

Abstract
Extensive studies have identified many growth factors and intracellular pathways that can promote neuronal survival after retinal injury, but the intrinsic survival mechanisms in the normal retina are poorly understood. Here we report that genetic ablation of Shp2 (Ptpn11) protein phosphatase resulted in progressive apoptosis of all retinal cell types. Loss of Shp2 specifically disrupted extracellular signal-regulated kinase (ERK) signaling in Müller cells, leading to Stat3 activation in photoreceptors. However, neither inactivation of Stat3 nor stimulation of AKT signaling could ameliorate the Shp2 retinal degeneration. Instead, constitutively activated Kras signaling not only rescued the retinal cell numbers in the Shp2 mutant but also functionally improved the electroretinogram recording (ERG). These results suggest that Shp2-mediated Ras–mitogen-activated protein kinase (Ras-MAPK) signaling plays a critical role in Müller cell maturation and function, which is necessary for the survival of retinal neurons.
INTRODUCTION
Neurodegenerative diseases, comprising a large portion of age-related human disorders, present a significant challenge for therapy, since most neuronal cells at adult stages, especially in the central nervous system, are postmitotic and unable to regenerate (7). Retinal degenerations (RD), including retinitis pigmentosa (RP), age-related macular degeneration (AMD), and glaucoma, are the leading neurodegenerative diseases in the human visual system, eventually leading to vision loss. RP and AMD are characterized by the loss of photoreceptor cells, while glaucoma results in the loss of retinal ganglion cells (RGCs). Although more than 200 genetic loci have been identified in human inherited retinal degenerations during the last 2 decades (12, 33), little is known regarding why and what kinds of intrinsic cell survival mechanisms are damaged in inherited or aging-induced retinal degeneration diseases (15, 33).
The adult retina in humans and mice share a prototypic laminar structure consisting of seven distinct cell types: six types of neurons (retinal ganglion cells, amacrine cells, bipolar cells, horizontal cells, and rod and cone photoreceptor cells) and one type of glia (Müller glia). Derived from a common retinal progenitor cell population during development, these cells eventually become organized into the proper retinal layers in a conserved fashion (11). Among these retinal cells, Müller glia are the only ones whose cell bodies span virtually the entire thickness of the retina from the outer nuclear layer (ONL) to the optic nerve fiber layer/inner limiting membrane layer (NFL/ILML), ensheathing every individual neuronal cell in the adult retina (1). Consistent with the structural relationships between Müller glia and other retinal cells, it is believed that Müller glia are critical for the survival and function of retinal neurons in both normal and pathogenic conditions, thus serving multiple functions carried out by astrocytes and oligodendrocytes in other parts of the central nervous system (1, 5, 13, 36).
How Müller glial cells protect retinal neurons from degeneration is currently under intense investigations. In both inherited and stress-induced retinal degeneration models, photoreceptors sharply increase the expression of endothelin 2 (End2), which is thought to induce Müller glial cells to secrete a number of neuroprotective factors, including leukemia inhibitory factor (LIF) and fibroblast growth factor 2 (FGF2) (10, 14, 27). This led to significant elevation of extracellular signal-regulated kinase (ERK), Stat3, and AKT phosphorylation in the retina (14, 32). In the LIF knockout mice, however, the neuroprotective function of Müller glial cells is lost and End2/FGF2/Stat3/AKT signaling is not upregulated (14). Further experiments showed that the receptor for LIF, gp130, is specifically required in the photoreceptors for neuroprotection but not in the Müller glial cells, suggesting that the Müller glia-derived LIF activates gp130 signaling in photoreceptors to induce protection (31). Recent studies also show that brain-derived neurotrophic factor (BDNF) indirectly promotes photoreceptor survival by stimulating TrkB signaling in Müller glial cells, further underscoring the central role of Müller glial cells in mediating neurotrophin signaling in photoreceptor protection (9).
Although ERK phosphorylation is one of the hallmarks of Müller glial cell activation in response to retinal injury, the functional importance of ERK signaling in the unstressed retina has not been explored in vivo. We have previously demonstrated that Shp2, a protein tyrosine phosphatase, is essential for Ras/ERK signaling in the establishment of retinal progenitor cell fate during optic vesicle patterning (3). The present work shows that Shp2 depletion after embryonic day 10.5 (E10.5) does not reduce retinal progenitor cell proliferation or perturb neuroretinal differentiation but leads to severe retinal degeneration and optic nerve dystrophy in the adult stages. In the Shp2 mutant, ERK signaling is specifically disrupted in Müller glial cells, while Stat3 but not AKT signaling is upregulated in photoreceptor cells. Finally, by generating compound mutants, we show that Kras signaling, but not AKT or Stat3 signaling, reverses the Shp2 retinal degeneration phenotypes both morphologically and functionally. These results establish Shp2-mediated Ras/mitogen-activated protein kinase (MAPK) signaling as the critical intrinsic survival mechanism for normal retina.
MATERIALS AND METHODS
Mice.
Shp2flox, Stat3flox, and LSL-KrasG12D mice have been previously described (19, 30, 37). Six3-Cre mice were kind gifts from Yasuhide Furuta (M.D. Anderson Cancer Center, Houston, TX) (6). α-Cre mice were kindly provided by Nadean Brown (Children's Hospital Research Foundation, Cincinnati, OH) and Ruth Ashery-Padan (Tel Aviv University, Tel Aviv, Israel) (18). Ptenflox mice were obtained from the Jackson Laboratory (Bar Harbor, ME) (8). All mice were maintained in mixed genetic backgrounds and were housed with normal 12-h light/12-h dark cycle. All experiments were performed in accordance with institutional guidelines.
Histology and cell counting.
Enucleated adult eyeballs with intact optic nerve were dissected to remove the cornea and the lens before overnight fixation in 4% paraformaldehyde (PFA) and progressive ethanol dehydration. After paraffin embedding, the samples were sectioned on a Leica 2125 microtome at a thickness of 10 μm and stained with hematoxylin and eosin (H&E) in a Leica ST5010 autostainer. For cell counting, only the retinal cross sections through the optic disk (optic nerve head) were selected for terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (5 sections per mouse) or immunohistochemistry (10 sections per mouse), and cells from at least three animals for each genotype at the specific stages were counted. Statistical significance was calculated using the Student t test.
Immunohistochemistry and Western blot.
The eyeball samples were fixed in 4% PFA overnight and cryosectioned before standard immunohistochemistry or TUNEL staining was performed as previously described (3, 24). For Shp2, phosphorylated ERK (phospho-ERK), phospho-Stat3, and phospho-AKT staining, we performed the tyramide signal amplification (TSA plus fluorescein system; PerkinElmer, Waltham, MA) procedure as described previously (3). Western blots on dissected retinas from adult mice were carried out as previously described (23). The antibodies used were anti-Shp2 (Sc-280; Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-ERK1/2 (catalog no. 4370; Cell Signaling Technology, Beverly, MA), anti-phospho-AKT (Ser473) (catalog no. 4060; Cell Signaling Technology), anti-phospho-Stat3 (Tyr705) (catalog no. 9145; Cell Signaling Technology), anti-Ki67 (catalog no. 550609; BD Pharmingen, San Diego, CA), anti-phospho-histone H3 (catalog no. 06-570; Upstate, Temecula, CA), anti-Brn3a (5A3.2; Chemicon, Billerica, MA), anticalbindin (D28K) (Sigma, St. Louis, MO), anti-glutamine synthetase (G2781) (Sigma), antirecoverin (SAB2101971) (Sigma), antirhodopsin (R5403) (Sigma), anti-NCAM/VC1.1 (NCAM stands for neural cell adhesion molecule (C0678) (Sigma), anti-GFAP (GFAP stands for glial fibrillary acidic protein) (Z0334; Dako, Carpinteria, CA), anti-NF-165 (2H3; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), and anti-Pax6 (PRB-278P; Covance, Berkeley, CA). Anti-Chx10 was kindly provided by Roderick McInnes (University of Toronto, Canada).
ERG.
Prior to electroretinogram (ERG) testing, 3-month-old mice were allowed to adapt to the dark overnight. Under dim red illumination, mice were anesthetized with an intraperitoneal injection of ketamine (70 mg/kg of body weight), xylazine (14 mg/kg), and acepromazine (1.2 mg/kg). Corneal anesthesia was achieved with a single drop of 0.5% proparacaine, and pupils were dilated with 1% tropicamide and 2.5% phenylephrine. The mice were placed on a heating pad maintained at 39°C inside a Ganzfeld dome. Platinum electrodes were gently placed on each cornea, and a small amount of 2.5% methylcellulose was applied to each eye. Platinum subdermal needle electrodes were inserted into the tail and forehead to serve as ground and reference electrodes, respectively. Signals were band pass filtered from 0.1 to 300 Hz and digitally sampled at 10 kHz. Flashes in the scotopic range were generated by a pair of cyan Luxeon K2 light-emitting diodes (LEDs) (λpeak = 505 nm; Δλ1/2 = 30 nm; Phillips Lumileds, San Jose, CA) wired in series. Square pulses of 0.5-ms duration and various currents were driven through the LEDs to create flashes of different intensities. At the lowest intensity, 25 responses were averaged with a delay of 4 s between each flash. As the intensity of the flash increased, fewer responses were averaged with a longer delay between flashes. Cone ERGs were obtained by the double-flash method using dual 1,500-W xenon flash bulbs (Novatron, Dallas, TX). Briefly, an initial conditioning flash saturates both rods and cones, followed 2 s later by a probe flash. Since cones recover faster than rods, the ERG recorded after the probe flash represents a cone-dominated response. For analysis of ERG waveforms, the A-wave was measured from baseline to trough of the initial negative deflection (unfiltered) and the B-wave was measured from the A-wave trough to the peak of the subsequent positive deflection (low-pass filtered; cutoff frequency [fc] = 60 Hz).
RESULTS
Acute retinal degeneration in Six3-Cre; Shp2flox/flox adult mice.
We previously reported that Shp2 was required for the early optic cup patterning at embryonic day 9.0 (E9.0), but the Six3-Cre-mediated depletion of Shp2 after E10.5 did not disrupt retinal development (3). Indeed, while the Six3-Cre driver could efficiently activate the R26R Cre reporter and ablate Shp2 in the central retina, the retinal progenitor genes Pax6, Sox2, and Chx10 and the retinal pigmented epithelium gene Mitf were all properly expressed in the Six3-Cre; Shp2flox/flox mutants at E14.5 (see Fig. S1 in the supplemental material). Using the M-phase marker phospho-histone H3 (pHH3) and the cell cycle marker Ki67, we did not observe any cell proliferation defects in the Six3-Cre; Shp2flox/flox mutants from the embryonic to early postnatal stages (see Fig. S2A to F in the supplemental material). By postnatal day seven (P7), retinal progenitor cells in the Shp2 mutant retina also exited the cell cycle appropriately as shown by the loss of pHH3 and Ki67 staining (see Fig. S2G and H in the supplemental material). As further demonstrated by a panel of retinal markers, retinal ganglion cells (Brn3a), amacrine cells (Pax6), bipolar cells (Chx10), horizontal cells (calbindin), Müller cells (glutamine synthetase), photoreceptor cells (recoverin and rhodopsin), and astrocytes (glial fibrillary acidic protein [GFAP]) were all present in similar densities in both the wild type and the Shp2 mutant (Fig. 1A and B; see Fig. S3 in the supplemental material).
Fig. 1.
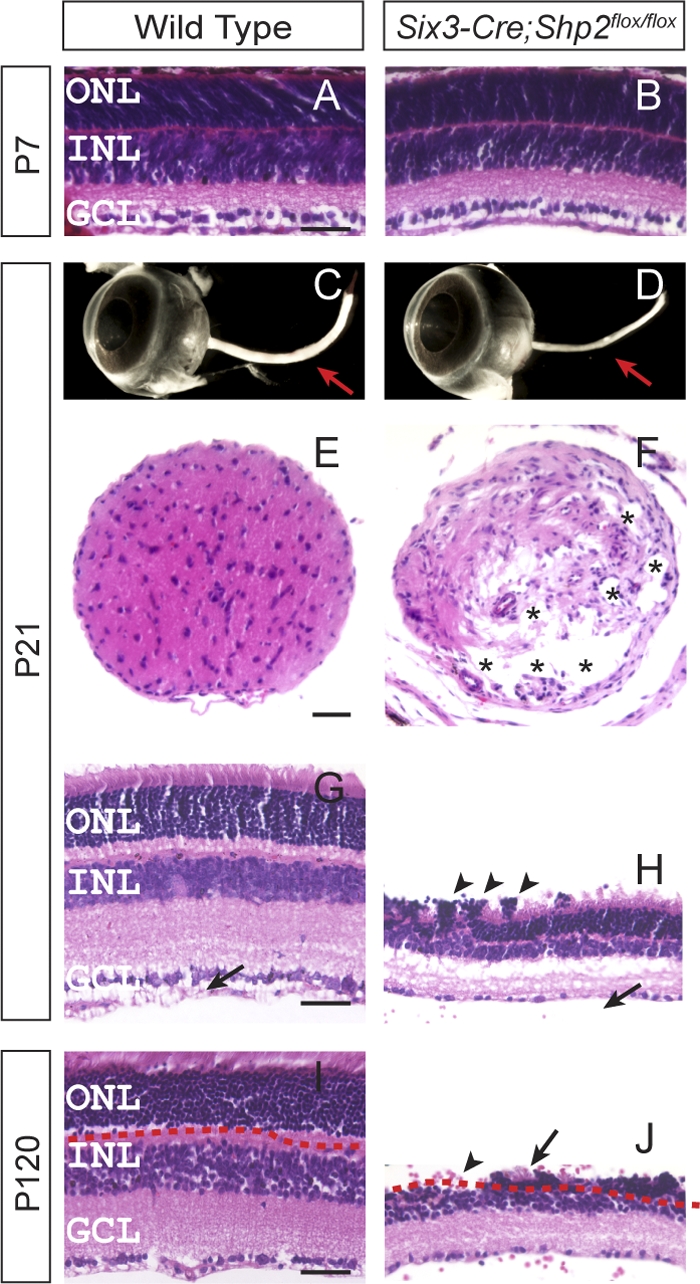
Severe degeneration of the retina and optic nerve in Six3-Cre; Shp2flox/flox adult mice. (A and B) Normal retinal histology in the Six3-Cre; Shp2flox/flox mutant at postnatal day 7 (P7). (C to F) In the Six3-Cre; Shp2flox/flox adult mice, the loss of optic nerve was observed in the dissected eyeballs (arrow in panel D) and in the transverse sections (asterisks indicate the vacuoles in the mutant optic nerve in panel F). (G to J) The Six3-Cre; Shp2flox/flox mutant retina sections were approximately 50% thinner than that of the wild type at P21 (H), and the outer nuclear layer (ONL) was completely absent in some retinal regions at P120 (the arrow in panel J indicates the neighboring residual photoreceptor cells). Notice the photoreceptor cell rosette (arrowheads in panel H) and the lack of the optic nerve fiber layer/inner limiting membrane layer (NFL/ILML) in the mutant retina (arrow in panel H). GCL, ganglion cell layer. Bars, 40 μm.
Despite its normal development, ocular defects in the Six3-Cre; Shp2flox/flox mutants became apparent at the weaning stage (P21) when the Shp2 mutant optic nerve appeared much thinner than the wild-type controls (Fig. 1C and D, arrows). In cross sections, the dystrophic optic stalk was disorganized with numerous vacuoles, indicating a loss of neural fibers (Fig. 1E and F, asterisks). Further histological analysis of the Shp2 mutant revealed drastic reduction in retinal thickness, formation of rosette structures in the outer nuclear layer (ONL) and absence of the optic nerve fiber layer/inner limiting membrane layer (NFL/ILML) (Fig. 1G and H, arrows and arrowheads). In contrast, none of the control animals (Six3-Cre or Six3-Cre; Shp2flox/+) exhibited any ocular phenotypes (Fig. 1G and data not shown). In mice on postnatal day 120, some regions of the ONL exhibited a complete loss of photoreceptor cells (Fig. 1I and J, arrow and arrowhead). Since Six3-Cre is known to be active in ventral forebrain, we also ablated Shp2 using a-Cre, which is exclusively expressed in peripheral retina (18). In the adult a-Cre; Shp2flox/flox mutants, we observed similar destruction of the retinal structure, including photoreceptor and ganglion cell loss (data not shown). Therefore, Shp2 deficiency resulted in rapid retinal degeneration in the adult animals.
Progressive cell apoptosis in the Shp2 mutant retina.
To determine whether retinal degeneration occurred through an apoptotic mechanism in the Six3-Cre; Shp2flox/flox mutants, we performed terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays. From E16 to P7, we observed similar levels of naturally occurring cell death in both the wild-type and Six3-Cre; Shp2flox/flox retinae (see Fig. S4A to C in the supplemental material). However, by P10 when cell apoptosis was diminishing in the wild-type controls, an increasing number of TUNEL-positive cells were observed in the Shp2 mutant retinae (see Fig. S4D to F in the supplemental material). Detailed quantification also revealed distinctive cell apoptosis dynamics in different retinal cell layers, with abnormal cell death in the ganglion cell layer (GCL) predominantly around P10, the inner nuclear layer (INL) from P10 to P14, and the outer nuclear layer around P21.
The extensive cell death in the Shp2 mutant was consistent with its pleiotropic degeneration. By immunostaining for cell-specific markers, we showed that there was roughly a 50% reduction in all retinal neurons and Müller glia in the P21 Shp2 mutants (Fig. 2A to F). The NF-165-positive nerve fibers in the NFL/ILML were lost, and the neural cell adhesion molecule (NCAM) (VC1.1)-staining inner plexiform layer (IPL) was significantly narrowed (Fig. 2G and H). As indicated by the upregulation of GFAP expression, the Shp2 mutant retina exhibited reactive Müller cell gliosis, a hallmark of retinal degeneration (Fig. 2I). By P120, all layers of the mutant retina were further reduced in thickness, with sparsely populated Müller cells and severely depleted photoreceptors (Fig. 2J and K and data not shown). These results suggest that Shp2 plays an undefined role in the maintenance of retinal neurons.
Fig. 2.

Shp2 depletion in Six3-Cre; Shp2flox/flox adult retina induced reduction in all retinal cell types and Müller cell gliosis. (A to F) Expression and relative fold changes of the retinal cell markers at P21. Brn3a stained for retinal ganglion cells, Pax6 for amacrine cells, Chx10 for bipolar cells, calbindin for horizontal cells, glutamine synthetase for Müller glial cells, recoverin for photoreceptors (rod and cone). The relative fold changes were determined by counting the number of cells and comparing them to the value for the wild type. All retina cell types were decreased by about 50% compared to wild type (bar graphs in panels A to F) (**, P ≤ 0.001). (G to I) We also observed the lack of optic nerve fiber by NF-165 staining (arrows in panels G), thinner inner plexiform layer (IPL) indicated by VC1.1 staining (for membranes of amacrine cells; brackets in panels H), and Müller cell gliosis by glial fibrillary acidic protein (GFAP) staining in the inner retina (arrows in panels I). (J and K) Expression of cell type markers in P120 Six3-Cre; Shp2flox/flox mutants. Note the fewer and disorganized Müller glial cells in the mutant (J) and a complete cell loss in some ONL regions (brackets in panels K). DAPI, 4′,6′-diamidino-2-phenylindole. Bar, 50 μm.
Dysregulation of ERK and Stat3 signaling in the Shp2 mutant.
To investigate the mechanism of retinal degeneration, we next examined the activities of the well-known neural survival pathways in the Shp2 mutants. Although strong during embryonic development, retinal extracellular signal-regulated kinase (ERK) signaling is known to be greatly diminished in the early postnatal stage, before rising again sharply after P8 (4). Interestingly, Western blotting of the P5 Six3-Cre; Shp2flox/flox retina did not detect any changes in phospho-ERK level, consistent with the idea that Shp2 is not required for the basal ERK signaling (Fig. 3A) (20). In the P21 Six3-Cre; Shp2flox/flox retina, however, there was strong upregulation of Stat3 phosphorylation, while ERK phosphorylation was sharply reduced (Fig. 3A). This is not due to a disproportional loss of specific cell types, because we have shown above that all retinal cell types degenerated to the similar extent (Fig. 2A to F). Furthermore, phospho-AKT levels remained unchanged (Fig. 3A).
Fig. 3.

Aberrant downstream signaling in the Six3-Cre; Shp2flox/flox retina. (A) Western blotting confirmed Shp2 depletion in the Six3-Cre; Shp2flox/flox retina at P5, but only by P21 did we observe the decrease in extracellular signal-regulated kinase (ERK) phosphorylation and the increase in Stat3 phosphorylation. The phospho-AKT level was unchanged. WT, wild type; pERK, phosphorylated ERK. (B) In the wild-type retina, Shp2 was ubiquitously expressed from the ganglion cell layer (GCL) to the inner nuclear layer (INL) but was almost absent in the outer nuclear layer (ONL). In the Six3-Cre; Shp2flox/flox retina, Shp2 was depleted in all layers. (C) Phospho-Stat3 was undetectable in the wild-type retina but strongly elevated in the Six3-Cre; Shp2flox/flox retina. Notice the scattered pattern of phospho-Stat3 staining (arrow). (D) The intensity of phospho-AKT staining was unchanged in the Six3-Cre; Shp2flox/flox retina. (E to H) In the wild-type retina, phospho-ERK (pERK) staining was predominantly observed in glutamine synthetase (GS)-positive Müller glial cells (arrows). The white boxes in panels E show the sections shown in panels F, G, and H. Such staining was lost in the Six3-Cre; Shp2flox/flox retina (arrowheads). Bar, 100 μm.
We next sought to investigate the role of Shp2 in neural survival pathways by examining their expression patterns. By immunofluorescence, Shp2 was predominantly found in the GCL and INL but not in the photoreceptor-containing ONL in the wild-type P21 retina, consistent with a previous report of Shp2 expression (Fig. 3B) (16). This was surprising, because the ONL was clearly disrupted by rosette structures in the Shp2-depleted mutant retina, suggesting that Shp2 may be indirectly required for photoreceptor cell survival (Fig. 3B). Further evidence for the non-cell-autonomous function of Shp2 came from phospho-Stat3 immunostaining, which was mostly observed in the disorganized photoreceptors (Fig. 3C, arrow). In contrast, the phospho-AKT pattern was unchanged in the Shp2 mutants (Fig. 3D). Using a tyramide-enhanced immunofluorescence technique, we also detected low but consistent phospho-ERK staining in the wild-type retina, and the staining appeared to span the Shp2-expressing GCL and INL regions. Further labeling with glutamine synthetase showed that phospho-ERK was present within the cell bodies of Müller glia in the wild-type retina, but such ERK activation was lost in the Six3-Cre; Shp2flox/flox mutant (Fig. 3E to H). Therefore, Shp2 deficiency induced Stat3 activation in photoreceptors but disrupted the basal level of Müller glia-specific ERK phosphorylation.
Stat3 depletion aggravated Shp2 retinal defects.
The ectopic Stat3 phosphorylation in the Six3-Cre; Shp2flox/flox mutant is particularly intriguing, because Shp2 has been shown to suppress Stat3 signaling by directly desphosphorylating Stat3 or its upstream kinase, JAK (21, 34). This led us to examine the biological significance of Stat3 activation in the Shp2 mutant retina. We generated Six3-Cre; Stat3flox/flox animals, which effectively abolished Stat3 signaling, but not ERK or AKT signaling, in the neural retina as shown by Western blots (Fig. 4A). Consistent with previous studies, ablation of Stat3 alone did not cause retinal degeneration (Fig. 4D and H) (22). However, as both ERK phosphorylation and Stat3 phosphorylation were downregulated in the Six3-Cre; Shp2flox/flox; Stat3flox/flox animal, the resulting retina was even thinner than that observed in the Six3-Cre; Shp2flox/flox mutant (Fig. 4E and I). Furthermore, the outer plexiform layer had largely disappeared, leaving the ONL and the INL merged into a single layer (Fig. 4E and I). These results suggest that Stat3 activation induced by Shp2 deficiency actually protected the retina from more profound damage.
Fig. 4.

Stat3 ablation aggravated Shp2 retinal defects. (A) Stat3 depletion in the Six3-Cre; Stat3flox/flox mutant retina did not affect ERK and AKT phosphorylation. In the Six3-Cre; Shp2flox/flox; Stat3flox/flox compound mutants, both Stat3 and ERK phosphorylation were disrupted. (B to E) Elimination of Stat3 did not cause an obvious retinal phenotype in the P21 Six3-Cre; Stat3flox/flox mutant, but it accelerated retinal degeneration in the Six3-Cre; Shp2flox/flox; Stat3flox/flox compound mutants compared to the Six3-Cre; Shp2flox/flox mutants. The sections were stained with hematoxylin and eosin (H&E). (F to I) Phospho-Stat3 was induced in the Six3-Cre; Shp2flox/flox mutant and abolished in the Six3-Cre; Shp2flox/flox; Stat3flox/flox mutant. The arrowheads in panel G indicate phospho-Stat3-positive cells. Bar, 40 μm.
Constitutive AKT signaling did not reverse Shp2 retinal degeneration.
Phosphoinositide 3-kinase (PI3K) signaling is another major neuroprotective pathway in retinal degeneration. We thus deleted the Pten gene, a negative regulator of PI3K signaling, to test whether increasing PI3K signaling could prevent the Shp2-deficient retina from degeneration. In the Six3-Cre; Ptenflox/flox mutants, Western blotting and immunostaining showed a significant increase in AKT phosphorylation, which is downstream of PI3K signaling, while phospho-ERK levels remained unchanged (Fig. 5A and L). Interestingly, we also observed a dramatic elevation of Stat3 phosphorylation, suggesting that Pten deletion may perturb normal retinal homeostasis. Indeed, the Six3-Cre; Ptenflox/flox mutant exhibited significant expansion of the inner plexiform layer and thickening of the optic nerve (Fig. 4D and H). However, in spite of the increased level of phospho-AKT throughout the retina, there was no attenuation of retinal degeneration in the Six3-Cre; Shp2flox/flox; Ptenflox/flox mutant (Fig. 5G, I, K, and M). Therefore, increasing PI3K signaling could not compensate for the loss of Shp2 function in retinal degeneration, demonstrating that Shp2 and PI3K signaling operated independently in retinal protection.
Fig. 5.

Pten mutation did not rescue Shp2 retinal defects. (A) Mutation in the gene encoding Pten, an upstream repressor of AKT signaling, resulted in ectopic AKT activation in the Six3-Cre; Shp2flox/flox; Ptenflox/flox mutant, but phospho-ERK levels remained suppressed. (B to E) The optic nerve became enlarged in the Six3-Cre; Ptenflox/flox mutant but remained thin in the Six3-Cre; Shp2flox/flox; Ptenflox/flox mutant. (F to I) At P21, Pten depletion led to thicker inner plexiform layers with displaced nuclei (brackets in panels H and I), but it failed to rescue retinal degeneration in the Six3-Cre; Shp2flox/flox; Ptenflox/flox mutant. (J to M) Phospho-AKT remained elevated in the Six3-Cre; Shp2flox/flox; Ptenflox/flox mutant. Bar, 40 μm.
Constitutive Kras signaling rescued the Shp2 retinal degeneration.
To determine the Shp2 downstream pathway in retinal degeneration, we next asked whether constitutive Kras signaling at physiologic levels could reverse the Shp2 phenotype. We tested this by crossing Six3-Cre mice with LSL-KrasG12D mice to induce retinal expression of activated KrasG12D under its endogenous promoter. This led to a modest increase in ERK but not AKT phosphorylation in the Six3-Cre; LSL-KrasG12D mutant, and no overt retinal phenotype was observed (Fig. 6A and data not shown). Quantitative Western blot analysis further showed that the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutant retina attained a level of phospho-ERK that was similar to that in the wild-type retina but had greatly reduced phospho-Stat3 levels compared to the Six3-Cre; Shp2flox/flox mutant retina (Fig. 6A). Remarkably, the optic nerve dystrophy was also reversed in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutant, which had a thick and well-organized optic stalk similar to the wild-type controls (Fig. 6B to G). In the neural retina, although Shp2 was still depleted in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutants, there was a marked recovery of phospho-ERK staining in Müller cells (Fig. 6H to M, arrows). Further histological analysis also showed normal retinal morphology without any overt degeneration (Fig. 6N to P). Even by P120, the optic nerve fiber layer/inner limiting membrane layer was still present in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutants, consistent with its normal optic nerve appearance (Fig. 6Q to S, arrows). Using molecular markers, we also showed that each retinal cell type is present in normal density in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutants (Fig. 6T). Therefore, constitutive Kras activation prevented retinal degeneration in the absence of Shp2.
Fig. 6.

Kras activation restored ERK phosphorylation and retinal morphology in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox compound mutants. (A) As shown by Western blotting, expression of the activated KrasG12D allele in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutant restored the phospho-ERK level to that of the wild type and suppressed phospho-Stat3 expression, while phospho-AKT levels remained unchanged. (B to G) The optic nerve morphology and histology was rescued in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutant. (H to M) While Shp2 staining remained absent in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutant, phospho-ERK expression was restored in the Müller glial cells (arrows). (N to S) The retinal histology in the P21 Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutant was comparable to that of the wild type. Notice the recovery of the optic nerve fiber layer/inner limiting membrane layer (arrows). (T) By cell counts, densities of all cell types recovered to the wild-type levels in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox compound mutants. Bar, 50 μm.
Finally, we performed electroretinograms (ERGs) to examine whether Kras activation rescued the Shp2 mutant functionally. To measure the rod-dominated scotopic ERG, 3-month-old dark-adapted mice were exposed to single flashes of increasing intensities (Fig. 7A). At the very bright flash of intensity (4.6 log photoisomerizations/rod), the saturated A-wave primarily reflected a rod response. The saturated A-wave amplitude was significantly reduced in the Six3-Cre; Shp2flox/flox mutants (mean ± standard error of the mean [SEM], 258.2 ± 25.2) compared to the wild type (582.7 ± 35.7) but was partially recovered in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutants (376.8 ± 26.7) (Fig. 7B). Similar functional rescue was observed in the dim flash series, where rod-mediated B-wave responses were consistently higher in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutants than in the Six3-Cre; Shp2flox/flox mutants throughout the scotopic range, while both the wild-type and Six3-Cre; LSL-KrasG12D animals exhibited normal responses (Fig. 7C and data not shown). To measure the cone ERG response, we used the double-flash method. Our results showed that the cone-mediated B-wave amplitude was also partially restored in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox mutants compared to the Six3-Cre; Shp2flox/flox mutants (Fig. 7D and E). These structural and functional studies not only supported the role of Shp2 in controlling Ras signaling but also demonstrated that retinal degeneration caused by aberrant receptor tyrosine kinase (RTK)-Shp2 signaling may be prevented by direct intervention in the Ras-MAPK pathway.
Fig. 7.

Functional recovery of the electrophysiological responses in the Six3-Cre; LSL-KrasG12D; Shp2flox/flox retina. (A to C) Scotopic (rod-dominated) electroretinogram recording (ERG) responses to single flashes of light. (A) Representative traces at four increasing flash energies; (B) representative saturated A-wave traces; (C), rod B-wave amplitude versus flash energy with Naka-Rushton fitted curves. (D and E) Double-flash ERGs to isolate cone responses. (D) Representative traces in response to second flash; (E) average cone B-wave amplitudes for each of the three cohorts. Three-month-old adult mice were used for the ERG tests. The means ± standard errors of the means (SEM) (error bars) are shown.
DISCUSSION
The normal retina in natural daylight conditions requires trophic factors for self-protection, but the intracellular signaling of these intrinsic neuroprotective mechanisms are far from understood (29). Although previous studies have shown that misexpression of dominant-negative Raf in the chick retina could induce apoptosis, genetic studies have so far failed to uncover any mutations in the Ras/Raf/MAP kinase pathway that have resulted in retinal degeneration (25). Furthermore, at least in explant culture, no abnormal retinal cell death was observed when ERK phosphorylation was inhibited pharmacologically (4). It has also been shown that mutation in insulin receptor substrate 2 (Irs2), a critical mediator of the insulin receptor/AKT pathway, could lead to photoreceptor death (35). However, without significant light damage, retinal degeneration has not been observed in insulin receptor and AKT knockouts (17, 26). Finally, as confirmed in this study, Stat3 signaling is also dispensable for retinal homeostasis under physiological conditions (22). These results thus raise questions regarding the identity of the intrinsic cell survival pathway in the unstressed retina.
We have now for the first time shown that Shp2 deletion abolished ERK phosphorylation in the neural retina, leading to extensive retinal cell death and degeneration. Although phospho-Stat3 was induced in the Shp2 mutant retina, unlike the widespread Shp2 ablation pattern, phospho-Stat3 appears to congregate around photoreceptor rosette structures. This suggests that Stat3 is unlikely a direct target of Shp2 in the retina. Moreover, we showed that, although Stat3 inactivation did not cause any overt retinal phenotype by itself, it exacerbated retinal degeneration when combined with a Shp2 deletion. Therefore, Stat3 activation appeared to be a general stress/injury response to Shp2 deletion-induced retinal degeneration, protecting retinal neurons from further damage (28). On the other hand, AKT phosphorylation was unchanged in the Shp2 mutant retina, and Pten-induced AKT signaling activation did not affect Shp2 retinal degeneration, demonstrating that there is little cross talk between Shp2 and AKT signaling during retinal degeneration. Finally, we showed that Kras-induced ERK signaling activation rescued the Shp2 mutant phenotype both morphologically and functionally. This result not only confirmed that Ras-ERK signaling is the downstream target of Shp2 in retinal survival but also provided a proof-of-principle example that precise targeting of relevant downstream signaling may be effective in preventing certain retinal degenerative diseases.
Our study further raises the question regarding the specific retinal cell type(s) that directly requires Shp2-Ras-ERK signaling. Although our immunostaining studies showed that the Shp2 protein was largely absent from the photoreceptor cell layer, photoreceptor cells nevertheless degenerated rapidly in the Shp2 mutant, suggesting that Shp2 may function nonautonomously in cells in photoreceptor cell survival. This was also consistent with the staining pattern of ERK phosphorylation, which was undetectable in the photoreceptor cells but was prominent in the Müller glial cells. Many studies have shown that the Müller glial cells are critical for the homeostasis of the normal retina, where they play important roles in energy metabolism, neurotransmitter recycling, and ion and water supply (2). Müller glial cells are also known to secrete neuroprotective factors in both normal and stressed conditions, and specific ablation of the Müller glial cells can lead to photoreceptor death and general retinal degeneration (5, 10, 14). These results suggest that Müller glial cell dysfunction may be a critical component of retinal degeneration in Shp2 mutants.
It should be noted that our results did not rule out a role of Shp2 in postnatal development of the retina. Although our Shp2 mutant retina appeared morphologically normal at P7, it degenerated rapidly from P10 to P21, a critical period of neuronal maturation. In fact, although constitutive Kras activation prevented retinal degeneration in the absence of Shp2, it only partially restored the ERG response. It is thus possible that Shp2 may be involved in the synaptic connection and pruning of retinal neurons, which was not completely rescued by Kras signaling. On the other hand, Shp2 ablation may also disrupt the maturation of Müller glial cells, which also occurs in the first 3 weeks after birth (2). This includes the ensheathment of retinal neurons, accumulation of specific enzymes, and changes in electrophysiological features, all of which are critical for Müller glial cells to maintain a healthy environment for retinal neuron survival. Further studies are required to determine the potential developmental phenotype in the Shp2 mutant and how it may contribute to the eventual retinal degeneration.
In summary, we would like to propose that Shp2 mediates a basal level of Ras-MAPK signaling in Müller glial cells during postnatal development and in the adult retina under normal physiological conditions. Although this Shp2-Ras-MAPK signaling is relatively low, it may be necessary for the Müller glial cells to carry out their normal functions in retinal homeostasis, which includes the maintenance of retinal architecture, supply of nutrients, clearance of neurotransmitters, and buffering of the ionic concentration. The lack of Shp2 signaling may also compromise the ability of Müller glial cells to release trophic factors to protect other retinal neurons from normal environmental stress. This intrinsic retinal survival pathway thus closely resembles the neuroprotective mechanism that is activated in the damaged retina, where Müller glial cells are known to secrete trophic factors, such as leukemia inhibitory factor (LIF), to promote protective Stat3 signaling in photoreceptor cells (14, 31). Therefore, Shp2 signaling may play equally important roles in retinal survival in both physiological and pathological conditions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yasuhide Furuta for the Six3-Cre mice, Roderick McInnes for the Chx10 antibody, and members of Zhang lab for discussions.
This work was supported by grants from NIH (grants EY017061 and EY018868 to X.Z.; grants EY004446, EY019908, and EY02520 [Core] to S.M.W.), the Retina Research Foundation (Houston), and Research to Prevent Blindness, Inc., to S.M.W.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
Published ahead of print on 16 May 2011.
REFERENCES
- 1. Bringmann A., et al. 2009. Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res. 28:423–451 [DOI] [PubMed] [Google Scholar]
- 2. Bringmann A., et al. 2006. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 25:397–424 [DOI] [PubMed] [Google Scholar]
- 3. Cai Z., Feng G.-S., Zhang X. 2010. Temporal requirement of the protein tyrosine phosphatase Shp2 in establishing the neuronal fate in early retinal development. J. Neurosci. 30:4110–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Donovan M., Doonan F., Cotter T. G. 2011. Differential roles of ERK1/2 and JNK in retinal development and degeneration. J. Neurochem. 116:33–42 [DOI] [PubMed] [Google Scholar]
- 5. Dubois-Dauphin M., et al. 2000. Early postnatal Müller cell death leads to retinal but not optic nerve degeneration in NSE-Hu-Bcl-2 transgenic mice. Neuroscience 95:9–21 [DOI] [PubMed] [Google Scholar]
- 6. Furuta Y., Lagutin O., Hogan B. L., Oliver G. C. 2000. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis 26:130–132 [PubMed] [Google Scholar]
- 7. Goldberg J. L., Barres B. A. 2000. The relationship between neuronal survival and regeneration. Annu. Rev. Neurosci. 23:579–612 [DOI] [PubMed] [Google Scholar]
- 8. Groszer M., et al. 2001. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294:2186–2189 [DOI] [PubMed] [Google Scholar]
- 9. Harada C., et al. 2011. Glia- and neuron-specific functions of TrkB signalling during retinal degeneration and regeneration. Nat. Commun. 2:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harada T., et al. 2000. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron 26:533–541 [DOI] [PubMed] [Google Scholar]
- 11. Harada T., Harada C., Parada L. F. 2007. Molecular regulation of visual system development: more than meets the eye. Genes Dev. 21:367–378 [DOI] [PubMed] [Google Scholar]
- 12. Hartong D. T., Berson E. L., Dryja T. P. 2006. Retinitis pigmentosa. Lancet 368:1795–1809 [DOI] [PubMed] [Google Scholar]
- 13. Jadhav A. P., Roesch K., Cepko C. L. 2009. Development and neurogenic potential of Müller glial cells in the vertebrate retina. Prog. Retin. Eye Res. 28:249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joly S., Lange C., Thiersch M., Samardzija M., Grimm C. 2008. Leukemia inhibitory factor extends the lifespan of injured photoreceptors in vivo. J. Neurosci. 28:13765–13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kennan A., Aherne A., Humphries P. 2005. Light in retinitis pigmentosa. Trends Genet. 21:103–110 [DOI] [PubMed] [Google Scholar]
- 16. Kinkl N., Hageman G., Sahel J., Hicks D. 2002. Fibroblast growth factor receptor (FGFR) and candidate signaling molecule distribution within rat and human retina. Mol. Vis. 14:149–160 [PubMed] [Google Scholar]
- 17. Li G., et al. 2007. Nonredundant role of Akt2 for neuroprotection of rod photoreceptor cells from light-induced cell death. J. Neurosci. 27:203–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marquardt T., Ashery-Padan R., Andrejewski N., Scardigli R., Guillemot F., Gruss P. 2001. Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105:43–55 [DOI] [PubMed] [Google Scholar]
- 19. Moh A., et al. 2007. Role of STAT3 in liver regeneration: survival, DNA synthesis, inflammatory reaction and liver mass recovery. Lab Invest. 87:1018–1028 [DOI] [PubMed] [Google Scholar]
- 20. Neel B. G., Gu H., Pao L. 2003. The ′Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28:284–293 [DOI] [PubMed] [Google Scholar]
- 21. Ohtani T., et al. 2000. Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3-and SHP2-mediated signals in immune responses. Immunity 12:95–105 [DOI] [PubMed] [Google Scholar]
- 22. Ozawa Y., et al. 2004. Downregulation of STAT3 activation is required for presumptive rod photoreceptor cells to differentiate in the postnatal retina. Mol. Cell. Neurosci. 26:258–270 [DOI] [PubMed] [Google Scholar]
- 23. Pan Y., Carbe C., Powers A., Feng G. S., Zhang X. 2010. Sprouty2-modulated Kras signaling rescues Shp2 deficiency during lens and lacrimal gland development. Development 137:1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pan Y., et al. 2008. Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development 135:301–310 [DOI] [PubMed] [Google Scholar]
- 25. Pimentel B., et al. 2000. c-Raf regulates cell survival and retinal ganglion cell morphogenesis during neurogenesis. J. Neurosci. 20:3254–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajala A., Tanito M., Le Y. Z., Kahn C. R., Rajala R. V. S. 2008. Loss of neuroprotective survival signal in mice lacking insulin receptor gene in rod photoreceptor cells. J. Biol. Chem. 283:19781–19792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rattner A., Nathans J. 2005. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J. Neurosci. 25:4540–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samardzija M., et al. 2006. Differential role of Jak-STAT signaling in retinal degenerations. FASEB J. 20:2411–2413 [DOI] [PubMed] [Google Scholar]
- 29. Stone J., et al. 1999. Mechanisms of photoreceptor death and survival in mammalian retina. Prog. Retin. Eye Res. 18:689–735 [DOI] [PubMed] [Google Scholar]
- 30. Tuveson D. A., et al. 2004. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5:375–387 [DOI] [PubMed] [Google Scholar]
- 31. Ueki Y., Le Y.-Z., Chollangi S., Muller W., Ash J. D. 2009. Preconditioning-induced protection of photoreceptors requires activation of the signal-transducing receptor gp130 in photoreceptors. Proc. Natl. Acad. Sci. U. S. A. 106:21389–21394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wahlin K. J., Campochiaro P. A., Zack D. J., Adler R. 2000. Neurotrophic factors cause activation of intracellular signaling pathways in Muller cells and other cells of the inner retina, but not photoreceptors. Invest. Ophthalmol. Vis. Sci. 41:927–936 [PubMed] [Google Scholar]
- 33. Wright A. F., Chakarova C. F., Abd El-Aziz M. M., Bhattacharya S. S. 2010. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11:273–284 [DOI] [PubMed] [Google Scholar]
- 34. Xu D., Q C. 2008. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 13:4925–4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yi X., et al. 2005. Insulin receptor substrate 2 is essential for maturation and survival of photoreceptor cells. J. Neurosci. 25:1240–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zack D. J. 2000. Neurotrophic rescue of photoreceptors: are Müller cells the mediators of survival? Neuron 26:285–286 [DOI] [PubMed] [Google Scholar]
- 37. Zhang E. E., Chapeau E., Hagihara K., Feng G. S. 2004. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc. Natl. Acad. Sci. U. S. A. 101:16064–16069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.