

Abstract
The mammalian target of rapamycin complex 1 (mTORC1) is a multiprotein signaling complex regulated by oncogenes and tumor suppressors. Outputs downstream of mTORC1 include ribosomal protein S6 kinase 1 (S6K1), eukaryotic translation initiation factor 4E (eIF4E), and autophagy, and their modulation leads to changes in cell growth, proliferation, and metabolism. Rapamycin, an allosteric mTORC1 inhibitor, does not antagonize equally these outputs, but the reason for this is unknown. Here, we show that the ability of rapamycin to activate autophagy in different cell lines correlates with mTORC1 stability. Rapamycin exposure destabilizes mTORC1, but in cell lines where autophagy is drug insensitive, higher levels of mTOR-bound raptor are detected than in cells where rapamycin stimulates autophagy. Using small interfering RNA (siRNA), we find that knockdown of raptor relieves autophagy and the eIF4E effector pathway from rapamycin resistance. Importantly, nonefficacious concentrations of an ATP-competitive mTOR inhibitor can be combined with rapamycin to synergistically inhibit mTORC1 and activate autophagy but leave mTORC2 signaling intact. These data suggest that partial inhibition of mTORC1 by rapamycin can be overcome using combination strategies and offer a therapeutic avenue to achieve complete and selective inhibition of mTORC1.
INTRODUCTION
Mammalian cells have evolved complex signaling networks to regulate and balance anabolic and catabolic processes. A central node in these networks is the mammalian target of rapamycin (mTOR), a kinase which senses the availability of nutrients and energy and integrates inputs from growth factors and stress signaling (11, 26, 46). mTOR is found in two multiprotein complexes, termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The two complexes contain common members such as mTOR, GßL, and deptor as well as mTORC1- and mTORC2-specific components such as raptor and rictor, respectively. The function of mTORC2 involves the regulation of cell survival via phosphorylation of Akt (38) and the modulation of actin cytoskeleton dynamics (19). mTORC1, on the other hand, promotes protein synthesis and cell growth by phosphorylating p70 ribosomal S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E-binding protein-1 (4EBP1) (27). mTORC1 also suppresses the initiation of autophagy presumably through phosphorylation of the Ulk1-mAtg13-FIP200 complex (12, 18, 20). Autophagy represents a major cellular degradation process that sequesters bulk cytosol into autophagosomes, which then fuse with lysosomes, where acidic hydrolases break down the lumenal content, recycle macromolecules, and provide the cytosol with free fatty acids and amino acids (47). In addition to bulk cytosol, low levels of basal autophagy clear damaged organelles and protein aggregates, thereby maintaining cellular homeostasis. Furthermore, autophagy can be induced by starvation or cytotoxic events to enhance cell survival when growth conditions are unfavorable. Pharmacological activation of autophagy represents an attractive strategy to enhance the clearance of aggregation-prone proteins and target various proteinopathies (35).
Identification of signaling events downstream of mTORC1 greatly profited from the discovery of rapamycin, a macrolide from Streptomyces hygroscopicus, which inhibits TOR in an allosteric manner (4, 5, 16, 36, 43). Rapamycin's mode of action involves the formation of an intracellular complex with the peptidyl-prolyl isomerase FKBP12 and binding to the FKBP12-rapamycin binding (FRB) domain of mTOR (6). This results in a conformational change in mTORC1 which is believed to alter and weaken the interaction with its scaffolding protein raptor, in turn impeding substrates such as S6K1 and 4EBP1 from accessing mTOR and being phosphorylated (15, 21, 30). Rapamycin and rapalogues such as RAD001 or CCI-779 have gained clinical relevance by inhibiting hyperactivation of mTOR associated with both benign and malignant proliferation disorders (9, 17). Recently, several novel small-molecule inhibitors of mTOR have been developed that target the kinase domain directly and act as catalytic, ATP-competitive inhibitors (10, 13, 42, 49). These catalytic mTOR inhibitors target mTORC1 and mTORC2 and are more effective inhibitors of mTORC1 than rapamycin because they modulate rapamycin-resistant mTORC1 outputs such as 4EBP1 phosphorylation at T37/46 (4EBP1-T37/46) and cap-dependent translation. Thoreen and colleagues report that the catalytic mTOR inhibitor Torin increases LC3 lipidation and green fluorescent protein (GFP)-LC3 punctum autophagy readouts in mouse embryonic fibroblasts and HeLa cells, whereas rapamycin has minor effects. This is surprising, given past reports showing that rapamycin modulates autophagy in a number of cell lines including hepatocytes, MCF-7, COS7, U87-MG, or primary neurons (1, 2, 31, 39, 41). Furthermore, autophagy activation by rapamycin clears aggregation-prone proteins such as mutant Huntingtin or α-synuclein in cell culture (33, 44), and CCI-779 reduces the aggregate load in a mouse model of Huntington's disease (34).
In this study, several cell lines were surveyed for autophagy activation in response to rapamycin and catalytic domain inhibitors. We show that rapamycin modulates autophagy in a cell-type-specific manner that correlates with the inherent stability of mTORC1. Sensitizing mTORC1 by partial raptor depletion or with nonefficacious concentrations of catalytic domain inhibitors allows rapamycin to robustly antagonize eukaryotic translation initiation factor 4E (eIF4E) and activate autophagy.
MATERIALS AND METHODS
Antibodies, inhibitors, and siRNA.
The following antibodies were used: anti-mTOR, antiraptor, antirictor, anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), anti-ß-tubulin, anti-eIF4G, anti-eIF4E, anti-4EBP1, anti-phospho-S6-S240/244 (S6 phosphorylated at S240/244), anti-phospho-S6K1-T389, anti-phospho-AKT-S473 (items 2972, 2280, 2114, 2118, 2128, 2498, 9742, 9452, 4838, 9234, and 9271, respectively; Cell Signaling Technology), anti-LC3 (NB100-2220; Novus), and anti-FKBP12 (PA1-026A; Pierce). The following inhibitors were used: bafilomycin A1 (Tocris), rapamycin (Calbiochem), RAD001 (Novartis), and Ku-0063794 and WYE-354 (Chemdea). All inhibitors were resuspended in dimethyl sulfoxide (DMSO; Calbiochem). The following On-Target small interfering RNA (siRNA) pools were purchased from Dharmacon: nontargeting (D-001810), human raptor (L-004107), and human rictor (L-016984). siRNA (25 nM) was reverse transfected using RNAiMax (Invitrogen) into cells plated in a six-well format.
Cell culture.
Cells were maintained in a humidified incubator at 37°C and 5% CO2. GP2-293 and H4 cells were grown in Dulbecco's modified Eagle's medium ([DMEM] 11995; Invitrogen) supplemented with 10% fetal bovine serum (FBS), RT112 cells in alpha-minimal essential medium ([αMEM] 12571; Invitrogen) supplemented with 18 mM d-glucose and 10% FBS, U2OS cells in McCoy's (16600; Invitrogen) supplemented with 10% FBS, SK-N-SH cells in Eagle's minimal essential medium ([EMEM] ATCC 30-2003) supplemented with 10% FBS, and HN10 cells (45) in DMEM (10564; Invitrogen) supplemented with 10% FBS.
Generation of mCherry-GFP-LC3-expressing cell lines.
Monomeric Cherry (mCherry)-GFP-LC3 was expressed from the pL(mCherry-GFP-LC3) construct (29), which is based on pLEGFP-C1 (Clontech) containing human LC3A [PCR-amplified from pCMV6-XL5(MAP1LC3A)] (NM_032514.2; Origene) and mCherry (Clontech) downstream and upstream of GFP, respectively. pL(mCherry-GFP-LC3) was packaged into retroviral particles by cotransfecting GP2-293 cells with pVPackVSV-G (where VSV-G is vesicular stomatitis virus G protein) (Stratagene) using Lipofectamine 2000 (Invitrogen). Retroviral supernatants were harvested at 72 h posttransfection, mixed with 8 μg/ml Polybrene (American Bioanalytical), and used to infect H4, RT112, U2OS, SK-N-SH, and HN10 cells. Stably transduced cells were selected using 250 to 500 μg/ml G418 (Invitrogen), and the bulk population of G418-resistant cells was maintained and used for experiments. G418 was omitted when cells were plated for experiments.
High-content imaging and analysis of autophagy and S6 phosphorylation.
H4, RT112, U2OS, SK-N-SH, and HN10 cells stably expressing mCherry-GFP-LC3 were plated into clear-bottom 384-well plates (Greiner) at a density of 2,000 to 4,000 cells/well in 30 μl of medium/well. One day after plating, cells were treated with compound for 4 h or 18 h by adding 10 μl/well DMEM containing 10% FBS and 4× concentrated compounds. For starvation, medium was aspirated, and cells were washed and incubated in 40 μl/well Earle's balanced salt solution (EBSS; HyClone). After treatments, cells were fixed for 1 h at room temperature by adding 10 μl/well 5× concentrated Mirsky's fixative (National Diagnostics) supplemented with 25 μg/ml Hoechst 33342 (Invitrogen). Cells were washed six times with Tris-buffered saline (TBS), and 384-well plates were subjected to automated epifluorescence microscopy using an InCell Analyzer 1000 (GE Healthcare). Four different fields were imaged in each well using ×20 magnification, which covered around 400 to 800 cells per well. Hoechst 33342 was excited at 360 nm and imaged using a 460- ± 40-nm emission filter and an exposure time of 150 ms. GFP was excited at 475 nm and imaged using a 535- ± 50-nM emission filter and an exposure time of 1,500 ms. mCherry was excited at 535 nm and imaged using a 620- ± 60-nm emission filter and an exposure time of 1,500 ms. Images were quantified using a Multi Target Analysis module of the InCell Investigator software (GE Healthcare). In brief, nuclei were identified based on the Hoechst 33342 staining, and cells were defined using a 4- to 8-μm collar. Cellular LC3 puncta were detected and quantified in the mCherry images using multi-top-hat segmentation and averaged per well. The positional information and mask of the mCherry-positive puncta were transferred onto the GFP image, and the fluorescence intensity inside the mask was measured (GFP intensity in mCherry-positive puncta) and averaged per well. Treatment-induced changes in GFP intensity in mCherry-positive puncta were calculated to depict autophagic flux. After imaging of mCherry-GFP-LC3, cells were incubated for 2 h at room temperature in blocking buffer (TBS supplemented with 0.1% Triton X-100 and 0.1% bovine serum albumin), stained overnight at 4°C with anti-phospho-S6-S240/244 antibody (diluted 1:300 in blocking buffer; 4838; Cell Signaling Technology) and for 2 h at room temperature with Cy5-conjugated goat anti-rabbit IgG (diluted 1:300 in blocking buffer; AP187S; Millipore). Cells were washed six times with TBS and 384-well plates were reimaged using an InCell Analyzer 1000. Hoechst 33342 and Cy5 images were acquired in two fields per well using a magnification of ×10. Cy5 was excited at 620 nm and imaged using a 700- ± 75-nm emission filter and an exposure time of 1,000 ms. Images were analyzed using the object intensity analysis module of the InCell Investigator software (GE Healthcare). In brief, Hoechst 33342 images were used to identify the nuclei, a 4- to 8-μm collar was used to define the cells, and Cy5 intensity was measured inside the cell collar and averaged per well. Cy5 baseline intensity was subtracted, and the fluorescence signal of the control treatment was set to 100%.
Analysis of compound synergy.
RAD001 and Ku-0063794 compound interaction was analyzed using the Bliss additivism or the highest single agent (HSA) model as described previously (3, 50). For each compound combination, relative effects on autophagy activation (LC3 puncta/cell) or inhibition of S6 phosphorylation (immunostaining intensity) were calculated by setting the effect of DMSO to 0% and the effect of the highest compound combination (12 μM Ku-0063794 plus 0.15 μM RAD001) to 100%. The Bliss additivism model calculates the difference (percent excess) between the actually measured effect of a given compound combination and the summed effect of the single doses. The HSA model calculates the difference between the actually measured effect of a given compound combination and the larger of the effects produced by the two single agents.
Cell lysis, cross-linking, and mTOR immunoprecipitation.
Whole-cell lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology) supplemented with protease inhibitor cocktail (Roche Applied Science), cleared by centrifugation at 16,000 × g for 10 min, normalized based on protein concentration (Bio-Rad protein assay), and boiled in NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) supplemented with 2% ß-mercaptoethanol. For cross-linking and mTOR immunoprecipitation, cells were grown in 10-cm dishes and treated for 18 h with 0.1% DMSO or 250 nM RAD001. Cross-linking was performed with 1 mg/ml dithiobis succinimidyl propionate (DSP; Pierce) as described previously (37). Cell monolayers were then washed with cold phosphate-buffered saline (PBS) and lysed in 40 mM HEPES, pH 7.5, 120 mM NaCl, 1 mM EDTA, 10 mM sodium pyrophosphate, 10 mM ß-glycerophosphate, 50 mM NaF, 1.5 mM Na3VO4, and 0.3% CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate) supplemented with protease inhibitor cocktail (Roche Applied Science). Lysates were cleared by centrifugation at 16,000 × g for 10 min and normalized based on protein concentrations (Bio-Rad protein assay). One-milligram lysate samples were incubated overnight at 4°C with 10 μl of anti-mTOR (2972; Cell Signaling Technology) before protein G Sepharose beads (GE Healthcare) were added for 1.5 h at 4°C. Beads were then washed three times with lysis buffer and boiled in NuPAGE LDS sample buffer (Invitrogen) supplemented with 2% ß-mercaptoethanol.
m7GTP cap assay.
Cells were grown in 10-cm dishes and treated with the inhibitors indicated in the figure legends. Cell monolayers were then washed with cold PBS and lysed in 10 mM KH2PO4/K2HPO4, pH 7.05, 5 mM EGTA, 10 mM MgCl2, 0.5% NP-40, 0.1% Brij35, 0.1% sodium deoxycholate supplemented with phosphatase, and protease inhibitor cocktails (Roche Applied Science). Lysates were cleared by centrifugation at 16,000 × g for 10 min and normalized based on protein concentrations (Bio-Rad protein assay). Five hundred micrograms of lysate samples was incubated with 7-methyl-GTP Sepharose beads (GE Healthcare) for 4 h at 4°C before beads were washed three times with lysis buffer and boiled in NuPAGE LDS sample buffer (Invitrogen) supplemented with 2% ß-mercaptoethanol.
Immunoblotting.
Protein samples were generally separated on NuPAGE 4 to 12% Bis-Tris gels using morpholinepropanesulfonic acid (MOPS) running buffer (Invitrogen). For the separation of 4EBP1 and LC3, NuPAGE 12% Bis-Tris gels were used. Proteins were transferred to nitrocellulose membranes (Invitrogen), probed with the primary antibodies indicated on the figures, and visualized using horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (AP307P; Millipore) and enhanced chemiluminescence (Pierce).
Fluorescence microscopy.
H4 mCherry-GFP-LC3 cells were grown on poly-d-lysine-coated four-well culture slides (BD Biosciences) and treated by adding inhibitors directly to the cell culture medium. For starvation, cells were washed and incubated in Earle's balanced salt solution (HyClone). Cell fixation was performed for 1 h at room temperature by adding 5× concentrated Mirsky's fixative (National Diagnostics) before cells were washed with PBS and embedded using ProLong Gold Antifade reagent containing 4′,6′-diamidino-2-phenylindole (DAPI; Invitrogen). Slides were analyzed with a Zeiss Axiovert 200 M fluorescence microscope using DAPI, fluorescein isothiocyanate (FITC), and Cy3 filter sets, and images were acquired using Zeiss AxioVision LE software. Adobe Photoshop was used to auto-contrast and overlay images.
RESULTS
Quantification of autophagic flux using an mCherry-GFP-LC3 cell-based assay.
To measure cellular autophagy in a quantitative and medium-throughput manner, we adopted an imaging-based approach that visualizes tandem fluorescently tagged LC3 proteins (23, 32). LC3, the mammalian homologue of yeast Saccharomyces cerevisiae Atg8, gets lipidated upon initiation of autophagy and redistributes from the cytosol to the phagophore membrane (22). We used human LC3A, N-terminally tagged with monomeric Cherry (mCherry) and the green fluorescent protein (GFP), two fluorophores that display different pH sensitivities. Upon exposure to the acidic environment of autolysosomes, mCherry fluorescence is stable while GFP fluorescence is readily quenched and lost. Hence, activation of autophagy is characterized by the redistribution of cytosolic mCherry-GFP-LC3 to autophagosomes and autolysosomes (apparent as puncta) and loss of GFP fluorescence in the autolysosomes. mCherry-GFP-LC3 was stably expressed in human glioblastoma H4 cells and validated using two well-characterized autophagy modulators, namely, treatment with the vacuolar ATPase-inhibitor bafilomycin A1 and starvation in Earle's balanced salt solution (EBSS). Bafilomycin neutralizes lysosomal acidification and stalls autophagy (25), whereas starvation inhibits mTORC1 and activates autophagy. Automated epifluorescence microscopy was combined with an image recognition algorithm to quantify cellular LC3 puncta (number of mCherry-positive puncta, which appear as yellow dots by microscopy, per cell) and autophagic flux (loss of GFP fluorescence in mCherry-positive puncta, which is detected as a decrease in yellow puncta by microscopy). Both EBSS and bafilomycin treatment significantly increased LC3 puncta after 4 h and 18 h (Fig. 1A). However, only EBSS starvation augmented autophagic flux, and a treatment time of 18 h was required to see a robust effect. Our quantification is in line with the increase in red and yellow puncta observed by fluorescence microscopy in starved and bafilomycin-treated cells, respectively (Fig. 1B). Western blot analysis of endogenous LC3 showed a starvation-induced decrease in LC3 levels (Fig. 1C), which is in agreement with enhanced autophagic flux. The time-dependent accumulation of LC3-II in bafilomycin-treated cells, on the other hand, is consistent with the inhibition of the autophagic process. Hence, imaging-based quantification of autophagic flux and cellular LC3 puncta mimics changes at the level of endogenous LC3 and can reliably unveil the status of the autophagic pathway.
Fig. 1.
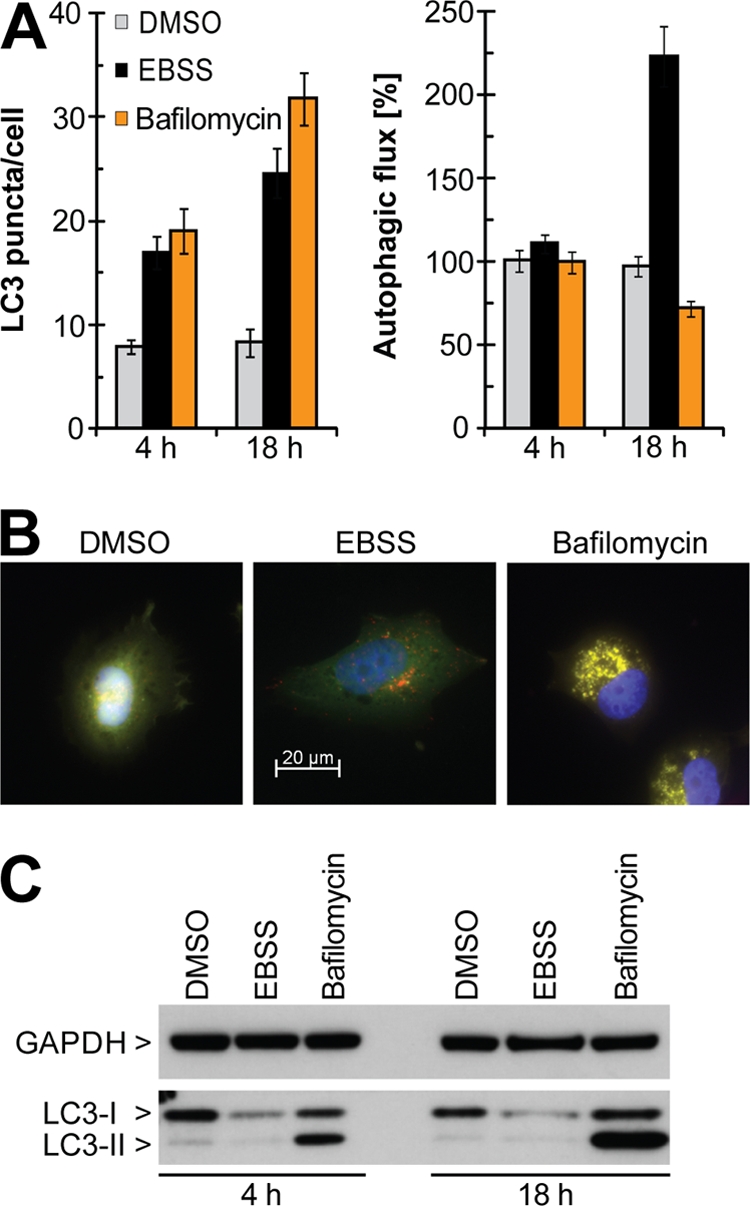
Quantitative visualization of autophagy using the mCherry-GFP-LC3 assay. H4 cells stably expressing mCherry-GFP-LC3 were treated for 4 h or 18 h with 0.1% DMSO or 100 nM bafilomycin or were starved in EBSS. (A) Cells were grown and treated in 384-well plates, fixed, stained with Hoechst, and subjected to automated epifluorescence microscopy, and autophagy was quantified using the image recognition algorithm described in Materials and Methods. Bars represent averaged data from 10 wells ± standard deviations. (B) Cells were grown in four-well culture slides, treated for 18 h, fixed, and subjected to fluorescence microscopy. Representative images are shown, and GFP (green), mCherry (red), and DAPI (blue) channels were merged. (C) Cells were grown and treated in 10-cm dishes and lysed, and endogenous GAPDH and LC3 were visualized by Western blot analysis.
Modulation of autophagy by mTOR inhibitors.
To what extent do different mTOR inhibitors modulate the autophagic pathway? To address this question, H4 mCherry-GFP-LC3 cells were treated for 18 h with various doses of the allosteric mTOR inhibitors rapamycin and RAD001 and the catalytic mTOR inhibitors Ku-0063794 (13) and WYE-354 (49). Rapamycin and RAD001 increased the number of LC3 puncta in the nanomolar range (Fig. 2A and D), and quantification of autophagic flux confirmed induction of autophagy. A concentration of 250 nM rapamycin or RAD001 robustly increased lipidation of endogenous LC3 in parental H4 cells (Fig. 2C). Ku-0063794 and WYE-354 required micromolar concentrations to activate autophagy but increased LC3 puncta and autophagic flux to a greater extent than the allosteric inhibitors (Fig. 2A and D). At saturating concentrations, all four compounds fully inhibited S6K1-T389 and ribosomal protein S6-S240/244 phosphorylation, while only the catalytic mTOR inhibitors targeted Akt-S473, an mTORC2 substrate site (Fig. 2B and C). We conclude that allosteric mTOR inhibitors do activate autophagy in H4 cells but are less efficacious than catalytic mTOR inhibitors.
Fig. 2.

The autophagy induction potential of mTOR inhibitors in H4 cells. H4 mCherry-GFP-LC3 cells were grown in 384-well plates and treated for 18 h with serial dilutions (2.8 pM to 12 μM) of rapamycin, RAD001, Ku-0063794, or WYE-354. (A) Cells were fixed and stained with Hoechst, and autophagy was quantified using high-content imaging and analysis. Each data point represents averaged data from three independent experiments ± standard deviation. (B) Cultures in 384-well plates were subjected to immunostaining against phosphorylated ribosomal protein S6-S240/244 using a phospho-specific primary and Cy5-conjugated secondary antibody. Cellular Cy5 signal was quantified using high-content imaging and analysis. Each data point represents averaged data from three independent experiments ± standard deviation. (C) Parental H4 cells were treated with the indicated compound for 18 h and lysed, and endogenous proteins were visualized by Western blot analysis. (D) H4 mCherry-GFP-LC3 cells were grown in four-well culture slides, treated with the indicated compound for 18 h, fixed, and subjected to fluorescence microscopy. Representative images are shown, and GFP (green), mCherry (red), and DAPI (blue) channels were merged. (E) H4 mCherry-GFP-LC3 cells were transfected with 25 nM nontargeting (NT), raptor, or rictor siRNA and lysed at 4 days posttransfection, and endogenous proteins were analyzed by Western blotting using the indicated primary antibodies. (F) H4 mCherry-GFP-LC3 cells were subjected to 3-day siRNA knockdown before 0.1% DMSO, 250 nM RAD001, or 3 μM Ku-0063794 was added for 4 h or 18 h. Autophagy was quantified in 384-well plates using high-content imaging and analysis. Bars represent averaged data from 20 wells ± standard deviation of a representative experiment.
Short-term treatments with rapamycin and RAD001 exclusively target mTORC1 (37) while Ku-0063794 and WYE-354 can inhibit both mTORC1 and mTORC2. To test the contribution of the two mTOR complexes to autophagy induction, we used a genetic approach and inhibited mTORC1 and mTORC2 by siRNA knockdown of raptor and rictor, respectively. Depletion of raptor and rictor was highly efficient in H4 mCherry-GFP-LC3 cells, and corresponding downstream signaling events were affected, namely, inhibition of S6K1-T389 phosphorylation for raptor and Akt-S473 phosphorylation for rictor knockdown (Fig. 2E). In terms of autophagy, silencing of raptor activated the pathway with a significant increase in LC3 puncta while knockdown of rictor had no effect (Fig. 2F). Interestingly, the addition of RAD001 to raptor-depleted cells resulted in autophagy activation to an extent seen with the catalytic mTOR inhibitor Ku-0063794. This effect was specific for the knockdown of raptor since cells transfected with nontargeting and rictor siRNA both showed only partial autophagy activation upon RAD001 treatment. This suggests that activation of autophagy does not depend on inhibition of mTORC2 but is rather determined by how strongly mTORC1 is inhibited, a finding that is in line with rapamycin being a poor autophagy agonist in mouse embryonic fibroblasts from wild-type as well as rictor knockout mice (42).
Rapamycin and RAD001 induce autophagy in a cell-type-specific manner.
We applied our image-based analysis of autophagy and mTORC1 activity to additional cell lines. In human bladder carcinoma RT112, human osteosarcoma U2OS, and human neuroblastoma SK-N-SH cells, cellular LC3 puncta were robustly induced by Ku-0063794 and WYE-354 (Fig. 3A and B). In contrast, rapamycin and RAD001 activated autophagy in RT112 and U2OS cells but not in SK-N-SH cells. Importantly, S6-S240/244 phosphorylation was equally sensitive to rapamycin and RAD001 in RT112, U2OS, and SK-N-SH cells (Fig. 3C). Autophagy was also indifferent to rapamycin and RAD001 in a second neuroblastoma cell line, HN10, despite the fact that S6-S240/244 phosphorylation was fully inhibited (Fig. 3).
Fig. 3.

Autophagy is indifferent to rapamycin and RAD001 in SK-N-SH and HN10 cells. RT112, U2OS, SK-N-SH, and HN10 cells stably expressing mCherry-GFP-LC3 were grown in 384-well plates and treated for 18 h with serial dilutions (11 pM to 12 μM) of rapamycin, RAD001, Ku-0063794, or WYE-354. Cells were fixed and stained with Hoechst, and autophagy was quantified using high-content imaging and analysis. (A) Absolute numbers of mCherry puncta quantified per cell. A representative experiment is shown, and each data point represents averaged data from four wells ± standard deviation. (B) Relative autophagy activation calculated as fold induction of mCherry puncta. Each data point represents averaged data from three independent experiments ± standard deviation. (C) Cultures in 384-well plates were subjected to immunostaining against phosphorylated ribosomal protein S6-S240/244 using a phospho-specific primary and Cy5-conjugated secondary antibody. Cellular Cy5 signal was quantified using high-content imaging and analysis. Each data point represents averaged data from three independent experiments ± standard deviation.
Why do rapamycin and RAD001 fail to induce autophagy in SK-N-SH and HN10 cells even though S6-S240/244 phosphorylation is fully inhibited? The fact that Ku-0063794 and WYE-354 activate autophagy in these cell lines argues that mTOR still acts as a key negative regulator of autophagy (Fig. 3). Since we found that activation of autophagy depends on how strong mTORC1 is inhibited (Fig. 2F), we hypothesized that rapamycin and RAD001 inhibit mTORC1 in SK-N-SH and HN10 cells to an extent which is efficacious enough to target the S6K1 effector pathway but not autophagy. Rapamycin and RAD001, in complex with their intracellular receptor FKBP12, bind mTOR and destabilize the interaction with raptor. We speculated that differences in FKBP12, mTOR, or raptor may account for the cell-type-specific effects of rapamycin and RAD001 on autophagy. We first compared expression levels of FKBP12, mTOR, and raptor in cell lines displaying either rapamycin-resistant autophagy (SK-N-SH and HN10) or rapamycin-sensitive autophagy (U2OS, H4, and RT112). Western blot analysis showed comparable expression levels of mTOR and raptor across all cell lines (Fig. 4A). FKBP12 levels varied between the cell lines but did not track with rapamycin sensitivity toward autophagy. To analyze composition and stability of the mTOR-raptor interaction, mTOR was immunoprecipitated from cells treated with or without the chemical cross-linker DSP. Cells were lysed in 0.3% CHAPS to preserve mTORC1 integrity (21). In cross-linked cells, similar amounts of raptor were recovered with mTOR in all five cell lines (Fig. 4B). In the absence of DSP, however, significantly more raptor was coimmunoprecipitated with mTOR in SK-N-SH and HN10 cells. This observation held true even after RAD001 treatment, where some raptor remained bound to mTOR in SK-N-SH and HN10 cells even though the total amount that coprecipitated with mTOR was drastically reduced. Hence, equal amounts of raptor interacted with mTOR in each cell line tested, but the mTOR-raptor interaction showed differential stability. In U2OS, H4, and RT112 cells, the raptor-mTOR interaction was unstable and lost in large part during cell lysis and mTOR immunoprecipitation. This unstable conformation may favor more complete inhibition of mTORC1 by allosteric inhibitors. In SK-N-SH and HN10 cells, RAD001 can destabilize mTORC1, yet residual raptor stays bound to mTOR under these conditions, enough to suppress autophagy.
Fig. 4.
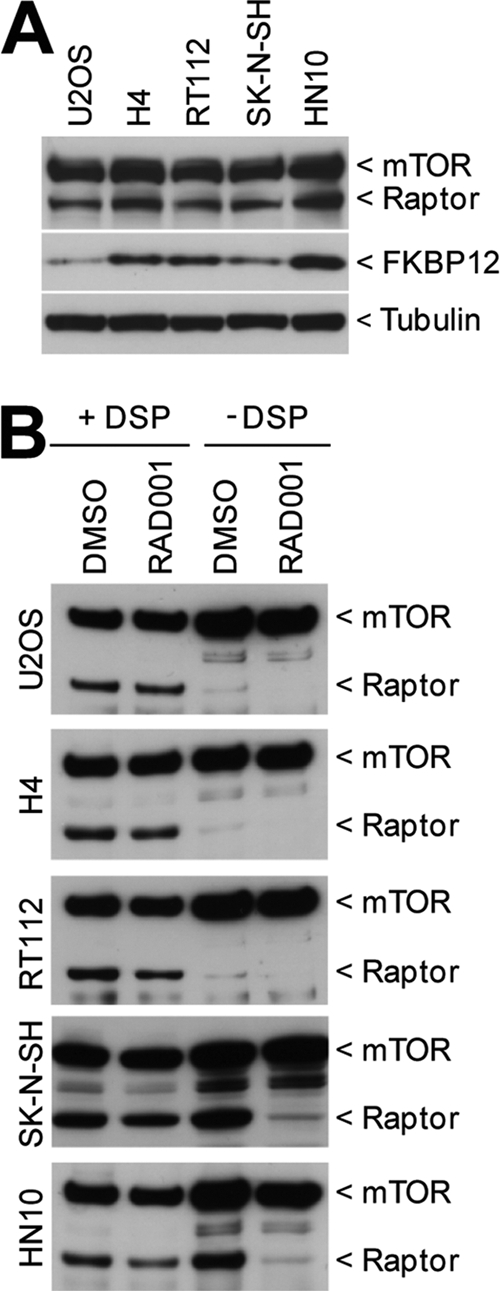
Autophagy induction potential of RAD001 correlates with mTOR complex 1 stability. (A) U2OS, H4, RT112, SK-N-SH, and HN10 mCherry-GFP-LC3 cell lines were lysed in RIPA buffer and analyzed by Western blotting using the indicated primary antibodies. (B) U2OS, H4, RT112, SK-N-SH, and HN10 mCherry-GFP-LC3 cell lines were treated for 18 h with 0.1% DMSO or 250 nM RAD001. Half of the dishes were subjected to chemical cross-linking using 1 mg/ml DSP. Cell lysates were prepared using 0.3% CHAPS, immunoprecipitated using anti-mTOR antibody, and analyzed by Western blot analysis.
Sensitizing autophagy and mTOR complex 1 to RAD001.
Our mTOR-raptor interaction data suggest that autophagy is indifferent to RAD001 in SK-N-SH and HN10 cells due to insufficient inhibition of mTORC1. Sensitization of mTORC1, therefore, should allow RAD001 to induce autophagy. In H4 cells, depletion of raptor indeed allowed RAD001 to fully activate autophagy, similar to a catalytic mTOR inhibitor (Fig. 2F). We tested if silencing of raptor can render autophagy sensitive to RAD001 in SK-N-SH mCherry-GFP-LC3 cells. Raptor siRNA was able to reduce raptor protein levels, but residual amounts were still detectable by Western blotting, and phosphorylation of S6K1-T389 was unaffected (Fig. 5A). In agreement with the incomplete knockdown of raptor, cellular LC3 puncta were unchanged (Fig. 5B). Nevertheless, addition of RAD001 to raptor-depleted cells was able to activate autophagy (Fig. 5B, compare RAD001 treatment in the nontargeting [NT] siRNA and raptor siRNA-treated cells), which indicates that a reduction in the number of mTOR-raptor complexes allows coupling of autophagy to mTORC1 in a RAD001-sensitive manner.
Fig. 5.

Sensitization of mTORC1 relieves autophagy from rapamycin resistance. (A) SK-N-SH mCherry-GFP-LC3 cells were transfected with nontargeting (NT) or raptor siRNA and lysed 4 days posttransfection, and endogenous proteins were analyzed by Western blotting using the indicated primary antibodies. (B) SK-N-SH mCherry-GFP-LC3 cells were subjected to 4-day siRNA knockdown before 0.1% DMSO, 250 nM RAD001, or 3 μM Ku-0063794 was added for 4 h. Autophagy was quantified in 384-well plates using high-content imaging and analysis. Bars represent averaged data from 20 wells ± standard deviation of a representative experiment. (C) SK-N-SH mCherry-GFP-LC3 cells were treated for 4 h with the indicated compounds and were either subjected to high-content analysis or Western blot analysis. (D) SK-N-SH mCherry-GFP-LC3 cells were grown in 384-well plates and were treated for 18 h with a 10-by-10 dose matrix of RAD001 and Ku-0063794, including a zero concentration point. Cellular LC3 puncta were quantified by high-content imaging and analysis, and autophagy activation is depicted relative to DMSO (set to 0%) and the highest compound combination (set to 100%). Mean values of two independent experiments are shown. (E) Plates were immunostained using a phospho-specific primary antibody against phosphorylated ribosomal protein S6-S240/244 and a Cy5-conjugated secondary antibody, and cellular Cy5 signal was quantified using high-content imaging and analysis. Inhibition of pS6-S240/244 is depicted relative to DMSO (set to 0%) and the highest compound combination (set to 100%). Mean values of two independent experiments are shown. The percent excess over Bliss additivism was determined for each compound concentration pair for autophagy activation (F) and pS6-S240/244 inhibition (G). Mean values of two independent experiments are shown. Note that negative, zero, and positive values indicate antagonism, additivism, or synergism, respectively.
To explore this further, mTORC1 was pharmacologically sensitized using a catalytic mTOR inhibitor at a concentration which does not activate autophagy. Interestingly, as a single agent 200 nM Ku-0063794 did not induce autophagy but allowed RAD001 to activate the autophagic pathway, as evidenced by increased cellular LC3 puncta as well as lipidation of endogenous LC3 (Fig. 5C). These data showed that RAD001 has the potential to activate autophagy in SK-N-SH cells but normally does not do so because mTORC1 is not sufficiently inhibited.
Since the combination of 250 nM RAD001 and 200 nM Ku-0063794 activated autophagy to a greater extent than the single doses, we examined whether their combination targets mTORC1 in a synergistic manner. The Bliss additivism model (3, 50) was used to analyze a 10-by-10 dose matrix of RAD001 and Ku-0063794. As a single agent, Ku-0063794 modulated both autophagy and pS6-S240/244 in the micromolar range, whereas RAD001 inhibited pS6-S240/244 in the nanomolar range but did not activate autophagy (Fig. 5D and E, where left columns and lowest rows reflect singe-agent activities). Doses of 0.19 μM, 0.38 μM, and 0.75 μM Ku-0063794 were able to sensitize autophagy to RAD001, and the Bliss additivism model suggested strong synergism (Fig. 5F). Importantly, synergism was observed only for RAD001 doses which also inhibited S6 phosphorylation as a single agent. The effect is twofold: RAD001 provokes a potency shift of Ku-0063794, whereas Ku-0063794 boosts the efficacy of RAD001. For inhibition of S6-S240/244 phosphorylation, synergy is restricted to a narrow dose range of RAD001 and Ku-0063794, likely due to the fact that both compounds are highly effective in inhibiting this readout as single agents (Fig. 5G). The highest single agent (HSA) model (3, 50) generated very similar synergistic values (data not shown), and we conclude that combining RAD001 and Ku-0063794 synergistically inhibits mTORC1 to activate autophagy.
Sensitizing eIF4E/4EBP1 to RAD001.
The 4EBP1-eIF4E effector pathway downstream of mTORC1 regulates cap-dependent translation and is reported to be resistant to rapamycin (10, 13, 49). Nonphosphorylated 4EBP1 binds to eIF4E on the m7GTP cap of mRNA molecules and inhibits cap-dependent translation by impeding the recruitment of eIF4G and other cap-binding proteins. Since autophagy can be activated by RAD001 when mTORC1 is sensitized, raptor depletion or low doses of Ku-0063794 were combined with RAD001 and tested in m7GTP bead pulldown assays. In SK-N-SH cells transfected with nontargeting siRNA, little 4EBP1 bound to m7GTP beads upon cell treatment with 250 nM RAD001, whereas 3 μM Ku-0063794 had a robust effect and displaced eIF4G (Fig. 6A). When raptor was depleted, 250 nM RAD001 readily recruited 4EBP1 to the m7GTP cap, displaced eIF4G, and mimicked the effect of the saturating dose of 3 μM Ku-0063794. A similar effect was observed when RAD001 was combined with nonefficacious doses of Ku-0063794. As single agents, 250 nM RAD001 and 200 nM Ku-0063794 were unable to significantly recruit 4EBP1 to the m7GTP cap, and eIF4G remained bound (Fig. 6B). Their combination, however, resulted in recruitment of 4EBP1 to m7GTP beads, with a consequent displacement of eIF4G, again very similar to the effect of 3 μM Ku-0063794. In contrast to 3 μM Ku-0063794, RAD001 combined with 200 nM Ku-0063794 did not target mTORC2, as evident by intact phosphorylation of Akt-S473 (Fig. 6B). We conclude that low doses of catalytic mTOR inhibitors can be combined with RAD001 to specifically and efficiently inhibit mTORC1, which allows for targeting of autophagy and 4EBP1, two mTORC1 effector pathways which are reported to be resistant to allosteric mTOR inhibitors such as rapamycin.
Fig. 6.

Sensitization of mTORC1 relieves eIF4E from rapamycin resistance. SK-N-SH mCherry-GFP-LC3 cells were subjected to 5-day siRNA knockdown before 0.1% DMSO, 250 nM RAD001, or 3 μM Ku-0063794 was added for 4 h. Cells were lysed, subjected to affinity isolation using m7GTP beads, and analyzed by Western blotting. (B) SK-N-SH mCherry-GFP-LC3 cells were treated for 4 h with the indicated compounds, and cell lysates were prepared, subjected to affinity isolation using m7GTP beads, and analyzed by Western blotting.
DISCUSSION
Over the last 20 years the use of rapamycin has enabled the study of TOR across a variety of eukaryotic species, and drug-like derivatives have been used as immunosuppressants and to treat patients with advanced kidney cancer (28). Importantly, it is now being recognized that rapamycin only partially inhibits mTORC1, whereas newly developed ATP-competitive antagonists block all outputs downstream of mTORC1 and mTORC2 (10, 13, 42, 49). The current study has broad implications for the use of both classes of mTOR inhibitors. First, more complete inhibition of mTORC1 can be achieved with rapamycin in cellular contexts where the stability of mTORC1 is inherently low. Second, approaches aimed at destabilizing mTORC1 might sensitize cells to rapamycin. Third, nonefficacious low concentrations of ATP-competitive mTOR inhibitors combined with saturating concentrations of rapamycin can robustly modulate autophagy and eIF4E. Finally, the mTORC2-Akt pathway is unaffected by this drug combination, suggesting a therapeutic approach to achieve efficient and selective inhibition of mTORC1.
TORC1 effector pathways were characterized in yeast as being rapamycin sensitive, but recent observations from several laboratories illustrate that the autophagy and eIF4E outputs downstream of mTORC1 are largely indifferent to rapamycin (10, 13, 42, 49). The present study suggests that autophagy should not be considered a rapamycin-resistant mTORC1 effector pathway since rapamycin does induce autophagy in some cell types. In cell lines where autophagy is inert to rapamycin, ATP-competitive inhibitors still lead to pathway activation, revealing that autophagy is regulated by mTOR function in this context. Moreover, since rapamycin is commonly used to investigate the role of mTOR upstream of autophagy (24, 35), lack of response to this drug does not necessarily imply that autophagy is uncoupled from mTOR. An off-target effect of Ku-0063794 and WYE-354 is unlikely since they represent two structurally distinct mTOR catalytic inhibitors and activate autophagy at concentrations paralleling inhibition of S6-S240/244 phosphorylation (Fig. 2B and 3). Rapamycin and rapalogues are commonly used at nanomolar concentrations in vitro, but Shor and colleagues report that CCI-779 can repress mTOR in an FKBP12-independent manner if used at a higher dose of 5 to 15 μM (40). In contrast to results with nanomolar concentrations, micromolar CCI-779 profoundly inhibits cell proliferation and protein synthesis in several tumor cells, suggesting greater inhibition of mTOR signaling. In the current study, differences in the autophagy-inducing ability of mid-nanomolar and micromolar concentrations were not observed (Fig. 2A and 3). Thus, the modest activation of autophagy by rapamycin in some cell lines cannot be maximized by using higher drug concentrations.
Allosteric and ATP-competitive mTOR inhibitors may modulate mTORC1 downstream signaling in a qualitatively different manner. For example, one cannot rule out the existence of mTORC1 subcomplexes with different cellular localizations and signaling functions which are accessible only to ATP-competitive inhibitors. However, it is intriguing that partial depletion of raptor or addition of a low concentration of ATP-competitive mTOR inhibitor causes autophagy and eIF4E readouts to become sensitive to rapamycin. Therefore, we suggest that the differential effects seen with the two classes of inhibitors is, rather, based on quantitative differences in mTORC1 inhibition and that near maximum inhibition of mTORC1 activity is required to fully activate autophagy and block the eIF4E complex. Rapamycin partially inhibits mTORC1 but does not achieve the level of inhibition which is required for modulation of autophagy and eIF4E/4EBP1. Maximal inhibition of mTORC1 can be achieved by saturating doses of catalytic domain inhibitors or by sensitizing mTORC1 to rapamycin by raptor knockdown (Fig. 5B and 6A) or nonefficacious doses of Ku-0063794 (Fig. 5C and 6B). With this model, the question arises of why S6K1 is so sensitive to allosteric mTOR inhibitors. The fact that autophagy and S6-S240/244 phosphorylation are modulated at similar concentrations of catalytic mTOR inhibitors (Fig. 2B and 3) argues against S6K1 requiring less stringent inhibition of mTORC1 to be targeted. One possibility is that binding of FKBP12-rapamycin to mTOR sterically impedes S6K1 from accessing mTORC1, as suggested in light of the recently solved structure of mTORC1 (48). Hindrance of substrate access by FKBP12-rapamycin could, indeed, explain why rapamycin inhibited phosphorylation of S6-S240/244 in all cell lines, independent of mTORC1 complex stability (Fig. 3 and 4). An alternative possibility, postulated by Choo and Blenis (7), is that differential binding affinities of S6K1 and 4EBP1 for mTORC1 might govern their sensitivity to rapamycin. Whereas 4EBP1 binds raptor relative tightly, S6K1 displays a lower affinity for raptor, and small disturbances in the confirmation of mTORC1 might be sufficient to fully inhibit S6K1 phosphorylation. The mechanism for how mTOR represses autophagy in mammalian cells is still vaguely defined but involves phosphorylation of the Ulk1-mAtg13-FIP200 complex, and overexpressed Ulk1 binds to raptor (12, 18, 20). Future studies will be needed to define the biophysical binding properties of Ulk1 for raptor and compare it to other mTORC1 substrates such as S6K1 and 4EBP1.
Pharmacological induction of autophagy is a promising approach to increase the clearance of aggregation-prone toxic protein species such as mutant Huntingtin or α-synuclein (33, 34, 44) and intracellular pathogens such as Mycobacterium tuberculosis (14). While rapamycin has shown some beneficial effects in cell culture and animal models of proteinopathies (34), the present work suggests that the full autophagy induction potential of rapamycin can be achieved by destabilizing mTORC1 or using low concentrations of ATP-competitive inhibitors along with rapamycin. Such combinations result in synergistic inhibition of mTORC1 and spare the mTORC2-Akt pathway. This last point is significant as Akt is an important regulator of glucose homeostasis in vivo (8), and adverse side effects from long-term changes in circulating and tissue glucose concentrations are undesirable. Factors that determine mTORC1 stability are largely unexplored but might involve additional binding partners or posttranslational modifications of mTOR or raptor. The identification of such factors could ultimately be used as positive and/or negative selection criteria when stratifying patients for rapamycin therapy. Finally, the observations above associated with combinations of rapamycin and ATP-competitive inhibitors are timely because the latter class of inhibitors has recently entered clinical trials for cancer.
ACKNOWLEDGMENTS
We thank Stephen Helliwell for comments and suggestions.
All authors (except D.J.K.) are employees of Novartis Pharmaceuticals.
D.J.K. was supported by NIH grant GM53396.
Footnotes
Published ahead of print on 16 May 2011.
REFERENCES
- 1. Blommaart E. F., Luiken J. J., Blommaart P. J., van Woerkom G. M., Meijer A. J. 1995. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 270:2320–2326 [DOI] [PubMed] [Google Scholar]
- 2. Boland B., et al. 2008. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J. Neurosci. 28:6926–6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borisy A. A., et al. 2003. Systematic discovery of multicomponent therapeutics. Proc. Natl. Acad. Sci. U. S. A. 100:7977–7982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown E. J., et al. 1994. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369:756–758 [DOI] [PubMed] [Google Scholar]
- 5. Chiu M. I., Katz H., Berlin V. 1994. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. U. S. A. 91:12574–12578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi J., Chen J., Schreiber S. L., Clardy J. 1996. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273:239–242 [DOI] [PubMed] [Google Scholar]
- 7. Choo A. Y., Blenis J. 2009. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 8:567–572 [DOI] [PubMed] [Google Scholar]
- 8. Crouthamel M. C., et al. 2009. Mechanism and management of AKT inhibitor-induced hyperglycemia. Clin. Cancer Res. 15:217–225 [DOI] [PubMed] [Google Scholar]
- 9. Dancey J. 2010. mTOR signaling and drug development in cancer. Nat. Rev. Clin. Oncol. 7:209–219 [DOI] [PubMed] [Google Scholar]
- 10. Feldman M. E., et al. 2009. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fingar D. C., Blenis J. 2004. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23:3151–3171 [DOI] [PubMed] [Google Scholar]
- 12. Ganley I. G., et al. 2009. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284:12297–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia-Martinez J. M., et al. 2009. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 421:29–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gutierrez M. G., et al. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766 [DOI] [PubMed] [Google Scholar]
- 15. Hara K., et al. 2002. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110:177–189 [DOI] [PubMed] [Google Scholar]
- 16. Heitman J., Movva N. R., Hall M. N. 1991. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253:905–909 [DOI] [PubMed] [Google Scholar]
- 17. Hidalgo M., Rowinsky E. K. 2000. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene 19:6680–6686 [DOI] [PubMed] [Google Scholar]
- 18. Hosokawa N., et al. 2009. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20:1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jacinto E., et al. 2004. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 6:1122–1128 [DOI] [PubMed] [Google Scholar]
- 20. Jung C. H., et al. 2009. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20:1992–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim D. H., et al. 2002. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–175 [DOI] [PubMed] [Google Scholar]
- 22. Kimura S., Fujita N., Noda T., Yoshimori T. 2009. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 452:1–12 [DOI] [PubMed] [Google Scholar]
- 23. Kimura S., Noda T., Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460 [DOI] [PubMed] [Google Scholar]
- 24. Klionsky D. J., et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klionsky D. J., Elazar Z., Seglen P. O., Rubinsztein D. C. 2008. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4:849–950 [DOI] [PubMed] [Google Scholar]
- 26. Laplante M., Sabatini D. M. 2009. mTOR signaling at a glance. J. Cell Sci. 122:3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma X. M., Blenis J. 2009. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10:307–318 [DOI] [PubMed] [Google Scholar]
- 28. Motzer R. J., et al. 2008. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372:449–456 [DOI] [PubMed] [Google Scholar]
- 29. Nicklin P., et al. 2009. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136:521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oshiro N., et al. 2004. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 9:359–366 [DOI] [PubMed] [Google Scholar]
- 31. Paglin S., et al. 2005. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 65:11061–11070 [DOI] [PubMed] [Google Scholar]
- 32. Pankiv S., et al. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145 [DOI] [PubMed] [Google Scholar]
- 33. Ravikumar B., Duden R., Rubinsztein D. C. 2002. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11:1107–1117 [DOI] [PubMed] [Google Scholar]
- 34. Ravikumar B., et al. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–595 [DOI] [PubMed] [Google Scholar]
- 35. Rubinsztein D. C., Gestwicki J. E., Murphy L. O., Klionsky D. J. 2007. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6:304–312 [DOI] [PubMed] [Google Scholar]
- 36. Sabatini D. M., Erdjument-Bromage H., Lui M., Tempst P., Snyder S. H. 1994. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78:35–43 [DOI] [PubMed] [Google Scholar]
- 37. Sarbassov D. D., et al. 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22:159–168 [DOI] [PubMed] [Google Scholar]
- 38. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 39. Sarkar S., et al. 2005. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170:1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shor B., et al. 2008. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 68:2934–2943 [DOI] [PubMed] [Google Scholar]
- 41. Takeuchi H., et al. 2005. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 65:3336–3346 [DOI] [PubMed] [Google Scholar]
- 42. Thoreen C. C., et al. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284:8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vézina C., Kudelski A., Sehgal S. N. 1975. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. (Tokyo) 28:721–726 [DOI] [PubMed] [Google Scholar]
- 44. Webb J. L., Ravikumar B., Atkins J., Skepper J. N., Rubinsztein D. C. 2003. α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278:25009–25013 [DOI] [PubMed] [Google Scholar]
- 45. Weiss A., Roscic A., Paganetti P. 2009. Inducible mutant Huntingtin expression in HN10 cells reproduces Huntington's disease-like neuronal dysfunction. Mol. Neurodegener. 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wullschleger S., Loewith R., Hall M. N. 2006. TOR signaling in growth and metabolism. Cell 124:471–484 [DOI] [PubMed] [Google Scholar]
- 47. Yang Z., Klionsky D. J. 2010. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22:124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yip C. K., Murata K., Walz T., Sabatini D. M., Kang S. A. 2010. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38:768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu K., et al. 2009. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 69:6232–6240 [DOI] [PubMed] [Google Scholar]
- 50. Zimmermann G. R., Lehar J., Keith C. T. 2007. Multitarget therapeutics: when the whole is greater than the sum of the parts. Drug Discov. Today 12:34–42 [DOI] [PubMed] [Google Scholar]