shed gp120 monomers. However, QUASI finds numerous positively selected variants on the non-neutralizing face of the inner domain. Assuming the "non-neutralizing" appellation is appropriate, how do mutations on this face provide selective advantage to the virus? The obvious answer is that mutations provide escape not from direct B-cell immunity but from other levels of immunity, such as T-cell immunity [including major histocompatibility complex (MHC) presentation] or indirect Ab immunity via Ab-dependent cellular cytotoxicity (where gp120 molecules found on infected cell surfaces are monomers).
To determine if HIV-1 viral sequences retain evidence that T-cell immunity is a significant selective force affecting HIV quasispeciation, we used QUASI to generate a selection map of HIV-1 RT (Fig. 4). Because RT is not a surface-expressed protein, it is not plausible that the positively selected variants of RT have been selected by direct B-cell immunity. A priori, RT quasispeciation could have been the result of neutral drift, but because QUASI finds that replacement mutations confer selective advantage on the virus, we reject the neutral drift hypothesis at the 22 RT codons. If T-cell immunity (including MHC presentation) is a selective pressure shaping RT quasispeciation, positively selected variants should be associated with T-cell epitopes. When known T-cell epitopes [48] are plotted on the RT selection map, the positively selected variants are found to localize significantly (Fisher's exact test) both with helper T-cell epitopes (p = 3.27 × 10-2) and CTL epitopes (p = 6.58 × 10-3). We conclude that T-cell immunity is a significant selection pressure shaping the quasispeciation of RT and presumably is a significant factor in the quasispeciation of other HIV proteins.
Figure 4.

Selection Map of HIV-1 RT. Positively selected variants are at those codons where one or more mutations from the consensus confer selective advantage on the virus (capitalized and listed below the consensus). Lowercase letters indicate mutations where the neutral drift hypothesis is not ruled out. Negatively selected variants are not shown. T-cell epitopes are colored yellow (helper T-cell epitopes) and red (CTL epitopes). Variants that confer AZT resistance are blue. Numbering is from the beginning of the RT protein. Epitope positions are taken from the Los Alamos HIV database epitope maps [48]. Known MHC associations of the epitopes are as follows: 1: A2, B61; 2: A28; 4: broad; 6: B35; 7: A2, A*0201; 8: B35, B*3501; 9: B51; 10:B7; 11: B35, B*3501, B7; 12: A3, A3.1, A11, A33, A*6801; 13: B35, B*3501; 14: A2; 15: A*0201; 18: B44; 19: B*5701; 21: DR5(11.01); 23: Bw62.
Thus, because positively selected gp120 variants found throughout gp120 may be selected by T-cell immunity, QUASI's finding that the non-neutralizing face includes positively selected variants is not at odds with models where the non-neutralizing face forms the gp120 trimer interface. Nor are QUASI's results incompatible with the silent face being silent to B-cell immunity. QUASI's finding that positively selected variants may be buried in the gp120 monomer is consistent with escape from T-cell immunity.
In addition to the selection pressure exerted by T-cell immunity, 3'-azido-3'-deoxythymidine (AZT) may also have provided selection pressure for RT quasispeciation in the sequences selection mapped by QUASI [59,51]. Indeed, QUASI identifies six of the eight mutations known to be associated with AZT resistance [52] as positively selected (N67, R70, W210, Y215, and F215) or possibly positively selected (L41) to HIV-1 (Fig. 4). The exceptions, two mutations of codon 219, are informative. Whereas mutations at other codons are necessary for high resistance to AZT, mutations at codon 219 are not, and codon 219 mutations arise late in infection after earlier mutations have already rendered RT resistant to AZT [53]. We conclude that the additional AZT resistance conferred by codon 219 mutations did not provide significant selective advantage to the profiled HIV viruses, possibly because HIV had already acquired the maximum effective AZT resistance selectable, in vivo, when mutations arose at this codon. Alternatively, the lysine at codon 219 may be important for proper in vivo RT function such that the advantage conferred by increased AZT resistance does not adequately compensate for impaired RT function. The RT sequences we analyze were taken from patients who either had no anti-RT treatment or were treated mainly with AZT (though some patients who were treated with AZT were also treated with 2',3'-dideoxyinosine) [49,50,51]. Therefore, we would predict that mutations associated with resistance to other anti-RT drugs should not be positively selected in the sequences QUASI analyzed. As predicted, QUASI identifies none of the 50 RT mutation associated with resistance to other drugs as positively selected (compare Figure 4 to the Los Alamos database [52]).
Conclusion
We have developed an algorithm for using sequence data to map the positively selected mutations of viral quasispecies. We have used this method to map the positively selected variants of influenza A HA, HIV-1 RT, and HIV-1 gp120. Other obvious targets for selection mapping are the hepatitis C and foot-and-mouth disease viruses. We believe that potentially the most illuminating use of selection mapping may be the comparison of viral subpopulations to determine which variants are advantageous under different selective pressures. For example, selection mapping of HIV isolates with different cellular tropisms will allow the determination of mutations that are positively selected depending on the host cell type. Also, we may use selection mapping to analyze HIV breakthrough infections to determine if vaccines prevented the HIV quasispecies from inhabiting normally advantageous regions of the quasispecies sequence space. Finally, we propose that the positively selected viral variants (as opposed to all viral variants) should be included in future, highly multivalent vaccines designed to compensate for B-cell-selected antigenic drift.
Materials and Methods
QUASI--the selection mapping algorithm
An executable version of the QUASI software is attached as an additional file (see additional file 1). Also attached are a users' manual (user.txt - see additional file 2) and a FASTA to QUASI file converter PERL script (F2Q.pl - additional file 3). Current versions of QUASI are available from the authors or may be accessed at the Los Alamos Influenza Sequence Database (http://www.flu.lanl.gov/).
For a set of viral nucleotide sequences, we determine the variants that confer selective advantage by measuring the empirical replacement to silent mutation ratio (R:S) of each possible amino acid replacement and then comparing this observed ratio to that which would be expected if mutation were unselected. An R:S that is found to be higher than expected indicates that the replacement mutation tested is positively selected, while a lower-than-expected observed R:S indicates that the tested replacement mutation is negatively selected.
Testing for an overabundance of replacements across a protein as a whole is a reasonable approach when only a few nucleotide sequences are available, but because a large number of mutated viral sequences are currently available, such aggregation is unnecessarily crude. Better are approaches that test for an overabundance of replacement mutations at individual codons [23,36,37,38]. However, these methods lump together replacement mutations and thus allow negatively selected mutations to conceal positively selected mutations and vice versa (e.g., replacement mutations at a codon may be negatively selected as a group despite the fact that one or more particular replacement mutations are positively selected).
To overcome these limitations, the QUASI algorithm does not test the overall R:S of the entire protein as an aggregate, nor does QUASI test the R:S of a codon to all its replacement mutations taken as a whole. Rather, QUASI tests the R:S of each particular replacement mutation at each codon. That is, QUASI measures the R:S of the mutations from a consensus codon towards each individual replacement amino. For example, if the consensus codon at a protein position were ttt (Phe), QUASI would test the R:Ss of all point mutations from ttt. One of these mutations is ttt→tat (Tyr). QUASI calculates the expected R:S for ttt→tat under the null hypothesis of neutral drift. The expected S is one because only one mutation (ttc) is silent, and the expected R is also one because only one point mutation of ttt (tat) codes for Tyr, so the expected R:S is one, in this case. If QUASI rejects the (Jukes-Cantor) neutral drift null hypothesis because the observed R:S is significantly higher than one, then QUASI classifies this replacement mutation (Tyr) as positively selected. Conversely, if QUASI rejects the null hypothesis because the observed R:S is significantly lower than one, then QUASI determines that this replacement mutation is negatively selected. QUASI performs this procedure for all replacement point mutations [e.g., in the example case, Tyr (tat), Ile (att), Leu (tta, ttg, and ctt), Val (gtt), Ser (tct), and Cys (tgt)].
In this paper, selection mapping is carried out independent of the underlying phylogeny. QUASI uses R:S to reject the null hypothesis that the mutational space surrounding the consensus codon is distributed randomly among all nine possible R or S point mutations (except stop codons, which are considered to be disallowed). This allows R:S calculations to be applied to viral sequences whose ancestral sequence is unclear or unknown. This is a both an advantage and a disadvantage over analyses that rely on phylogeny. Phylogeny is difficult to determine accurately and uniquely, and relying on phylogeny ignores the persistence of positively selected replacement mutations (the major effect of selection). On a practical level, using phylogeny to reconstruct viruses' mutational histories and then using intuited mutations leaves one with insufficient data to determine positively-selected codons [36] unless, as some have done, one assumes observed drift is neutral and then tests for codons where selection is more positive than average [23,38]. The significance problem can be compounded when one is looking for independent occurrences of particular mutations; often, there simply has not been enough sequence evolution in HIV or influenza to map positively selected variants if the retention of positively selected mutations is ignored. The drawback of ignoring phylogeny is a potentially high false positive rate (see below).
Empirical R:S is compared to neutral R:S by means of a two-sided test of the binomial distribution. For each codon, we test the null hypothesis that all nine point mutants are equally probable. The quotient p = R/(R+S) is the probability of a replacement mutation at this codon if each nucleotide is equally mutable and each of the three mutational targets at that codon are equally likely. The numerator, R, is the number of point mutations that lead from the consensus codon to the target amino acid. The chance of observing r replacement mutations is given by the binomial distribution, 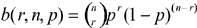 , where n is the number of codons providing data for this position. To form a two-sided test, we sum all terms b(kn,,p) such that b(kn,,p) is not greater than b(rn, p,), wherek is in the set (0,...,n) and r is the number of observed replacement mutations. In other words, we sum the chances of all events that are no more likely than that of the observation. If this sum, α, is small (e.g., not greater than 0.05), we reject the null hypothesis at the α level of significance.
, where n is the number of codons providing data for this position. To form a two-sided test, we sum all terms b(kn,,p) such that b(kn,,p) is not greater than b(rn, p,), wherek is in the set (0,...,n) and r is the number of observed replacement mutations. In other words, we sum the chances of all events that are no more likely than that of the observation. If this sum, α, is small (e.g., not greater than 0.05), we reject the null hypothesis at the α level of significance.
Working example
We analyze the following scenario as a working example (Table 1).
Table 1.
| Amino Acid | Mutation(s) | Obs. | Exp. R:S | Obs. R:S | Selection |
| Phe (silent) | ttt→ttc | 10 | na | na | na* |
| Tyr | ttt→tat (Tyr) | 24 | 1:1 | 24:10 | positive (p = 0.02) |
| Ile | ttt→att | 1 | 1:1 | 1:10 | negative (p = 0.01) |
| Leu | ttt→tta or | 3 | 3:1 | 3:10 | negative (p = 0.0001) |
| ttt→ttg or | |||||
| ttt→ctt | |||||
| Val | ttt→gtt | 4 | 1:1 | 4:10 | neutral (p = 0.18) |
| Ser | ttt→tct | 23 | 1:1 | 23:10 | positive (p = 0.04) |
| Cys | ttt→tgt | 0 | 1:1 | 0:10 | negative (p = 0.002) |
*neutral by definition
The consensus codon is given as ttt (Phe; Table 1, column 1). Each observed mutation is also given (Table 1, columns 1-3)
Because we know the frequency of silent mutations (given as 10 in this example; Table 1, column 3), we also know the expected R:S for each replacement mutation (Table 1, column 4). That is, if selection is neutral for any particular replacement mutation, we can calculate the incidence of each replacement mutation we expect to observe (by looking at a table of the genetic code).
Using the given frequency of each mutation observed, we also know what the observed R:S is in each case (Table 1, column 5).
Now we use a two-tailed test of the binomial distribution to determine if each observed R:S is significantly different from the corresponding expected R:S (Table 1, column 6). In some cases, the differences are significant, in which case the appropriate replacement mutation is assigned positive or negative selection (positive if the observed R:S is larger than expected and negative if the observed R:S is lower than expected). Otherwise, the selection is assigned to be neutral drift.
The QUASI algorithm thus indicates at this exemplary codon that both tyrosine and serine are positively selected; isoleucine, leucine, and cystine are negatively selected; and the selective advantage or disadvantage of valine is indistinguishable from neutral drift. Any other ttt codon will have its own selective pressures assessed in a similar but independent testing procedure.
Minimum sequences
For each possible replacement mutation, a minimum number of mutations will need to be observed for a selective event to be detected. This minimum number differs depending on the consensus codon and the level of significance. We have calculated the minimum number of mutations needed to achieve the 5% significance level. At the lower bound of this range, only 2 replacement mutations will be required to detect positive selection for any replacement mutation from cta or ctg. Any replacement mutation from cta or ctg has an expected R:S of 1:4, and thus an observed R:S of 2:0 will be sufficient to reject neutral drift in favor of positive selection. Conversely, detecting negative selection at a cta or ctg codon is difficult. At the upper bound of the range, a minimum of 17 observed mutations are required to detect negative selection at such a codon (0:17 is significantly lower than 1:4). For the most-typical codon, the expected R:S is 1:3. For these modal codons, at least 3 mutations must be observed to detect positive selection (i.e., if observed R:S = 3:0). At the same most-typical codon, 12 mutations are required, at a minimum, to detect negative selection (i.e., if observed R:S = 0:12). Because identification of positive selection is generally the goal, the QUASI algorithm appears to have a practical advantage over extant selection detectors, which require either many more mutations or a biased expectation of neutral drift in order to detect positive selection.
False-positive testing
False positives are likeliest when drift is completely neutral (e.g., as was found by Suzuki and Gojobori [36]). One may estimate the maximum frequency of false positives by testing simulated sequences generated under neutral drift parameters. We used the EVOLVER program of the PAML package [54] to generate sequences drifting under neutral Jukes-Cantor evolution. For each simulation, we generated 300 related sequences of length 999 and with average branch lengths varying in 0.1 length intervals from 0.1 to 1.0; each parameter set was used to generate 10 sets of 300 sequences. False positive percentages [Fig. 5; false positive percentage = false positives / (false positives + neutral drift variants)] were extremely low (∼ 2%) for relatively long branch lengths (0.1-1.0). Extremely high false positive rates (up to 70%) were found for extremely short branch lengths (maximum at 0.001). As accurate branch lengths are calculated using maximum likelihood phylogeny, these branch lengths may be used to find the appropriate false-positive percentage. For instance, if the branch length were 0.01 [as appears appropriate for HA (unpublished observation)], then the maximum false positive rate would be estimated at 32%. We then use the calculated neutral drift frequency (from QUASI) as an estimator for the maximum false positives. If (as we report) HA is found to have 5 neutral drift variants, then we would expect a maximum of 2.3 false-positives.
Figure 5.
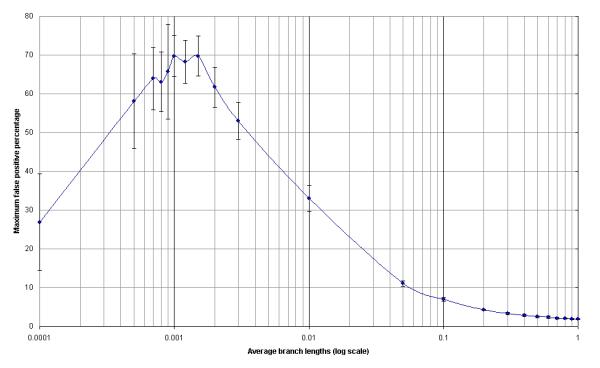
Maximum Expected False Positives. The number of false positives are given for various branch lengths for sequences evolved under neutral drift parameters.
Selection mapping
QUASI presents its results in the following format:
1. The consensus amino acids are written in capital letters.
2. Beneath each consensus amino acid are written in capital letters all variants determined to be positively selected (in descending order of frequency).
3. The negatively selected variants are not shown.
4. In lowercase letters and interspersed according to their frequencies among the positively selected variants are variants where the neutral drift null hypothesis cannot be rejected with the given sequence data. As a reasonable but arbitrary cut-off, we include apparently unselected variants if they are among the 2H+σ most frequent variants, where H is the Shannon information content of the site and σ is the standard error of its estimation [55]. 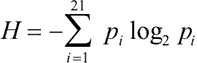 is the ith fraction of amino acids at the site (the alignment gap is counted as a 21st amino acid). For the Shannon calculation, alignment gaps are considered distinct from "no data" gaps (artifacts of indeterminate sequencing or sequence fragment overlaps; such data absences are excluded from calculation).
is the ith fraction of amino acids at the site (the alignment gap is counted as a 21st amino acid). For the Shannon calculation, alignment gaps are considered distinct from "no data" gaps (artifacts of indeterminate sequencing or sequence fragment overlaps; such data absences are excluded from calculation).
Sequences
Nucleotide sequences were downloaded from GenBank at the NIH. Sequences were included only if they were isolated in the field and were not obviously pseudogenes (sequences containing premature stop codons were removed from consideration). We analyzed 310 sequences of human-infective influenza A (H3 serotype), 6,151 HIV-1 gp120 sequences, and 400 HIV-1 RT sequences. All sequences were pre-aligned with PILEUP [56] and/or DIALIGN2 [57] then hand-corrected.
Abbreviations
HIV, human immunodeficiency virus; HA, hemagglutinin; Ab, antibody; gp120, glycoprotein 120; RT, reverse transcriptase; R:S, replacement to silent mutation ratio; CD4BS, CD4 binding site epitope; CD4i, CD4-induced epitope; MHC, major histocompatibility complex; AZT, 3'-azido-3'-deoxythymidine.
Supplementary Material
Acknowledgments
Acknowledgments
We thank M. Weigert for inspiring this work and advising us throughout the project; L. Enquist, B. Foley, B. Korber, C. Macken, and M. Nowak for informative discussions; C. Benedict, F. Brard, L. Enquist, B. Foley, W. Gerhard, W. Hendrickson, P. Kwong, P. McNutt, R. Mehr, P. Seiden, M. Shannon, J. Wadsack, M. Weigert, and V. Zumbrunn for commenting on the manuscript prior to submission; J. Goodman for his excellent technical support; and J. Wadsack for her continuing support (without which this work would have been impossible).
Contributor Information
Jeffrey J Stewart, Email: jjs@alumni.princeton.edu.
Perry Watts, Email: wattsp@dca.net.
Samuel Litwin, Email: litwin@euclid.fccc.edu.
References
- Holland JJ, de la Torre JC, Steinhauer DA. RNA virus populations as quasispecies. Curr Top Microbiol Immunol. 1992;176:1–20. doi: 10.1007/978-3-642-77011-1_1. [DOI] [PubMed] [Google Scholar]
- Smith DB, McAllister J, Casino C, Simmonds P. Virus 'quasispecies': making a mountain out of a molehill? J Gen Virol. 1997;78:1511–1519. doi: 10.1099/0022-1317-78-7-1511. [DOI] [PubMed] [Google Scholar]
- Domingo E, Martinez-Salas E, Sobrino F, de la Torre JC, Portela A, Ortin J, Lopez-Galindez C, Perez-Brena P, Villanueva N, Najera R, VandePol S, DePolo N, Holland J. The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance-a review. Gene. 1985;40:1–8. doi: 10.1016/0378-1119(85)90017-4. [DOI] [PubMed] [Google Scholar]
- Domingo E, Holland J, Biebricher C, Eigen M. Quasi-species: the concept and the word. Molecular Basis of Virus Evolution Edited by Gibbs AJ, Calisher C, García-Arenal F. pp. Cambridge: Cambridge University Press; 1995. pp. 181–191.
- Duarte EA, Novella IS, Weaver SC, Domingo E, Wain-Hobson S, Clarke DK, Moya A, Elena SF, de la Torre JC, Holland JJ. RNA virus quasispecies: significance for viral disease and epidemiology. Infect Agents Dis. 1994;3:201–214. [PubMed] [Google Scholar]
- Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 1990;8:737–771. doi: 10.1146/annurev.iy.08.040190.003513. [DOI] [PubMed] [Google Scholar]
- Taboga O, Tami C, Carrillo E, Nunez JI, Rodriguez A, Saiz JC, Blanco E, Valero ML, Roig X, Camarero JA, Andreu D, Mateu MG, Giralt E, Domingo E, Sobrino F, Palma EL. A large-scale evaluation of peptide vaccines against foot-and-mouth disease: lack of solid protection in cattle and isolation of escape mutants. J Virol. 1997;71:2606–2614. doi: 10.1128/jvi.71.4.2606-2614.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman PW, Gray AM, Wrin T, Vennari JC, Eastman DJ, Nakamura GR, Francis DP, Gorse G, Schwartz DH. Genetic and Immunologic Characterization of Viruses Infecting MN-rgp120-Vaccinated Volunteers. J Inf Dis. 1997;176:384–397. doi: 10.1086/514055. [DOI] [PubMed] [Google Scholar]
- Webster RG, Laver WG, Air GM, Schild GC. Molecular mechanisms of variation in influenza viruses. Nature. 1982;296:115–121. doi: 10.1038/296115a0. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Caton AJ, Gerhard W. Selection of influenza A virus adsorptive mutants by growth in the presence of a mixture of monoclonal antihemagglutinin antibodies. J Virol. 1986;57:623–628. doi: 10.1128/jvi.57.2.623-628.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard W, Yewdell J, Frankel M. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature. 1981;290:713–717. doi: 10.1038/290713a0. [DOI] [PubMed] [Google Scholar]
- Lambkin R, McLain L, Jones SE, Aldridge SL, Dimmock NJ. Neutralization escape mutants of type A influenza virus are readily selected by antisera from mice immunized with whole virus: a possible mechanism for antigenic drift. J Gen Virol. 1994;75:3493–3502. doi: 10.1099/0022-1317-75-12-3493. [DOI] [PubMed] [Google Scholar]
- Cleveland SM, Taylor HP, Dimmock NJ. Selection of neutralizing antibody escape mutants with type A influenza virus HA-specific polyclonal antisera: possible significance for antigenic drift. Epidemiol Infect. 1997;118:149–154. doi: 10.1017/S0950268896007303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney SD, Matthews TJ, Robey WG, Lynn DL, Robert-Guroff M, Mueller WT, Langlois AJ, Ghrayeb J, Petteway SR, Jr, Weinhold KJ, Fischinger PJ, Wong-Staal F, Gallo RC, Bolognesi DP. HTLV-III/LAV-neutralizing antibodies to an E. coli-produced fragment of the virus envelope. Science. 1986;234:1392–1395. doi: 10.1126/science.2431482. [DOI] [PubMed] [Google Scholar]
- Goudsmit J, Debouck C, Meloen RH, Smit L, Bakker M, Asher DM, Wolff AV, Gibbs CJ, Jr, Gajdusek DC. Human immunodeficiency virus type 1 neutralization epitope with conserved architecture elicits early type-specific antibodies in experimentally infected chimpanzees. Proc Natl Acad Sci U S A. 1988;85:4478–4482. doi: 10.1073/pnas.85.12.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaherian K, Langlois AJ, McDanal C, Ross KL, Eckler LI, Jellis CL, Profy AT, Rusche JR, Bolognesi DP, Putney SD, Matthews TJ. Principal neutralizing domain of the human immunodeficiency virus type 1 envelope protein. Proc Natl Acad Sci U S A. 1989;86:6768–6772. doi: 10.1073/pnas.86.17.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, Anderson RM, McLean AR, Wolfs TF, Goudsmit J, May RM. Antigenic diversity thresholds and the development of AIDS. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- Wolinsky SM, Korber BT, Neumann AU, Daniels M, Kunstman KJ, Whetsell AJ, Furtado MR, Cao Y, Ho DD, Safrit JT. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science. 1996;272:537–542. doi: 10.1126/science.272.5261.537. [DOI] [PubMed] [Google Scholar]
- Delwart EL, Pan H, Sheppard HW, Wolpert D, Neumann AU, Korber B, Mullins JI. Slower evolution of human immunodefiency virus type 1 quasispecies during progression to AIDS. J Virol. 1997;71:7498–7508. doi: 10.1128/jvi.71.10.7498-7508.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashov VV, Kuiken CL, Goudsmit J. Intrahost human immunodeficiency virus type 1 evolution is related to length of the immunocompetent period. J Virol. 1995;69:6911–6916. doi: 10.1128/jvi.69.11.6911-6916.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Schacker T, Musey L, Shriner D, McElrath MJ, Corey L, Mullins JI. Divergent patterns of progression to AIDS after infection from the same source: human immunodeficiency virus type 1 evolution and antiviral responses. J Virol. 1997;71:4284–4295. doi: 10.1128/jvi.71.6.4284-4295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshan S, Dickover RE, Korber BT, Bryson YJ, Wolinsky SM. Human immunodeficiency virus type 1 genetic evolution in children with different rates of development of disease. J Virol. 1997;71:663–677. doi: 10.1128/jvi.71.1.663-677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM, Leiter JM, Li XQ, Palese P. Positive Darwinian evolution in human influenza A viruses. Proc Natl Acad Sci U S A. 1991;88:4270–4274. doi: 10.1073/pnas.88.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell DP. Positive selection and rates of evolution in immunodeficiency viruses from humans and chimpanzees. Proc Natl Acad Sci U S A. 1996;93:3284–3288. doi: 10.1073/pnas.93.8.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Gojobori T. Evolutionary mechanisms and population dynamics of the third variable enveloperegion of HIV within single hosts. Proc Natl Acad Sci U S A. 1997;94:1264–1269. doi: 10.1073/pnas.94.4.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Diaz RS, Ho DD, Mosley JW, Busch MP, Mayer A. Host-specific driving force in human immunodeficiency virus type 1 evolution in vivo. J Virol. 1997;71:2555–2561. doi: 10.1128/jvi.71.3.2555-2561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss M, Rodrigo AG, Gosink JJ, Learn GH, de Vange Panteleeff D, Martin HLJ, Bwayo J, Kreiss JK, Overbaugh J. Evolution of envelope sequences from the genital tract and peripheral blood of women infected with clade A human immunodeficiency virus type 1. J Virol. 1998;72:8240–8251. doi: 10.1128/jvi.72.10.8240-8251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R, Gupta P, Learn GHJ, Rodrigo AG, Rinaldo CRJ, Gorry MC, Mullins JI, Nara PL, Ehrlich GD. Evolution of human immunodeficiency virus type 1 envelope sequences in infected individuals with differing disease progression profiles. Virology. 1998;241:251–259. doi: 10.1006/viro.1997.8996. [DOI] [PubMed] [Google Scholar]
- Ida S, Gatanaga H, Shioda T, Nagai Y, Kobayashi N, Shimada K, Kimura S, Iwamoto A, Oka S. HIV type 1 V3 variation dynamics in vivo: long-term persistence of non-syncytium-inducing genotypes and transient presence of syncytium-inducing genotypes during the course of progressive AIDS. AIDS Res Hum Retroviruses. 1997;13:1597–1609. doi: 10.1089/aid.1997.13.1597. [DOI] [PubMed] [Google Scholar]
- Cichutek K, Merget H, Norley S, Linde R, Kreuz W, Gahr M, Kurth R. Development of a quasispecies of human immunodeficiency virus type 1 in vivo. Proc Natl Acad Sci U S A. 1992;89:7365–7369. doi: 10.1073/pnas.89.16.7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Li W-H, Tanimura M, Sharp PM. Rates and dates of divergence between AIDS virus nucleotide sequences. Mol Biol Evol. 1988;5:313–330. doi: 10.1093/oxfordjournals.molbev.a040503. [DOI] [PubMed] [Google Scholar]
- Simmonds P, Balfe P, Ludlam CA, Bishop JO, Brown AJ. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J Virol. 1990;64:5840–5850. doi: 10.1128/jvi.64.12.5840-5850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenow M, Huet T, Saurin W, Kwok S, Sninsky J, Wain-Hobson S. HIV-I isolates are rapidly evolving quasispecies: evidence for viral mixtures and preferred nucleotide substitutions. J AIDS. 1989;2:344–352. [PubMed] [Google Scholar]
- Bonhoeffer S, Holmes EC, Nowak MA. Causes of HIV diversity. Nature. 1995;376:125. doi: 10.1038/376125a0. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Gojobori T. A method for detecting positive selection at single amino acid sites. Mol Biol Evol. 1999;16:1315–1328. doi: 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- Nielsen R. The ratio of replacement to silent divergence and tests of neutrality. J Evol Biol. 1997;10:217–231. doi: 10.1007/s000360050019. [DOI] [Google Scholar]
- Bush RM, Fitch WM, Bender CA, Cox NJ. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol. 1999;16:1457–1465. doi: 10.1093/oxfordjournals.molbev.a026057. [DOI] [PubMed] [Google Scholar]
- Graham CM, Warren AP, Thomas DB. Do antigenic drift residues in influenza hemagglutinins of the H3 subtype qualify as contact sites for MHC class II interaction? Int Immunol. 1992;4:917–922. doi: 10.1093/intimm/4.8.917. [DOI] [PubMed] [Google Scholar]
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the Evolution of Human Influenza A. Science. 1999;286:1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3Å resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Wiley DC, Skehel JJ. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- Ho DD, McKeating JA, Li XL, Moudgil T, Daar ES, Sun NC, Robinson JE. Conformational epitope on gp120 important in CD4 binding and human immunodeficiency virus type 1 neutralization identified by a human monoclonal antibody. J Virol. 1991;65:489–493. doi: 10.1128/jvi.65.1.489-493.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Sodroski J. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J Virol. 1996;70:1863–1872. doi: 10.1128/jvi.70.3.1863-1872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B, Moore J, Brander C, Koup R, Haynes B, Walker B. HIV Molecular Immunology Database. 1997.
- Wildemann B, Haas J, Ehrhart K, Wagner H, Lynen N, Storch-Hagenlocher B. In vivo comparison of zidovudine resistance mutations in blood and CSF of HIV-1-infected patients. Neurology. 1993;43:2659–2663. doi: 10.1212/wnl.43.12.2659. [DOI] [PubMed] [Google Scholar]
- Wong JK, Ignacio CC, Torriani F, Havlir D, Fitch NJS, Richman DD. In vitro compartmentalization of human immunodeficiency virus: evidence from the examination of pol sequences from autopsy tissues. J Virol. 1997;71:2059–2071. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng NN, Hurren L, Neilan BA, Cooper DA, Delaney SF, McQueen PW. Sequence analyses of the reverse transcriptase region of HIV type 1 isolates from Sydney, Australia. AIDS Res Hum Retroviruses. 1996;12:1731–1732. doi: 10.1089/aid.1996.12.1731. [DOI] [PubMed] [Google Scholar]
- Mellors JW, Schinazi RF, Larder BA. Mutations in retroviral genes associated with drug resistance. Human Retroviruses and AIDS. Edited by Meyers G, Korber B, Foley B, Jeang K, Mellors JW, Wain-Hobson S. Los Alamos: Los Alamos National Laboratory; 1996.
- Boucher CA, O'Sullivan E, Mulder JW, Ramautarsing C, Kellam P, Darby G, Lange JM, Goudsmit J, Larder BA. Ordered appearance of zidovudine resistance mutations during treatment of 18 human immunodeficiency virus-positive subjects. J Infect Dis. 1992;165:105–110. doi: 10.1093/infdis/165.1.105. [DOI] [PubMed] [Google Scholar]
- Yang Z. Phylogenetic Analysis by Maximum Likelihood (PAML). Phylogenetic Analysis by Maximum Likelihood (PAML), 1.3 ed. City; 1997.
- Shannon CE. The mathematical theory of communication. Urbana: University of Illinois Press; 1949.
- GCG Wisconsin Package. Wisconsin Package, 9.1 ed. City: Genetics Computer Group; 1997.
- Morgenstern B, Frech K, Dress A, Werner T. DIALIGN: Finding local similarities by multiple sequence alignment. Bioinformatics. 1998;14:290–294. doi: 10.1093/bioinformatics/14.3.290. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Xu X, Hall HE, Allen JR, Regnery HL, Cox NJ. Comparison of 10 influenza A (H1N1 and H3N2) haemagglutinin sequences obtained directly from clinical specimens to those of MDCK cell- and egg-grown viruses. J Gen Virol. 1993;74:2513–2518. doi: 10.1099/0022-1317-74-11-2513. [DOI] [PubMed] [Google Scholar]
- Kodihalli S, Justewicz DM, Gubareva LV, Webster RG. Selection of a single amino acid substitution in the hemagglutinin molecule by chicken eggs can render influenza A virus (H3) candidate vaccine ineffective. J Virol. 1995;69:4888–4897. doi: 10.1128/jvi.69.8.4888-4897.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A. Graphical Representation and Analysis of Surface Properties (GRASP). Graphical Representation and Analysis of Surface Properties (GRASP). City: Columbia University; 1992.
- Starcich BR, Hahn BH, Shaw GM, McNeely PD, Modrow S, Wolf H, Parks ES, Parks WP, Josephs SF, Gallo RC, et al. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell. 1986;45:637–648. doi: 10.1016/0092-8674(86)90778-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.