

Abstract
Ferritin is a cytosolic protein that stores excess iron, thereby protecting cells from iron toxicity. Ferritin-stored iron is believed to be utilized when cells become iron deficient; however, the mechanisms underlying the extraction of iron from ferritin have yet to be fully elucidated. Here, we demonstrate that ferritin is degraded in the lysosome under iron-depleted conditions and that the acidic environment of the lysosome is crucial for iron extraction from ferritin and utilization by cells. Ferritin was targeted for degradation in the lysosome even under iron-replete conditions in primary cells; however, the mechanisms underlying lysosomal targeting of ferritin were distinct under depleted and replete conditions. In iron-depleted cells, ferritin was targeted to the lysosome via a mechanism that involved autophagy. In contrast, lysosomal targeting of ferritin in iron-replete cells did not involve autophagy. The autophagy-independent pathway of ferritin delivery to lysosomes was deficient in several cancer-derived cells, and cancer-derived cell lines are more resistant to iron toxicity than primary cells. Collectively, these results suggest that ferritin trafficking may be differentially regulated by cell type and that loss of ferritin delivery to the lysosome under iron-replete conditions may be related to oncogenic cellular transformation.
INTRODUCTION
Iron is an essential nutrient for almost all living organisms. Iron plays important roles in a number of cellular processes, such as oxygen transport, energy production, and DNA repair (2). However, iron is highly reactive and can generate damaging reactive oxygen species that damage cellular components. Therefore, iron metabolism in the cell is tightly regulated (20).
In mammalian cells, iron homeostasis is maintained by compensatory regulation of iron uptake and storage depending on the availability of iron. Ferritin is the major iron storage protein in mammals. Ferritin forms a three-dimensional protein shell consisting of 24 protein subunits that can store up to 4,500 atoms of iron (19). Two isoforms of ferritin, ferritin heavy chain (H chain) and light chain (L chain), cooperate in storing iron in the ferritin shell. The production of ferritin H and L chains is regulated by iron availability at the posttranscriptional level through interactions between an RNA stem-loop structure, the iron-responsive element (IRE), and a small family of IRE-binding proteins, the iron regulatory proteins (IRPs) (43, 47). An IRE is located in the 5′ untranslated region of the ferritin H and L chain mRNAs. Under iron-depleted conditions, IRPs bind to the IREs of the ferritin H and L mRNAs and inhibit their translation, thereby preventing further storage of iron. Under conditions of sufficient iron, IRPs dissociate from the ferritin mRNA IREs, and ferritin synthesis is increased. The increased levels of ferritin store excess iron under iron-replete conditions, thereby preventing damage to cellular components due to iron toxicity (4).
While the mechanism of iron-mediated regulation of ferritin expression has been well defined, comparatively little is known regarding the fate of the iron that is stored by ferritin. Iron can be extracted from the ferritin shell in vitro by iron chelators and reducing agents (7), a process that involves regulation of the gating of ferritin pores (24, 44). Recent evidence suggests that the utilization of iron stored in the ferritin shell involves ferritin degradation. Several reports have indicated that ferritin is degraded in response to bacterial infections (33), the presence of an antitumor agent (31), and iron deficiency (25). Recently, De Domenico et al. showed that the overexpression of ferroportin, an iron exporter located in the plasma membrane, renders ferritin susceptible to degradation by the proteasome (13). The same group subsequently showed that the iron chelator deferoxamine (Dfo) specifically induces autophagy, resulting in ferritin degradation in the lysosome (14). Lysosomal degradation of ferritin has been reported under various conditions (25, 32, 33, 42). However, while multiple protein degradation mechanisms have been implicated in ferritin turnover, the role of these degradation systems in the extraction and utilization of ferritin iron stores has not been adequately investigated.
In the current study, we investigated the mechanism of iron utilization from ferritin stores by examining the fate of ferritin under iron-replete and iron-deficient conditions in mouse embryonic fibroblasts (MEFs). Ferritin was degraded via lysosomal degradation when iron was scarce, and macroautophagy (hereinafter referred as autophagy) contributed to this process. Autophagy-deficient MEFs became iron deficient more readily than wild-type (WT) MEFs when cells were shifted from iron-rich to iron-scarce conditions, indicating that the lysosomal degradation of ferritin plays a crucial role in the utilization of ferritin-stored iron. The acidic environment of the lysosome was also crucial for iron extraction from ferritin under iron-deficient conditions, as treatment of MEFs with bafilomycin A1, a vacuolar ATPase inhibitor that increases lysosomal pH, rendered cells iron deficient. Under iron-rich conditions, ferritin was also degraded through a mechanism that was distinct from autophagy. These results indicate that iron stored in ferritin is extracted in the lysosome for utilization by cells and that this process is an important component of cellular iron homeostasis. The results also strongly suggest that ferritin is degraded in the lysosome regardless of the iron status of the cell. Iron released from lysosomes might be stored in newly synthesized ferritin under iron-replete conditions as a mechanism of protecting cells from iron toxicity. A more detailed understanding of the delivery of ferritin to lysosomes under iron-replete conditions may provide mechanistic insight into the generation of hemosiderin complexes, which are insoluble iron-rich granules located in membranous compartments that are often found under conditions of iron overload. Moreover, the findings that ferritin is fairly stable under iron-replete conditions in several cancer cells and that cancer-derived cell lines are more resistant to iron toxicity than primary cells indicate that the fate of ferritin iron may be differentially regulated depending on the cell type. Thus, the loss of ferritin delivery to lysosomes under iron-rich conditions may be related to oncogenic cellular transformation.
MATERIALS AND METHODS
Cell culture.
WT, Atg5 knockout (KO), and Atg7 KO MEFs, HeLa, MCF-7, and Hepa1-6 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (JRH Biosciences), 100 IU/ml penicillin G, and 100 μg/ml streptomycin. Primary MEFs from WT and Atg7 KO mice were isolated from mice at embryonic day 13.5 (E13.5). Splenic single-cell suspensions (splenocytes) were prepared from C57BL/6 mice. All animal experiments were approved by the Animal Research Committee of the Graduate School of Medicine, Osaka University (Osaka, Japan). To establish Fpn tet-on MEFs, the open reading frame (ORF) of human Fpn was amplified by reverse transcriptase (RT)-PCR from HepG2 cell mRNA. The Fpn cDNA with a C-terminal hemagglutinin (HA) tag (Fpn-HA) was subcloned into pTRE2 (Clontech). A single clone that expressed Fpn-HA in a doxycycline (DOX)-dependent manner was selected. For growth under iron-deficient conditions, MEFs or HeLa cells were treated with 25 μM to 100 μM deferoxamine (Dfo) (Desferal; Novartis), 300 μM bathophenanthroline disulfonate (BPS; Sigma), or 30 μM deferasirox (Dfx) (Exjade; Novartis). Cells were treated with 10 μg/ml or 100 μg/ml of ferric ammonium citrate (FAC) to induce iron-replete conditions.
Establishment of Lamp2A KD cells.
Oligonucleotides for knockdown (KD) of Lamp2A were cloned into the pSuper vector (Ambion). The following oligonucleotides were used: #1 sense, 5′-GATCCCCGACTGCAGTGCAGATGAAGTTCAAGAGACTTCATCTGCACTGCAGTCTTTTTGGAAA-3′; #1 antisense, 5′-AGCTTTTCCAAAAAGACTGCAGTGCAGATGAAGTCTCTTGAACTTCATCTGCACTGCAGTCGGG-3′; #2 sense, 5′-GATCCCCCTGCAATCTGATTGATTATTTCAAGAGAATAATCAATCAGATTGCAGTTTTTGGAAA-3′; and #2 antisense, 5′-AGCTTTTCCAAAAACTGCAATCTGATTGATTATTCTCTTGAAATAATCAATCAGATTGCAGGGG-3′. The region targeted by the small interfering RNA (siRNA) was exon 8a of the Lamp2A gene, as described previously (36). These vectors were transfected into Atg5 KO MEFs, and a single clone that effectively knocked down Lamp2A was selected.
Antibodies and reagents.
Affinity-purified anti-horse ferritin, anti-human spleen ferritin, and anti-ferritin H chain were obtained from Sigma, Rockland, and Santa Cruz Biotechnology, respectively. The anti-IRP2 monoclonal antibody (3B11) has been described previously (5). Anti-Atg5 was purchased from Cell Signaling, anti-β-actin and antitubulin were from Santa Cruz Biotechnology, anti-Lamp2A was from Sigma, and anti-HA was from Covance. E64d, pepstatin A, and lactacystin were purchased from the Peptide Institute. DOX, 3-methyladenine (3-MA), and bafilomycin A1 were obtained from WAKO. Cycloheximide (CHX) was purchased from Calbiochem.
siRNA.
Double-stranded siRNA for human Atg5 was prepared by Sigma as follows: sense, 5′-GGAUCAACUAUUUGCCUGATT-3′, and antisense, 5′-UCAGGCAAAUAGUUGAUCCTT-3′. Mission siRNA universal negative control (Sigma) was used as the control siRNA. siRNAs were transfected using RNAiMax (Invitrogen). siRNA and RNAiMax were mixed in Opti-MEM (Invitrogen) and incubated for 20 min at room temperature to allow complex formation. The mixtures were added to wells, and HeLa cells were then plated into the wells.
Immunoblot analysis.
Cells were lysed in lysis buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Complete EDTA-free protease inhibitor; Roche). Nucleus and membrane fractions were removed by centrifugation. Lysates were separated by SDS–PAGE, and proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with the appropriate primary antibody, followed by incubation with a horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG (GE Healthcare). Immunoreactive proteins were visualized using the SuperSignal immunoblotting detection system (Pierce) and an LAS3000 scanning system (Fuji Film).
35S pulse-chase analyses.
Cells were pretreated with 10 μg/ml (MEFs) or 100 μg/ml (HeLa cells) FAC and then cultured for 30 min in methionine- and cysteine-deficient DMEM containing 10% dialyzed fetal bovine serum (FBS), after which they were pulse-labeled with 3.7 MBq/ml of EXPRE35S35S (Perkin Elmer Life Science) for 30 min. After removal of the EXPRE35S35S-containing culture medium, the cells were cultured in DMEM supplemented with 10% FBS for various periods of time. Cell lysates were subjected to immunoprecipitation using antiferritin, and the immune complexes were separated by 14% SDS-PAGE. Radiolabeled proteins were visualized by autoradiography and quantified with a BAS5000 Bioimaging system (Fuji Film).
RNA gel shift assays.
Cells were lysed in an RNA gel shift lysis buffer containing 1% Triton X-100, 25 mM Tris-HCl (pH 7.4), 40 mM KCl, and protease inhibitors (10 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride) on ice. Nuclei and membrane fractions were removed by centrifugation. RNA-gel shift assays were performed as described previously (23). Briefly, 15 μg of cell lysate was incubated with a 32P-labeled IRE probe for 10 min on ice. The mixture was separated by electrophoresis on a 10% nondenaturing polyacrylamide–Tris-borate-EDTA (TBE) gel. Signals were visualized with a BAS5000 Bioimaging system (Fuji Film).
Immunofluorescence microscopy.
Cells were grown on collagen-coated coverslips. After fixation in 2% paraformaldehyde–PBS and permeabilization with 0.1% saponin–PBS, the cells were incubated with the appropriate primary antibody, followed by incubation with an Alexa Fluor 488-conjugated anti-rabbit or anti-mouse secondary antibody (Molecular Probes). To visualize lysosomes, cells were stained with LysoTracker Red (Molecular Probes) for 1.5 h. To detect the lysosomal marker Lamp1, cells on coverslips were incubated with anti-Lamp1 (Santa Cruz Biotechnology), followed by incubation with an Alexa Fluor 647-conjugated anti-rat secondary antibody (Molecular Probes). Images were obtained using a confocal microscope (FV1000; Olympus).
Iron staining.
Cells were fixed with 2% paraformaldehyde–PBS, incubated in Perl's solution [1% K4Fe(CN)6 and 1% HCl] for 30 min, and then incubated with 0.05% diaminobenzidine (DAB) for 15 min. Hydrogen peroxide (final concentration, 1%) was added, and the cells were incubated for an additional 30 min. For coimmunostaining, cells were first stained with Perl's solution and DAB and then incubated with anti-Lamp1, followed by incubation with an Alexa Fluor 647-conjugated anti-rat secondary antibody.
TEM-EDX.
Transmission electron microscopy–energy-dispersive X-ray (TEM-EDX) analysis was performed by Tokai Electron Microscopy. Cells were cultured on molybdenum grids (Nissin EM), and samples were prepared for microscopy by rapid-freezing and freeze-substitution fixation. Cells were rapidly frozen in liquid propane and then exposed to acetone containing 2.5% distilled water at −80°C for 1 day. Samples were dehydrated through anhydrous acetone and then infiltrated with LR White resin (London Resin). Polymerization was carried out at 50°C for 24 h. Ultrathin sections of uniform thickness were prepared using an ultramicrotome (2088 Ultrotome V; LKB) equipped with a diamond knife. Sections were mounted on copper grids and stained with 2% uranyl acetate and lead staining solution (Sigma). TEM-EDX analysis was performed using a transmission electron microscope (JEM-2010; JEOL) equipped with an EDX analyzer (NORAN Voyager III; NORAN Instruments) at an accelerating voltage of 200 kV. The electron beam was irradiated for 100 s, and elemental peak data were analyzed with an EDX analyzer.
RESULTS
Ferritin produced under iron-rich conditions is degraded in the lysosome under iron-depleted conditions.
Utilization of iron stored in the ferritin shell has been proposed as a cellular response to iron-depleted conditions (4). To investigate whether degradation of ferritin is involved in the utilization of ferritin iron stores, we examined whether ferritin synthesized under iron-rich conditions was degraded when cells were shifted to iron-depleted conditions. Immortalized MEFs were pretreated with FAC to induce ferritin synthesis and then cultured in the presence of the iron chelator Dfo. The levels of ferritin decreased under iron-depleted conditions (Fig. 1 A). To confirm that the decrease of ferritin in response to Dfo was the result of protein degradation, ferritin levels were monitored by pulse-chase analysis. MEFs were pretreated with FAC before being pulse-labeled with [35S]methionine and [35S]cysteine, and time-dependent changes in the level of 35S-labeled ferritin were assessed in the presence of Dfo. Ferritin was degraded in Dfo-treated MEF cells (Fig. 1B). Furthermore, this decrease in ferritin was suppressed by the lysosomal protease inhibitors E64d and pepstatin A (E64d-pepstatin A) but not by the proteasome-specific inhibitor lactacystin (Fig. 1C). These results indicate that the ferritin produced under iron-rich conditions is degraded via lysosomal degradation in Dfo-treated cells.
Fig. 1.

Ferritin is degraded via the lysosome under iron-starved conditions. (A) Decreased levels of ferritin in Dfo-treated MEFs. MEFs pretreated with FAC were cultured with Dfo for the indicated times. Cell lysates were analyzed by immunoblotting (IB) with antiferritin or anti-β-actin. (B) Ferritin is degraded in Dfo-treated cells. MEFs pretreated with FAC were pulse-labeled with EXPRE35S35S in the presence of Dfo. Cells were cultured in medium containing Dfo and harvested at the indicated times. Antiferritin immunoprecipitates were visualized with a BAS 5000 imager. The upper band is the ferritin L chain (L), and the lower band is the ferritin H chain (H). The amount of ferritin L chain was quantified. (C, D) Ferritin is degraded in the lysosome in MEFs treated with iron chelators. MEFs pretreated with FAC were cultured in the presence of indicated inhibitor(s) or dimethyl sulfoxide (DMSO) as the vehicle control, together with Dfo (C) or Dfx or BPS (D) for 6 h. Cell lysates were analyzed as described for panel A. (E) DOX-induced expression of Fpn-HA. Fpn tet-on MEFs were incubated in the presence or absence of DOX, followed by immunostaining with anti-HA. Scale bar, 10 μm. Dic, differential interference contrast image. (F) Ferritin is degraded in the lysosome under Fpn-induced iron-deficient conditions. Fpn tet-on MEFs pretreated with FAC were cultured in the presence or absence of DOX for the indicated periods. Cell lysates were probed as described for panel A.
Iron depletion of MEFs was induced by various mechanisms, and ferritin levels were then examined under each set of conditions. After pretreatment with FAC, iron starvation was induced by treatment with Dfx, a membrane-permeable iron chelator, or BPS, a membrane-impermeable iron chelator, in the presence or absence of protease inhibitors (Fig. 1D). Ferritin levels were reduced in Dfx- and BPS-treated cells in the absence of protease inhibitors; however, cotreatment with E64d-pepstatin A but not lactacystin inhibited the Dfx- and BPS-induced decreases in ferritin. Ferritin levels were examined in MEFs engineered to express the iron exporter ferroportin (Fpn tet-on MEFs) in response to DOX. We first confirmed that Fpn was expressed on the plasma membrane in the presence of DOX (Fig. 1E). Under these conditions, the level of ferritin decreased in the absence of protease inhibitors, and the addition of E64d-pepstatin A but not lactacystin inhibited the decrease in ferritin (Fig. 1F). These results indicate that ferritin is degraded primarily in the lysosome under conditions of Fpn-induced iron deficiency. Thus, ferritin synthesized under iron-replete conditions is subsequently degraded via the lysosome under iron-starved conditions in MEFs.
Role of autophagy in lysosomal targeting and degradation of ferritin in iron-depleted MEFs.
Autophagy is a process whereby cytosolic components are delivered to the lysosome for degradation (38). To assess the role of autophagy in lysosomal targeting of ferritin under iron-starved conditions, MEFs were first pretreated with FAC and then cultured in the presence of Dfo (Fig. 2 A), in the presence or absence of 3-MA, an inhibitor of autophagy. The level of ferritin was drastically reduced in cells treated with Dfo alone, and the addition of 3-MA suppressed the Dfo-mediated decrease in ferritin. Since 3-MA is not autophagy specific (39), we also examined the lysosomal targeting and degradation of ferritin in autophagy-deficient MEFs. Atg5 has been implicated in the early stages of autophagosome formation, and virtually no autophagy is observed in Atg5 knockout (KO) MEFs (30). Atg5 KO and WT MEFs were pretreated with FAC, and the levels of ferritin were then assessed over time in the presence of Dfo. The Dfo-mediated decrease of ferritin was markedly attenuated in Atg5 KO MEFs (Fig. 2B). Similar results were observed for Atg5 KO MEFs treated with Dfx and BPS (Fig. 2C and D). To confirm that autophagy was involved in the degradation of ferritin under iron-starved conditions, we repeated the pulse-chase analysis of ferritin levels using Atg5 KO and WT MEFs (Fig. 2E). The degradation of ferritin under iron-starved conditions was strongly attenuated in Atg5 KO MEF cells compared to the level of degradation in WT MEFs. Dfo-mediated degradation was also markedly attenuated in MEF cells lacking Atg7, in which autophagosome formation and autophagy are almost completely abolished (29) (Fig. 2F). These results clearly indicate that autophagy plays an important role in the lysosomal targeting of ferritin in iron-starved MEFs.
Fig. 2.

Autophagy plays a role in lysosomal ferritin degradation in iron-deficient MEFs. (A) Suppression of Dfo-induced ferritin degradation by 3-MA. MEFs pretreated with FAC were cultured in Dfo in the presence or absence of 3-MA for the indicated times. (B to D) The reduction in ferritin in response to iron chelators is suppressed in autophagy-deficient cells. WT or Atg5 KO MEF cells pretreated with FAC were cultured in the presence of Dfo (B), Dfx (C), or BPS (D) for the indicated times. For the experiments whose results are shown in panels A to D, cell lysates were probed as described in the legend to Fig. 1A. (E, F) Attenuation of Dfo-induced ferritin degradation in autophagy-deficient MEFs. WT and Atg5 KO (E) or Atg7 KO (F) MEFs were pulse-labeled as described in the legend to Fig. 1B. The amounts of ferritin L chain (upper bands) in WT and Atg5 KO (E) or Atg7 KO (F) MEFs were quantified. Error bars show standard deviations. *, P < 0.05.
Lysosomal acidification is crucial for the extraction of iron from ferritin.
The degradation of ferritin was delayed in autophagy-deficient MEFs. We next probed whether lysosomal degradation of ferritin was involved in the utilization of ferritin-stored iron. The binding of IRP1 and IRP2 to IREs is induced by iron depletion (43, 47), and IRE-binding activity is regarded as a sensor for iron availability in cells (27). Atg7 KO MEFs were pretreated with FAC to allow iron to accumulate. Cells were then treated with Dfo, and the IRE-binding activities of IRPs were assessed by RNA gel shift assays (Fig. 3 A). The IRE-binding activity of IRP1 and IRP2 was induced in a time-dependent manner in WT and Atg7 KO MEFs in response to Dfo. However, the induction of IRE-binding activity of both IRPs was much more rapid in Atg7 KO cells than in WT cells. Since IRP2 is regulated via iron-dependent degradation and the IRE-binding activity of IRP2 is correlated with the amount of the protein, IRP2 is also a sensor for cellular iron availability (22, 23). Immunoblot analysis confirmed that, after the shift to Dfo, the amount of IRP2 increased rapidly in Atg7 KO MEFs compared to the levels in WT MEFs (Fig. 3B). These results strongly indicate that iron is extracted from ferritin in the lysosome and that the extracted iron is used by MEFs under iron-starved conditions.
Fig. 3.

Lysosomal acidification is crucial for the extraction of ferritin iron. (A, B) Accelerated IRE binding by IRP1 and IRP2 (A), and IRP2 levels (B) in Atg7 KO MEFs. WT and Atg7 KO MEFs pretreated with FAC were cultured in the presence of Dfo for the indicated times. The IRE-binding activities of IRP1 and IRP2 (A) and the levels of IRP2 (B) were assessed. The amount of IRP2 is quantified (B). (C) Stabilization of ferritin in cells treated with inhibitors of lysosomal proteases. MEFs pretreated with FAC were cultured in the presence of the indicated inhibitor(s) or DMSO for the indicated times. Cell lysates were probed as described in the legend to Fig. 1A. pep A, pepstatin A; baf A, bafilomycin A1. (D) Localization of ferritin to lysosomes in cells treated with lysosomal inhibitors. MEFs pretreated with FAC were cultured as described for panel C for 4 h and coimmunostained with antiferritin and anti-Lamp1. Higher-magnification views of the boxed areas are shown in the insets. Scale bar, 10 μm. (E) Accelerated accumulation of IRP2 in MEFs treated with bafilomycin A1. MEFs cultured as described for panel C were probed as described for panel B. The amounts of IRP2 were quantified. (F, G) Colocalization of iron and Lamp1 in MEFs treated with E64d-pepstatin A, bafilomycin A1, or bafilomycin A1 and E64d-pepstatin A. MEFs pretreated with FAC were cultured in the presence of the indicated inhibitor(s) and BPS for 4 h. Cells were stained with Perl's solution and DAB, followed by immunostaining with anti-Lamp1. Scale bar, 10 μm. (F). Iron-positive lysosomes were counted in 10 E64d-pepstatin A-treated cells, 11 bafilomycin A1-treated cells, and 11 E64d-pepstatin A- and bafilomycin A1-treated cells. The percentages of iron-positive lysosomes are shown. Error bars show standard deviations. *, P < 0.01 (G). (H, I) Accumulation of iron in lysosomes in bafilomycin A1-treated MEFs. (H) MEFs were treated as described for panel F, and EDX spectra were recorded at 200 kV. Representative spectra of selected lysosomes (inset, marked by a circle) are shown. Scale bar, 500 nm. Absorption peaks are labeled with symbols of the elements detected. (I) The intensities of Fe(Kα) peaks in nine lysosomes from E64d-pepstatin A- or eight lysosomes from bafilomycin A1-treated cells are shown. Horizontal bars show the means. *, P < 0.05. (J) Increased pH in bafilomycin A1-treated MEFs. MEFs were treated as described for panel F and incubated with LysoTracker Red for the last 1.5 h of incubation, followed by immunostaining with anti-Lamp1. Higher-magnification views of the boxed areas are shown in the insets. Scale bar, 10 μm.
To address the mechanism of iron extraction from ferritin in the lysosome, WT MEFs were pretreated with FAC and then treated with either E64d-pepstatin A or bafilomycin A1 in the presence of an iron chelator (BPS) for various periods of time (Fig. 3C). Ferritin levels decreased in the presence of BPS, and this BPS-induced decrease in ferritin was suppressed in the presence of E64d-pepstatin A or bafilomycin A1. Immunofluorescence staining showed the absence of ferritin in control cells (without protease inhibitor), whereas ferritin colocalized with Lamp1 in cells treated with E64d-pepstatin A or bafilomycin A1 (Fig. 3D). These results suggest that lysosomal degradation of ferritin is suppressed in the presence of E64d-pepstatin A or bafilomycin A1. As an indicator of iron status, we evaluated changes in cellular IRP2 levels over time (Fig. 3E). The level of IRP2 in MEF cells treated with E64d-pepstatin A was similar to the level in control cells. In contrast, IRP2 was rapidly induced in cells treated with bafilomycin A1. These results suggest that bafilomycin A1 but not E64d-pepstatin A attenuates iron release from lysosomes, although ferritin degradation was inhibited to a similar extent by both treatments.
Bafilomycin A1 also inhibits the acidification of endosomes and suppresses iron uptake by cells. In iron-replete cells, bafilomycin A1 blocks the iron-dependent degradation of IRP2 (data not shown) (22, 23). To determine whether bafilomycin A1 inhibited the utilization of ferritin iron stores, the presence of iron in lysosomes was probed by Perl's-DAB staining (Fig. 3F and G). The accumulation of ferric iron was more pronounced in bafilomycin A1-treated cells than in E64d-pepstatin A-treated or control cells. Cotreatment with bafilomycin A1 and E64d-pepstatin A did not further enhance ferric iron accumulation in lysosomes in comparison to bafilomycin A1 treatment alone (Fig. 3G). Furthermore, Perl's-DAB-positive signals in bafilomycin A1-treated, as well as in bafilomycin A1- and E64d-pepstatin A-treated cells, overlapped with the lysosomal protein Lamp1 (Fig. 3F). To confirm this apparent accumulation of iron in the lysosomes of bafilomycin A1-treated cells, we used transmission electron microscopy–energy-dispersive X-ray (TEM-EDX) analysis (Fig. 3H). Electron-dense particles were clearly observed in the lysosomes of bafilomycin A1-treated cells but not E64d-pepstatin A-treated MEFs. The structures of the lysosomal membranes in each cell were not as sharp in the TEM-EDX images as in standard TEM due to differences in tissue fixation. However, the EDX energy spectra confirmed that iron was more highly concentrated in the lysosomes in bafilomycin A1-treated MEFs than in E64d-pepstatin A-treated MEFs (Fig. 3I). The acidic organelle-sensitive probe LysoTracker confirmed that bafilomycin A1 raised the lysosomal pH in MEFs (Fig. 3J). These results strongly indicate that it is the acidic environment and not the degradation of ferritin per se that is crucial for iron extraction from ferritin in the lysosome.
Ferritin is degraded in lysosomes even under iron-replete conditions in MEFs.
Ferritin was targeted to and degraded in lysosomes when cells were starved of iron, although ferritin degradation was dispensable for the utilization of iron stores. It has been hypothesized that ferritin is degraded under iron-deficient conditions but not iron-replete conditions due to the presence of an adequate supply of iron (46). The amount of ferritin did not decrease in iron-replete MEFs (Fig. 4A). To investigate ferritin turnover under iron-rich conditions, we conducted a pulse-chase analysis of ferritin in iron-replete MEFs. Ferritin was degraded in FAC-treated cells as efficiently as in Dfo-treated cells (Fig. 4B). Cycloheximide (CHX) decay analysis revealed that the degradation of ferritin could be blocked by E64d-pepstatin A but not by lactacystin under iron-rich conditions (Fig. 4C), similar to the results under iron-depleted conditions (Fig. 1C). The results of immunofluorescence staining confirmed that ferritin was localized to the lysosome in E64d-pepstatin A-treated MEFs under the iron-replete conditions (Fig. 4D). These results clearly indicate that ferritin is degraded in the lysosome under both iron-replete and iron-depleted conditions in MEFs. However, while lysosomal targeting of ferritin in iron-depleted cells was mediated mainly by autophagy (Fig. 2), degradation of ferritin was not significantly affected in autophagy-defective Atg5 KO or Atg7 KO MEFs under iron-replete conditions (Fig. 4E and F). Cytosolic components can be delivered to the lysosome by several transport pathways in addition to autophagy. Chaperone-mediated autophagy (CMA) delivers cytoplasmic components to lysosomes by recognition of the lysosomal membrane receptor Lamp2A in a chaperone-dependent manner (11). To probe whether CMA is involved in ferritin degradation under iron-replete conditions, we knocked down Lamp2A in Atg5 KO MEFs. The level of expression of Lamp2A in knockdown (KD) cells was less than ∼15% of that in control cells (Fig. 4G). CHX decay analysis revealed that the degradation of ferritin under iron-rich conditions was not affected by KD of Lamp2A (Fig. 4H), which suggests that CMA is not involved in ferritin degradation. However, 3-MA significantly suppressed the degradation of ferritin in iron-replete MEFs (Fig. 4I). These results suggest that ferritin is transported to and degraded in the lysosomes regardless of the iron status of the cells. The mechanism of lysosomal targeting of ferritin differed between cells under iron-depleted and iron-replete conditions in that autophagy was a major pathway of lysosomal targeting of ferritin in iron-depleted MEFs, whereas ferritin was targeted to the lysosome by a 3-MA-sensitive pathway that was independent of autophagy and CMA in iron-replete cells.
Fig. 4.

Ferritin is degraded in the lysosome in MEFs under iron-replete conditions. (A) Ferritin is not decreased in iron-replete MEFs. MEFs pretreated with FAC were maintained with FAC for the indicated periods. (B) Ferritin in MEFs under iron-replete conditions is degraded at a rate comparable to its rate of degradation in iron-depleted MEFs. MEFs pretreated with FAC were pulse-labeled in the presence of Dfo or FAC. Antiferritin immunoprecipitates from radiolabeled lysates were quantified using a BAS 5000 imager. The amount of ferritin L chain under each condition is presented in the lower panel. (C) Ferritin is degraded in the lysosome under iron-replete conditions. MEFs pretreated with FAC for 16 h were treated with CHX and the indicated inhibitor(s) in the presence of FAC for 6 h. (D) Colocalization of ferritin with Lamp1 in cells treated with lysosomal inhibitors. MEFs were treated with FAC and E64d-pepstatin A or DMSO or left untreated for 6 h. Cells were stained with antiferritin and anti-Lamp1. Higher-magnification views of the boxed areas are shown in the insets. Scale bar, 10 μm. (E, F) Ferritin degradation is not delayed in autophagy-deficient MEFs under iron-rich conditions. WT and Atg5 KO (E) or Atg7 KO (F) MEFs were pulse-labeled as described for panel B. The amounts of ferritin L chain in WT and Atg5 KO (E) or Atg7 KO (F) MEFs are shown in the lower panel. (G) Establishment of Lamp2A KD Atg5 KO MEFs. The amounts of Lamp2A and β-actin in control and Lamp2A KD cells were assessed. (H) Ferritin degradation is not delayed in Lamp2A KD Atg5 KO MEFs under iron-replete conditions. MEFs pretreated with FAC were cultured in the presence of FAC and CHX for the indicated times. (I) 3-MA attenuated ferritin degradation under iron-replete conditions. MEFs pretreated with FAC were incubated with FAC and CHX in the presence or absence of 3-MA for the indicated times. For the experiments whose results are shown in panels A, C, H, and I, cell lysates were probed as described in the legend to Fig. 1A. Error bars show standard deviations.
In primary cells, ferritin is degraded in the lysosome regardless of the iron status.
The MEFs used in this study were established from C57BL/6 mice by transformation with the simian virus 40 (SV40) large T (LT) antigen. To determine whether ferritin turnover was similarly regulated in primary cells, iron-dependent changes in the amount of ferritin in primary MEFs were assessed (Fig. 5A). The levels of ferritin decreased in a time-dependent manner in iron-depleted primary MEFs but not in iron-replete cells, similar to the results for the transformed MEFs described above. Pulse-chase analysis revealed that ferritin is degraded in FAC-treated primary MEFs as efficiently as in Dfo-treated MEFs (Fig. 5B). The degradation of ferritin was suppressed by the addition of E64d-pepstatin A but not lactacystin in both Dfo- and FAC-treated primary MEFs (Fig. 5C and D). Ferritin was also degraded in the presence of Dfo and FAC in primary mouse splenocytes (Fig. 5E and F). These results demonstrate that ferritin is targeted to the lysosome for degradation regardless of the iron status in primary cells as well as in immortalized MEFs. We also assessed the role of autophagy in lysosomal targeting of ferritin in primary MEFs from Atg7 KO mice. The degradation of ferritin was delayed in primary Atg7 KO MEFs under iron-depleted conditions (Fig. 5G). However, under iron-replete conditions, ferritin was degraded as effectively as in primary MEFs from WT mice (Fig. 5H). Treatment with 3-MA suppressed this decrease of ferritin under iron-replete conditions (Fig. 5I). These results confirmed that ferritin is targeted to and degraded in the lysosomes in primary MEFs regardless of the iron status of the cells and that autophagy plays a role in lysosomal targeting under iron-deficient conditions. Similarly, in primary cells, the lysosomal targeting of ferritin under iron-rich conditions was distinct from that under iron-depleted conditions. Thus, there are two pathways of lysosomal targeting and degradation of ferritin depending on the iron status of the cell. Under iron-depleted conditions, targeting is mediated primarily by autophagy, whereas under iron-rich conditions, targeting occurs predominantly via a 3-MA-sensitive pathway that is independent of autophagy.
Fig. 5.

In primary cells, ferritin is degraded in the lysosome regardless of the iron status. (A) Decreased levels of ferritin in Dfo- but not FAC-treated primary MEFs. Primary MEFs pretreated with FAC were cultured in the presence of Dfo or FAC for the indicated times. (B) Ferritin is degraded under both iron-replete and iron-depleted conditions in primary MEFs. Primary MEFs were pulse-labeled, and antiferritin immunoprecipitates from labeled cells were quantified using a BAS 5000 imager. The amounts of ferritin L chain are shown in the lower panel. (C, D) Ferritin is degraded in the lysosome under both iron-depleted (C) and iron-replete (D) conditions in primary MEFs. MEFs pretreated with FAC were cultured in the presence of Dfo (C) or FAC and CHX (D) together with the indicated inhibitor(s). (E) Decreased levels of ferritin in Dfo- but not FAC-treated splenocytes. Primary splenocytes pretreated with FAC were cultured in the presence of Dfo or FAC for the indicated times. (F) Ferritin is degraded under both iron-replete and iron-depleted conditions in splenocytes. Primary splenocytes pretreated with FAC were cultured in the presence of Dfo or FAC and CHX for the indicated times. (G, H) Ferritin degradation is delayed in primary Atg7 KO MEFs under iron-depleted but not iron-replete conditions. Primary WT or Atg7 KO MEFs were cultured as described for panel E or F for the indicated times. (I) 3-MA attenuated ferritin degradation under iron-replete conditions in primary MEFs. Primary WT MEFs were cultured as described for panel F in the presence or absence of 3-MA. For the experiments whose results are shown in panels A and C to I, cell lysates were probed as described in the legend to Fig. 1A.
Ferritin is not degraded under iron-rich conditions in cancer-derived cell lines.
In transformed and primary MEFs, ferritin was degraded regardless of the iron status of the cell. In contrast, the results of previous studies suggest that ferritin is degraded under iron-deficient conditions but not iron-replete conditions (46). To investigate whether the degradation of ferritin under iron-replete conditions was cell-type specific, we examined ferritin turnover in the HeLa cervical cancer cell line. HeLa cells were pretreated with FAC to increase ferritin synthesis and then cultured under iron-depleted or iron-replete conditions (Fig. 6A). The level of ferritin decreased in the presence of Dfo but not in the presence of FAC, as was observed in transformed MEFs and primary cells. We confirmed that ferritin was degraded in the lysosome in iron-depleted HeLa cells, because ferritin degradation was blocked by E64d-pepstatin A but not lactacystin (Fig. 6B). However, pulse-chase analysis in HeLa cells revealed that ferritin was degraded in the presence of Dfo, as observed in MEFs, whereas ferritin degradation was barely detectable in the presence of FAC (Fig. 6C). This result prompted us to examine whether ferritin is degraded under iron-replete conditions in other cancer-derived cell lines. Ferritin was degraded under iron-starved conditions but was stable under iron-replete conditions in a human breast cancer cell line, MCF-7 (Fig. 6D and E), and in a mouse hepatoma cell line, Hepa1-6 (Fig. 6F and G). These results suggest that ferritin is degraded only under iron-depleted conditions in these cancer-derived cell lines. Ferritin was delivered to the lysosome for degradation by autophagy in iron-starved primary cells (Fig. 2 and 5). In HeLa cells, the Dfo-mediated decrease in ferritin was suppressed by an inhibitor of autophagy, 3-MA (Fig. 6H). Since 3-MA also suppressed ferritin degradation in iron-replete MEFs, the involvement of autophagy in Dfo-mediated degradation of ferritin was examined by knocking down Atg5 in HeLa cells. Dfo-mediated degradation of ferritin was indeed suppressed in Atg5 KD HeLa cells (Fig. 6I). These results clearly indicate that ferritin turnover is differentially regulated in primary cells and cancer-derived cells. Ferritin was degraded under iron-depleted conditions in both primary and cancer-derived cells, mediated primarily by autophagy. However, ferritin was also degraded under iron-replete conditions in primary cells but not in cancer-derived cells.
Fig. 6.

Ferritin turnover in cancer-derived cell lines. (A) Decreased levels of ferritin in Dfo- but not FAC-treated HeLa cells. HeLa cells were cultured as described in the legend to Fig. 5A. (B) Ferritin is degraded in the lysosome in iron-depleted HeLa cells. HeLa cells pretreated with FAC were cultured in the presence of the indicated inhibitor(s) or DMSO, together with Dfo. (C) Ferritin is degraded under iron-depleted but not iron-replete conditions in HeLa cells. HeLa cells were pulse-labeled, and antiferritin immunoprecipitates from radiolabeled lysates were quantified using a BAS 5000 imager. The amounts of ferritin are shown in the lower panel. Error bars show standard deviations. *, P < 0.05. (D) Decreased levels of ferritin in Dfo- but not FAC-treated MCF-7 cells. MCF-7 cells were cultured as described in the legend to Fig. 5E. (E) Ferritin is degraded under iron-depleted but not iron-replete conditions in MCF-7 cells. MCF-7 cells were cultured as described in the legend to Fig. 5F. (F) Decreased levels of ferritin in Dfo- but not FAC-treated Hepa1-6 cells. Hepa1-6 cells were cultured as described in the legend to Fig. 5E. (G) Ferritin is degraded under iron-depleted but not iron-replete conditions in Hepa1-6 cells. Hepa1-6 cells were cultured as described in the legend to Fig. 5F. (H) Suppression of Dfo-induced ferritin degradation by 3-MA. HeLa cells pretreated with FAC were cultured in the presence or absence of 3-MA together with Dfo for the indicated times. For the experiments whose results are shown in panels A, B, and D to H, cell lysates were probed as described in the legend to Fig. 1A. (I) Ferritin degradation is delayed by Atg5 KD in HeLa cells under iron-depleted conditions. HeLa cells transfected with control or Atg5 siRNA were pretreated with FAC and cultured in the presence of Dfo, followed by immunoblotting with antiferritin, anti-Atg5, or anti-β-actin.
Difference in iron susceptibility between primary and cancer-derived cells.
Ferritin is degraded under iron-rich conditions in primary cells but not in cancer-derived cells. We examined the differences in iron toxicity between these cell types. HeLa, MCF-7, Hepa1-6, transformed MEF, and primary MEF cells were cultured in the presence or absence of 100 μg/ml FAC for 48 h, and the relative numbers of viable cells were assessed. As shown in Fig. 7A, the numbers of iron-treated cancer-derived cells (HeLa, MCF-7, and Hepa1-6 cells) were almost identical to the numbers of control cells. In contrast, the numbers of iron-treated transformed and primary MEFs were much lower than the numbers of cells cultured in the absence of iron. The decrease in cell number may be attributed to iron-induced cytotoxicity (Fig. 7B). These results suggest that the loss of ferritin degradation under iron-replete conditions has protective effects against iron toxicity.
Fig. 7.

Differences in iron susceptibility between primary and cancer-derived cells. Cells were cultured in the presence or absence of FAC for 48 h. (A) Relative cell numbers are the ratios of the cell numbers in the presence of FAC to the cell numbers in the absence of FAC. Error bars show standard deviations. (B) Cells cultured in the presence or absence of FAC for 48 h are shown.
DISCUSSION
While iron is an essential nutrient, it is also toxic to cells. In mammalian cells, excess iron is stored in the cytosolic iron storage protein ferritin as a mechanism of protection from iron toxicity (21). Iron stored in ferritin is redox inactive, which mitigates its toxic effects on cellular components (4). Under conditions of iron starvation, ferritin iron stores are extracted and used by cells for a wide variety of biological processes (4). In the present study, we showed that iron is extracted from ferritin after the protein is delivered to the lysosome in iron-starved MEFs and that the acidic environment of the lysosome, not protein degradation in the organelle, plays a crucial role in iron extraction from ferritin.
Several groups have reported that lysosomal degradation of ferritin is crucial for the utilization of ferritin iron stores in a number of different settings (25, 33, 42) and plays a central role in iron extraction from ferritin. In the current study, bafilomycin A1, a vacuolar ATPase inhibitor that increases lysosomal pH, inhibited iron release from lysosomes more profoundly than lysosomal protease inhibitors (Fig. 3F). The activity of lysosomal proteases is sensitive to pH (49), which may explain why ferritin degradation was inhibited to a similar extent by bafilomycin A1 and the protease inhibitors (Fig. 3C and D). These results suggest that the degradation of ferritin per se may not be sufficient for iron release from the lysosome and that an acidified environment is more crucial than protein degradation for iron extraction from ferritin. The importance of an acidic environment in the lysosome may be attributed to the increased solubility of ferric ion at low pH. The solubility of ferric iron is very low at neutral pH (∼10−18 M at pH 7.0) (9); ferric iron is more soluble in low-pH than in high-pH environments (18). Thus, the acidic environment of the lysosome may affect the solubilization of ferric iron from mineralized iron in ferritin (31). Alternatively, low pH may be critical for iron release from the lysosome but not from the ferritin shell. Previous studies have reported that ferric iron delivered via transferrin is reduced to ferrous iron (Fe2+) by the ferric reductase Steap3 and is then transported into the cytosol from the endosome via the ferrous transporter DMT1 (37, 40). A similar mechanism may exist for iron transport from lysosomes into the cytosol, wherein the activity of a ferric reductase or ferrous transporter may be inhibited at high pH.
When an iron-depleted state was induced in MEFs through several different pathways, including overexpression of the Fpn iron exporter and treatment with the membrane-permeable chelator Dfx, ferritin was degraded in the lysosome in response to each treatment (Fig. 1D and F). De Domenico et al. reported that ferritin is degraded in the lysosome in cells treated with Dfo, which is only weakly membrane permeant, and accumulates in lysosomes (14). In contrast to our results, they also reported that ferritin is degraded by the proteasome in the cytosol under conditions of iron depletion induced either by Fpn overexpression or treatment with Dfx. In addition, iron extraction from ferritin precedes proteasomal degradation of the protein (13). The reasons for these discrepancies are not clear, but they may be due to differences in the cell types used in each study. We used primary and immortalized MEFs in the present study, whereas De Domenico and colleagues carried out their studies using HEK293T cells. An alternative explanation may be the presence of a mammalian siderophore, the existence of which was recently demonstrated (6, 15). The ferritin pore structure and rates of ferritin iron release are regulated by iron chelators in vitro (35). Knockdown of Bdh2, an enzyme that is necessary for the synthesis of a mammalian siderophore, resulted in cytosolic iron accumulation and increased ferritin levels, which suggests that the action of a mammalian siderophore may be a key factor in the extraction of ferritin iron stores. Additionally, the site of siderophore-mediated iron extraction may be the cytosol. HEK293T cells are a kidney-derived cell line, and the expression of Bdh2 is much higher in the kidney than in any other organ (15). Thus, variable levels of siderophore could influence the dynamics of ferritin turnover in different settings and in different cells. An additional possibility relates to the type of lysosomal inhibitor used to examine ferritin turnover. De Domenico et al. used chloroquine to inhibit lysosomal proteases (13, 14), whereas in the current study, we used E64d-pepstatin A. Chloroquine is a weak base, and we have observed that chloroquine also inhibits the degradation of IRP2, as does bafilomycin A1, even when excess iron is added to the culture medium (data not shown). Chloroquine, however, does not inhibit the proteasomal degradation of IκBα in tumor necrosis factor alpha (TNF-α)-stimulated cells (8; data not shown). These results suggest that chloroquine may inhibit iron uptake into cells, since it counteracts endosomal acidification, which is critical for the release of iron from transferrin (12). Thus, the use of chloroquine may have suppressed iron uptake from the extracellular environment, which may have affected the results of De Domenico and colleagues.
Our observation that ferritin is delivered to the lysosome even under iron-rich conditions in primary cells may provide mechanistic insights into the generation of hemosiderin. Hemosiderin was initially described histologically as an insoluble iron-rich granule that localized to membranous compartments (34). The protein components of the hemosiderin complex are poorly characterized but may include apoferritin and degradation products of apoferritin. Although the process of hemosiderin formation remains poorly defined, it has been hypothesized that hemosiderin is derived from intralysosomal aggregation and the degradation of ferritin (9). The level of hemosiderin in cells is clearly higher under conditions of iron overload, including hemochromatosis (34). In the current study, while ferritin was targeted to and degraded in the lysosome, even under iron-rich conditions (Fig. 4B), not all of the ferritin in the cell was rapidly degraded and partially degraded products of ferritin were observed (Fig. 8). Under conditions of iron overload, it is possible that the lysosomes are unable to completely digest ferritin and efficiently export iron to the cytosol. Partial degradation of ferritin has been observed when ferritin is isolated from iron-loaded rat livers (3). Given that the acidic environment of the lysosome was crucial for iron extraction from ferritin, as well as ferritin degradation, an increased pH in the lysosome may be associated with hemosiderin accumulation. Further biochemical studies should help to elucidate the precise mechanism of hemosiderin production.
Fig. 8.
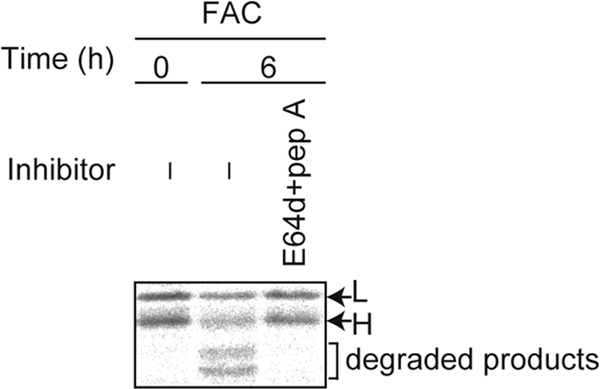
Ferritin is not completely digested in the lysosome. MEFs were pulse-labeled, and antiferritin immunoprecipitates from radiolabeled lysates were quantified using a BAS 5000 imager.
When starved of iron, cells utilize ferritin iron stores via a mechanism that involves the release of iron from ferritin subsequent to the targeting of the protein shell to lysosomes. We have shown that the ferritin produced under iron-rich conditions was targeted to the lysosome for degradation and that autophagy played a major role in the lysosomal targeting of ferritin under iron-starved conditions. Lysosomal targeting seems to be crucial for iron release from ferritin, because autophagy-deficient cells became iron-deficient more readily than WT MEFs (Fig. 3A). Since ferritin production is suppressed under iron-depleted conditions (45), iron released from the lysosome into the cytosol, possibly through a lysosomal iron exporter, such as TRPML1 (16), is available for use by the cell (Fig. 9A).
Fig. 9.

Proposed mechanism for the fate of iron extracted from ferritin in the lysosome. (A) Ferritin is targeted to the lysosome by autophagy under iron-depleted conditions in primary cells. Iron released from ferritin in the lysosome is transported to the cytoplasm and used by cells because ferritin production is suppressed under iron-depleted conditions in primary cells. This may also be the case in cancer-derived cells. (B) Ferritin is targeted to the lysosome by an autophagy-independent pathway that is 3-MA sensitive. In primary cells, iron released from ferritin in the lysosome is transported to the cytoplasm and sequestered in newly synthesized ferritin to prevent iron toxicity under iron-replete conditions. In cancer-derived cells, ferritin is not degraded, and excess iron is stably and safely stored by ferritin under iron-rich conditions.
Ferritin was delivered to the lysosome under iron-replete conditions in primary and transformed MEF cells (Fig. 2 and 5), whereas in cancer-derived cells, ferritin is not degraded under iron-replete conditions (Fig. 6). In primary cells, iron is not stably sequestered in ferritin and iron is continuously released from the lysosome into the cytosol, which enables cells to utilize iron readily when they become iron deficient (Fig. 9B, Primary cells). In contrast, cancer-derived cells may not readily access ferritin-stored iron, since ferritin is not degraded under iron-rich conditions (Fig. 9B, Cancer-derived cells). Therefore, the degradation of ferritin under iron-rich conditions may be a mechanism for fine-tuning iron availability, which may be beneficial for primary cells.
We also observed that cancer-derived cells are resistant to iron toxicity, although primary cells are sensitive (Fig. 7). In primary cells, iron is continuously released from ferritin and must be sequestered into newly synthesized ferritin to protect the cells from iron toxicity. Thus, under high-iron conditions, primary cells may not be able to sequester excess iron completely and excess iron which cannot be stored in ferritin may provoke iron toxicity. However, since excess iron is tightly sequestered in cancer-derived cells, the amount of excess iron is minimal even under high-iron conditions and cancer-derived cells are protected from iron toxicity. Therefore, loss of ferritin delivery to the lysosome under iron-rich conditions may protect against cell death induced by iron-induced oxidative insults. We have shown that the delivery of ferritin to the lysosome under iron-rich conditions was mediated by an autophagy-independent pathway (Fig. 4I). In addition to autophagy (macroautophagy), several putative transport pathways for the delivery of cytosolic components to the lysosome have been reported, including CMA and microautophagy (10). Lamp2A is the lysosomal membrane receptor for CMA (11). CMA is not involved in ferritin degradation by knocking down Lamp2A in MEFs, which is consistent with the observation that ferritin delivery to the lysosome in iron-replete MEFs was inhibited by 3-MA, since CMA is insensitive to 3-MA (17). The molecular mechanisms of microautophagy have yet to be clarified, but the process may be sensitive to 3-MA. Ferritin can be transported into lysosomes in vitro by microautophagy (1), suggesting that this mechanism may function in intact cells as well. A priority for future studies will be identifying the pathway of ferritin delivery to lysosomes under iron-rich conditions, because our results strongly suggest that this pathway is defective in cancer-derived cells but remains intact in primary cells, raising the possibility that the loss of this pathway is related to oncogenic transformation.
Our observations also provide mechanistic insight into selective autophagy, since autophagy-mediated lysosomal targeting of ferritin was observed only in iron-depleted cells (Fig. 2). Although autophagy has been regarded as a nonselective degradation pathway for cytosolic components (48), recent studies have identified adaptor proteins that selectively target protein substrates to the lysosome via autophagy (26, 28, 41). The results of the current study support the presence of selective autophagy adaptor(s) for ferritin. First, ferritin was delivered to lysosomes in HeLa cells under iron-depleted but not iron-replete conditions. Second, ferritin was delivered to the lysosome under both iron-rich and iron-starved conditions in MEFs; however, autophagy did not play a major role in ferritin targeting under iron-replete conditions. These observations suggest the presence of specific adaptor proteins that target ferritin for delivery to the lysosome via autophagy only under iron-depleted conditions. The identification of the adaptor proteins involved in the recognition of ferritin under iron-depleted conditions will shed additional light on the differential regulation of lysosomal targeting of ferritin under iron-depleted and iron-replete conditions.
ACKNOWLEDGMENTS
We thank Haruto Ishikawa for valuable suggestions and members of the Iwai Laboratory for stimulating discussions. We also thank Novartis Company for providing Exjade.
This work was partly supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology in Japan to K.I.
Footnotes
Published ahead of print on 28 March 2011.
REFERENCES
- 1. Ahlberg J., Glaumann H. 1985. Uptake microautophagy and degradation of exogenous proteins by isolated rat-liver lysosomes—effects of pH, ATP, and inhibitors of proteolysis. Exp. Mol. Pathol. 42:78–88 [DOI] [PubMed] [Google Scholar]
- 2. Andrews N. C. 2005. Molecular control of iron metabolism. Best Pract. Res. Clin. Haematol. 18:159–169 [DOI] [PubMed] [Google Scholar]
- 3. Andrews S. C., Treffry A., Harrison P. M. 1987. Siderosomal ferritin. The missing link between ferritin and haemosiderin? Biochem. J. 245:439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arosio P., Ingrassia R., Cavadini P. 2009. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 1790:589–599 [DOI] [PubMed] [Google Scholar]
- 5. Ashizuka M., et al. 2002. Novel translational control through an iron-responsive element by interaction of multifunctional protein YB-1 and IRP2. Mol. Cell. Biol. 22:6375–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bao G. H., et al. 2010. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat. Chem. Biol. 6:602–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chasteen N. D. 1998. Ferritin. Uptake, storage, and release of iron. Metal Ions Biol. Syst. 35:479–514 [PubMed] [Google Scholar]
- 8. Chen Z. J., et al. 1995. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 9:1586–1597 [DOI] [PubMed] [Google Scholar]
- 9. Crichton R. R. (ed.). 2009. Iron metabolism: from molecular mechanisms to clinical consequences. John Wiley & Sons Ltd., Chichester, United Kingdom [Google Scholar]
- 10. Cuervo A. M. 2004. Autophagy: many paths to the same end. Mol. Cell. Biochem. 263:55–72 [DOI] [PubMed] [Google Scholar]
- 11. Cuervo A. M. 2010. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol. Metab. 21:142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dautry-Varsat A., Ciechanover A., Lodish H. F. 1983. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. U. S. A. 80:2258–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Domenico I., et al. 2006. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 25:5396–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Domenico I., Ward D. M., Kaplan J. 2009. Specific iron chelators determine the route of ferritin degradation. Blood 114:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Devireddy L. R., Hart D. O., Goetz D. H., Green M. R. 2010. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 141:1006–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong X. P., et al. 2008. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455:992–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finn P. E., Mesires N. T., Vine M., Dice J. F. 2005. Effects of small molecules on chaperone-mediated autophagy. Autophagy 1:141–145 [DOI] [PubMed] [Google Scholar]
- 18. Fox L. E. 1988. Solubility of colloidal ferric hydroxide. Nature 333:442–444 [Google Scholar]
- 19. Harrison P. M., Arosio P. 1996. Ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275:161–203 [DOI] [PubMed] [Google Scholar]
- 20. Hentze M. W., Muckenthaler M. U., Andrews N. C. 2004. Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–297 [DOI] [PubMed] [Google Scholar]
- 21. Hentze M. W., Muckenthaler M. U., Galy B., Camaschella C. 2010. Two to tango: regulation of mammalian iron metabolism. Cell 142:24–38 [DOI] [PubMed] [Google Scholar]
- 22. Iwai K., et al. 1998. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc. Natl. Acad. Sci. U. S. A. 95:4924–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwai K., Klausner R. D., Rouault T. A. 1995. Requirements for iron-regulated degradation of the RNA-binding protein, iron regulatory protein-2. EMBO J. 14:5350–5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin W., Takagi H., Pancorbo B., Theil E. C. 2001. “Opening” the ferritin pore for iron release by mutation of conserved amino acids at interhelix and loop sites. Biochemistry 40:7525–7532 [DOI] [PubMed] [Google Scholar]
- 25. Kidane T. Z., Sauble E., Linder M. C. 2006. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 291:C445–C455 [DOI] [PubMed] [Google Scholar]
- 26. Kirkin V., et al. 2009. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33:505–516 [DOI] [PubMed] [Google Scholar]
- 27. Klausner R. D., Rouault T. A., Harford J. B. 1993. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell 72:19–28 [DOI] [PubMed] [Google Scholar]
- 28. Komatsu M., et al. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163 [DOI] [PubMed] [Google Scholar]
- 29. Komatsu M., et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuma A., et al. 2004. The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036 [DOI] [PubMed] [Google Scholar]
- 31. Kurz T., Terman A., Gustafsson B., Brunk U. T. 2008. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta 1780:1291–1303 [DOI] [PubMed] [Google Scholar]
- 32. Kwok J. C., Richardson D. R. 2004. Examination of the mechanism(s) involved in doxorubicin-mediated iron accumulation in ferritin: studies using metabolic inhibitors, protein synthesis inhibitors, and lysosomotropic agents. Mol. Pharmacol. 65:181–195 [DOI] [PubMed] [Google Scholar]
- 33. Larson J. A., Howie H. L., So M. 2004. Neisseria meningitidis accelerates ferritin degradation in host epithelial cells to yield an essential iron source. Mol. Microbiol. 53:807–820 [DOI] [PubMed] [Google Scholar]
- 34. Lauffer R. B. (ed.). 1992. Iron and human disease. CRC Press, Boca Raton, FL [Google Scholar]
- 35. Liu X., Theil E. C. 2005. Ferritins: dynamic management of biological iron and oxygen chemistry. Acc. Chem. Res. 38:167–175 [DOI] [PubMed] [Google Scholar]
- 36. Massey A. C., Kaushik S., Sovak G., Kiffin R., Cuervo A. M. 2006. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U. S. A. 103:5805–5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKie A. T. 2005. A ferrireductase fills the gap in the transferrin cycle. Nat. Genet. 37:1159–1160 [DOI] [PubMed] [Google Scholar]
- 38. Mizushima N. 2007. Autophagy: process and function. Genes Dev. 21:2861–2873 [DOI] [PubMed] [Google Scholar]
- 39. Mizushima N., Yoshimori T., Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohgami R. S., et al. 2005. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 37:1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pankiv S., et al. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145 [DOI] [PubMed] [Google Scholar]
- 42. Radisky D. C., Kaplan J. 1998. Iron in cytosolic ferritin can be recycled through lysosomal degradation in human fibroblasts. Biochem. J. 336(Pt. 1):201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rouault T. A. 2006. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2:406–414 [DOI] [PubMed] [Google Scholar]
- 44. Takagi H., Shi D., Ha Y., Allewell N. M., Theil E. C. 1998. Localized unfolding at the junction of three ferritin subunits. A mechanism for iron release? J. Biol. Chem. 273:18685–18688 [DOI] [PubMed] [Google Scholar]
- 45. Torti F. M., Torti S. V. 2002. Regulation of ferritin genes and protein. Blood 99:3505–3516 [DOI] [PubMed] [Google Scholar]
- 46. Truty J., Malpe R., Linder M. C. 2001. Iron prevents ferritin turnover in hepatic cells. J. Biol. Chem. 276:48775–48780 [DOI] [PubMed] [Google Scholar]
- 47. Wallander M. L., Leibold E. A., Eisenstein R. S. 2006. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim. Biophys. Acta 1763:668–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang Z., Klionsky D. J. 2010. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. 1991. Bafilomycin-A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein-degradation in lysosomes of cultured-cells. J. Biol. Chem. 266:17707–17712 [PubMed] [Google Scholar]