

Abstract
Signal transduction through mitogen-activated protein kinases (MAPKs) is implicated in growth and proliferation control through translation regulation and involves posttranslational modification of translation initiation factors. For example, convergent MAPK signals to Mnk1 lead to phosphorylation of eukaryotic translation initiation factor 4E (eIF4E), which has been linked to malignant transformation. However, understanding the compound effects of mitogenic signaling on the translation apparatus and on protein synthesis control remains elusive. This is particularly true for the central scaffold of the translation initiation apparatus and ribosome adaptor eIF4G. To unravel the effects of signal transduction to eIF4G on translation, we used specific activation of protein kinase C (PKC)-Ras-Erk signaling with phorbol esters. Phospho-proteomic and mutational analyses revealed that eIF4G1 is a substrate for PKCα at Ser1186. We show that PKCα activation elicits a cascade of orchestrated phosphorylation events that may modulate eIF4G1 structure and control interaction with the eIF4E kinase, Mnk1.
INTRODUCTION
In eukaryotes, protein synthesis is controlled mainly via restrictive initiation. Translation initiation occurs upon recruitment of mRNAs to 40S ribosomal subunits. Conventionally, this requires eukaryotic translation initiation factor 4F (eIF4F), a multipartite complex consisting of the m7G-cap binding protein eIF4E, the RNA helicase eIF4A, and the central scaffolding protein eIF4G, binding to the canonical 5′ m7G-cap structure on mRNAs (12). eIF4G, through interaction with eIF3 (16), provides the link to the 43S preinitiation complex.
Protein synthesis control is a fundamental homeostatic function of cells that is intricately linked to extracellular stimuli driving cell growth and proliferation. Hence, key mitogenic signal transduction pathways converge on translation initiation factors. Signaling to the phosphoinositide-3 kinase (PI3K) pathway activates the mammalian target of rapamycin (mTOR), which is the kinase for eIF4E-binding proteins (4E-BPs) (27). mTOR-catalyzed 4E-BP phosphorylation promotes eIF4E binding to eIF4G and, thus, cap-dependent translation (3). Similarly, the association of eIF4G with eIF3 and, thus, the 43S preinitiation complex, may be controlled by mTOR (14). The extracellular signal-regulated kinase 1 and 2 (Erk1/2) and p38 mitogen-activated protein kinases (MAPKs) converge on the MAPK signal integrating kinase (Mnk1), which phosphorylates eIF4E on Ser209 (8, 41). Mnk1 recognizes eIF4E through binding eIF4G (28), which is strongly responsive to MAPK activation of Mnk1 (35).
All mitogenic signal-controlled effects on translation machinery involve regulated associations with eIF4G. However, little is known about its role in these processes. Due to eIF4G's central function in ribosome recruitment, this impairs understanding of the adaptive responses of the translation apparatus to mitogenic signal transduction. eIF4G carries a multitude of confirmed phosphorylation sites that respond to mitogenic signaling. The first systematic analysis of eIF4G phosphorylation identified three major serum-responsive sites (29). Later, large-scale phospho-proteome analyses identified at least 12 additional sites (6, 10, 26, 32). Intriguingly, a majority of phosphorylation sites (including the initial three mitogen-responsive candidates) cluster in a defined region, structurally characterized as the interdomain linker due to its position in between HEAT (Huntingtin, elongation factor 3, a subunit of protein phosphatase 2A, and target of rapamycin) (23) domains 1 and 2 of eIF4G (22). The identity of kinases for any of the many confirmed eIF4G phosphorylation sites, the upstream signaling pathways responsible for their activation, and the physiological consequences of eIF4G phosphorylation are unknown.
We provide evidence that stimulation of serum-starved cells with the phorbol esters 12-O-tetradecanoylphorbol-13-acetate (TPA) and 12-O-(N-methylanthranyl)-phorbol-13-acetate (sapintoxin D [STD]) induces phosphorylation at Ser1186 and Ser1232 of eIF4G. Consideration of kinase consensus sites, coimmunoprecipitation (co-IP) assays, in vitro phosphorylation assays, studies with specific inhibitors, and mutational analyses of eIF4G suggest that Ser1186 is directly phosphorylated by PKCα. Protein-protein interaction studies with eIF4G harboring targeted amino acid substitutions suggested that phosphorylation of Ser1186 modulates the binding of eIF4G to Mnk1 and, thus, may be involved in regulating eIF4E phosphorylation.
MATERIALS AND METHODS
Cell lines, DNA transfections, and Tet-inducible cell lines.
HEK293 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and nonessential amino acids and were transfected with 10 μg of plasmid DNA using 25 μl of Lipofectamine 2000 (Invitrogen) per 15-cm plate. Sixteen hours posttransfection, the medium was changed to the starvation medium (without FBS). After 24 h of serum starvation, the cells were treated with kinase inhibitors and/or activators, harvested, and lysed as described below. Stable cell lines expressing tetracycline (Tet)-inducible eIF4G1 variants were established from Flip-In T-Rex HEK293 cells (Invitrogen) according to the manufacturer's instructions. Stable cell lines were maintained in DMEM supplemented with 10% FBS, nonessential amino acids, hygromycin B (100 μg/ml; Mediatech), and Blasticidin S HCl (15 μg/ml; Invitrogen). Cells were grown to ∼80% confluence, Tet induced, and starved for 24 h in serum-free medium and treated with kinase inhibitors and/or activators, harvested, and lysed as described below.
eIF4G1 fragments and mutants.
c-myc and Flag epitopes were inserted into NheI/HinDIII and XhoI/XbaI sites of pcDNA3.1 (Invitrogen) using annealed complementary oligonucleotide pairs 1/2 and 3/4, respectively (Table 1). eIF4G truncation variants were generated by PCR amplification of corresponding fragments with primers 5 to 11 (see Fig. 3A) (primers are listed in Table 1), which were ligated into the HinDIII and XhoI sites of the dual-tag cassette. Ala/Glu mutations of Ser1186 and/or Ser1188 (Ser1186/1188) were introduced using a QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) with primers 12 to 21 (Table 1). To establish stable Tet-inducible cell lines, eIF4G-e and its variants containing c-myc and Flag tags were subcloned into pcDNA5/FRT/TO (Invitrogen) from the corresponding pcDNA3.1 expression constructs. A recombinant glutathione S-transferase (GST)-tagged C-terminal fragment of eIF4G1 (GST-Ct) was purified as described before (18).
Table 1.
Oligonucleotide primers used in this study
| Primer |
Sequence (5′–3′)a | |
|---|---|---|
| No. | Nameb | |
| 1 | c-myc (+) | CTAGCACCATGGAGCAGAAACTCATCTCTGAAGAGGATCTGA |
| 2 | c-myc (−) | AGCTTCAGATCCTCTTCAGAGATGAGTTTCTGCTCCATGGTG |
| 3 | flag (+) | TCGAGGATTACAAGGATGACGACGATAAGAT |
| 4 | flag (−) | CTAGATCTTATCGTCGTCATCCTTGTAATCC |
| 5 | 683-Ct (+) | TTAAGCTTGGGCCCCCAAGGGGTGG |
| 6 | 1133 (−) | TTCTCGAGTACCGCTTGTTGAAGGGC |
| 7 | 1085 (+) | GTAAGCTTTTTGCACCTGGAGGGCGACTG |
| 8 | 1245 (+) | GTAAGCTTAAATCCAAGGCTATCATTGAGG |
| 9 | 1600 (−) | TTCTCGAGGTTGTGGTCAGACTCCTCCTC |
| 10 | 1412 (−) | TTCTCGAGCTGGCCTTCAGGTAGAAATTCC |
| 11 | ΔMnk1 (−) | TTCTCGAGGGCTGTGACAGATTTAAGGG |
| 12 | S1186A (+) | GCTACCAAGCGGGCCTTCAGCAAGGAAGTGG |
| 13 | S1186A (−) | CCACTTCCTTGCTGAAGGCCCGCTTGGTAGC |
| 14 | S1188A (+) | CAAGCGGAGCTTCGCCAAGGAAGTGGAGG |
| 15 | S1188A (−) | CCTCCACTTCCTTGGCGAAGCTCCGCTTG |
| 16 | S1186/88 S2A2 (+) | GCTACCAAGCGGGCCTTCGCCAAGGAAGTGG |
| 17 | 1186/88 S2A2 (−) | CCACTTCCTTGGCGAAGGCCCGCTTGGTAGC |
| 18 | 1186/88 S2E2 (+) | GCTACCAAGCGGGAATTCGAAAAGGAAGTGG |
| 19 | 1186/88 S2E2 (−) | CCACTTCCTTTTCGAATTCCCGCTTGGTAGC |
| 20 | S1232A (+) | CCTACCCCCAGTGGCCCCCCTGAAGGCG |
| 21 | S1232A (−) | CGCCTTCAGGGGGGCCACTGGGGGTAGG |
Restriction sites used for cloning are underlined, and mutated nucleotides are in boldface.
(+), forward (5′) primer; (−), reverse (3′) primer.
Fig. 3.

Mapping the PKCα phosphorylation site in eIF4G with C-terminal truncation variants. (A) Schematic representation of the eIF4G isoforms and C-terminal eIF4G fragments used in this study. All proteins are tagged on either terminus with Myc or Flag tags. CTMCΔMnk1 lacks the C-terminal 15 amino acids conveying Mnk1 binding activity. (B) Phosphorylation of eIF4G(Ser1148/Ser1232) in Tet-induced HEK293eIF4G-b cells (eIF4G-b) or in HEK293 cells transiently transfected with the CTMC fragment (CTMC). HEK293eIF4G-b cells were Tet induced and serum starved for 24 h and treated for 60 min with DMSO (0) or 200 nM TPA. HEK293 cells were transfected for 16 h, serum starved for 24 h, and treated for 60 min with DMSO (0) or 200 nM TPA. Cell lysates were subjected to Flag-IP followed by immunoblotting with the indicated antibodies. The experiment was repeated twice, and the results of a representative assay are shown. M, molecular mass. (C) TPA-induced phosphorylation by PKCα, PKCα-binding and Mnk1-binding of CTMC, CTMCΔMnk, CTC or CTM in Tet-induced/transfected HEK293 cells expressing C-terminal eIF4G fragments as indicated. Cells and cell lysates were processed as described for panel B. Experiments described in panels B and C were repeated three times with similar results; representative assays are shown. ND, not done.
Kinase inhibitors and activators.
Inhibitors of PKC (bis-indolylmaleimide 1 [Bis1]; Tocris), MEK1 (UO126; Cell Signaling), and Mnk1/2 (CGP57380; Sigma) and PKC activators 12-O-tetradecanoyl-phorbol-13-acetate (TPA; Sigma) and 12-O-(N-methylanthranyl)-phorbol-13-acetate (sapintoxin D; Enzo Life Sciences) were dissolved in dimethyl sulfoxide (DMSO) and used at the concentrations indicated in Results. Tetracycline (1 mg/ml) was dissolved in ethanol.
Immunoprecipitation, immunoblotting, and antibodies.
Cell lysates were prepared using polysomal lysis buffer (PLB) consisting of 10 mM HEPES, pH 7.4, 100 mM KCl, 5 mM MgCl2, 0.5% Igepal CA-630, NP-40 (Sigma), and 3 mM dithiothreitol (DTT) as described previously (35). Prior to IP, anti-Flag M2-agarose beads (Sigma) were washed twice in NT-2 solution (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 0.05% NP-40) and blocked with 1% bovine serum albumin (BSA) in the same buffer for 1 h at 4°C. The blocking solution was removed, and 30 μl of beads was incubated overnight at 4°C with cell lysates containing 6.5 mg of total protein in 1 ml of PLB. Then, beads with immunoprecipitated proteins were washed five times with 0.9 ml of NT-2 solution, and 75 μl of NuPAGE 2× loading buffer (Invitrogen) was added to each sample. Proteins were subjected to SDS-PAGE followed by blotting onto nitrocellulose membrane as described previously (35). Membranes were blocked with 5% BSA in phosphate-buffered saline–Tween 20 (PBST) solution and probed with phospho-specific antibodies for PKCα phosphorylated at Thr638 [p-PKCα(Thr638)], phospho-Ser substrate of PKC [p-substrate (PKC)], p-mTOR(Ser2448), p-Erk1/2(Thr202/Y204), p-eIF4G1(Ser1148), p-eIF4E(Ser209), p-S6(Ser235/236), p-S6(Ser240/244) (Cell Signaling), and p-eIF4G1(Ser1232) (Novus Biologicals) or antibodies for c-myc (Sigma), PKC (α, βII, ζ, and μ isoforms), mTOR, Erk1/2, Mnk1, S6, eIF4E, eIF4A, or N-terminal or C-terminal eIF4G1 [eIF4G1(Nt)/(Ct)] (Cell Signaling). Immunoblots were developed using SuperSignal West Pico or Femto ECL Kits (Thermo Scientific). For kinetic analyses, p-eIF4G(Ser1232), p-substrate (PKC), p-PKCα, p-Erk1/2, and p-eIF4E immunoblot signals were analyzed by chemiluminescence with a FluorChem FC2 imaging system (Cell Biosciences) and AlphaEase FC program.
In vitro phosphorylation assay and mass spectrometry.
Small-scale in vitro phosphorylation reactions were performed in 30 μl of mixture containing kinase buffer (25 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM DTT), 200 μM CaCl2, 20 μg/ml phosphatidyl serine, 20 μg/ml diacyl glycerol (DAG) analogue 1,2-dioctanoylglycerol (both Sigma), 1.5× Halt phosphatase inhibitor cocktail (Thermo Scientific), 100 μM ATP, and 10 μg of recombinant GST-Ct. The reaction mixture was incubated at room temperature with different amounts (0.5 or 1 μg) of recombinant GST-PKCα (Cell Sciences, Canton, MA) for various times. Aliquots taken at 0 min (before PKCα was added) and after 1, 2, or 4 h of incubation at 37°C were tested by immunoblotting with eIF4G(Ct) and p-substrate (PKC) antibodies. Large-scale in vitro phosphorylation for phospho-proteomic analyses was performed with 50 μg of recombinant GST-Ct. Two reaction mixtures, one of which contained 2 μg of recombinant GST-PKCα and a negative control, were incubated at room temperature for 4 h. A suspension (100 μl) of glutathione-Sepharose 4B (GE Healthcare) was blocked with 1% BSA in NT-2 solution for 1 h at 4°C and added to each sample. After a 2-h incubation at 4°C, the beads were washed twice with 1 ml of NT-2 solution with 1.5× Halt phosphatase inhibitor cocktail and twice with 1 ml of 50 mM NH4HCO3 and then subjected to phospho-proteomic analysis. Identification of residues phosphorylated in GST-Ct in vitro was performed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) at the Duke University Proteomics Core Facility. Phosphorylated and control GST-Ct samples were digested with the highest grade trypsin (Promega) and injected into high-pressure liquid chromatography nano-Acquity UPCL (Waters) for phospho-enrichment. Separated peptides were eluted directly into a Synapt G2 High Definition mass spectrometer. Phosphorylated peptides were identified using an Ultima quadrupole time-of-flight (Q-TOF) instrument. Stable isotope labeling with amino acids in cell culture (SILAC) quantification and peptide identification were accomplished using Rosetta Elucidator, version 3.2 (Rosetta Biosoftware), and Mascot, version 2.2 (Matrix Sciences).
RESULTS
Phorbol ester induces eIF4G phosphorylation.
Many studies on signaling pathways involved in translation regulation employed insulin or serum stimulation of cells and, accordingly, revealed events susceptible to the mTOR inhibitor rapamycin (14, 15, 29). Our approach is based on selective activation of the PKC family with phorbol esters (Fig. 1A). The main downstream effect of PKC activation is stimulation of Erk1/2 (Fig. 1B). However, activation of mTORC1 via Rsk1 upon phorbol ester-induced Erk1/2 stimulation has also been reported (Fig. 1A) (30).
Fig. 1.

eIF4G phosphorylation via PKC-Ras-Erk activation. (A) Schematic representation of signaling pathways and protein kinase inhibitors investigated in this research. TPA/STD-dependent stimulation of PKC activates Mnk1 and eIF4E phosphorylation. (B) Inhibition of TPA-dependent eIF4G phosphorylation by Bis1 and UO126. Tet- or mock-induced, serum-starved HEK293eIF4G-b cells were pretreated for 1 h with 4 μM Bis1 or 10 μM UO126. Cells were stimulated for 15 min with DMSO (0) or 200 nM TPA. Cell lysates were subjected to immunoblotting (left panel) or Flag-IP followed by immunoblotting (right panel). The experiment was repeated three times with similar results. A representative assay is shown. M, molecular mass. (C) Cell lysates were treated as described in panel B and processed for immunoblotting [eIF4G(Ser1148/1232)] or Flag-IP/immunoblotting [p-substrate (PKC)], followed by quantitative chemiluminescence measurements (see Materials and Methods). The values represent fold increases over untreated cells. The experiment was repeated twice with similar results. A representative assay is shown. (D) Substrate consensus of Erk1/2 (37) compared with the eIF4G sequence (aa 1229 to 1235) around the proposed substrate site at Ser1232. Values in parentheses indicate the relative selectivity for the amino acids. Bold letters in the consensus represent strongly preferred amino acids. Bold letters in the eIF4G sequence indicate positions identical to the consensus.
To enable biochemical assays, we utilized HEK293 cell lines with tetracycline (Tet)-inducible expression of eIF4G1 (referred to here as eIF4G [HEK293eIF4G]) carrying N-terminal (myc) and C-terminal (Flag) tags (35). Amino acid numbering used in this report refers to the eIF4G-a isoform (4). This isoform is poorly expressed, possibly due to a proline-rich N terminus; therefore, studies shown employ the eIF4G-b or -e isoforms, which are generated from initiation codons 40 amino acids (aa) or 196 aa downstream of the eIF4G-a variant, respectively (4).
To investigate the response of eIF4G to PKC activation, confirmed and potential phosphorylation sites were probed by immunoblotting of total lysates or Flag-IP after TPA stimulation of serum-starved, Tet-induced HEK293eIF4G-b cells (Fig. 1B). Tet induction led to efficient Myc-eIF4G-Flag expression (Fig. 1B). TPA stimulation for 15 min produced potent phosphorylation of Erk1/2 (Fig. 1B, left panel) in accordance with the predicted pathway activation (Fig. 1A). We consistently failed to detect significant changes of mTORC1(Ser2448) phosphorylation upon TPA treatment (Fig. 1B, left panel). Yet phosphorylation of eIF4G(Ser1148), which was shown to respond to the mTOR inhibitor rapamycin (29), was modestly increased (∼1.5-fold) by TPA, and this response was abolished by the PKC inhibitor bisindolylmaleimide 1 (Bis1) (Fig. 1B and C).
In contrast, eIF4G(Ser1232) phosphorylation showed a potent, 6-fold response to TPA, which was blocked by Bis1 and UO126 pretreatment (Fig. 1B and C). The context of Ser1232 in eIF4G, consistent with the Erk1/2 consensus (Fig. 1D), indicates preference for Pro residues at the −2 and +1 positions and for small hydrophobic amino acids (Leu and Val) at the −1 position (37). Only cyclin-dependent kinases and MAPKs preferentially phosphorylate Ser-Pro-oriented peptides (39), indicating their possible involvement in eIF4G(Ser1232) phosphorylation.
To investigate whether eIF4G is a direct target of PKC family kinases, we carried out Flag-IPs from samples shown in Fig. 1B (right panel) and quantified the chemiluminescent signal (Fig. 1C). First, we compared phosphorylation of eIF4G(Ser1148) in lysates before and after Flag-IP. Our data revealed a similar phosphorylation pattern for Ser1148 in both instances (Fig. 1B). Second, we used antibodies that recognize a phospho-Ser substrate of PKC [p-substrate (PKC)] with phospho-Ser residues surrounded by Arg/Lys at the −2 and +2 positions and a hydrophobic/aromatic residue at the +1 position (44). Interestingly, p-substrate (PKC) antibodies detected a product of identical size with eIF4G-b in the Flag-IP (Fig. 1B, right panel). This signal was strongly responsive to TPA stimulation (∼3.3-fold) and Bis1 inhibition but not UO126 treatment, suggesting a possible presence of a PKC phosphorylation site in eIF4G (Fig. 1C).
PKCα phosphorylates eIF4G.
TPA activates classic (α, β, and γ), novel (ε, σ, η, and θ), and μ PKC isoforms (11), and Bis1 preferably inhibits the α- and β-isoforms of PKC (42). To determine PKC isoform involvement in eIF4G phosphorylation, we tested Flag-IP of eIF4G from Tet-induced, TPA-stimulated HEK293eIF4G-b cells for coprecipitation of PKC proteins (Fig. 2A). This test specifically detected the PKCα isoform but not the PKCβII, -μ, or -ζ variant (Fig. 2A). This suggests that PKCα may phosphorylate eIF4G, an observation that is also supported by data with the PKCα-specific phorbol ester STD (see below). To confirm recognition of eIF4G by the p-substrate (PKC) antibody, we deliberately overexposed immunoblots of Flag-IPs from HEK293eIF4G-b cells stained with diverse eIF4G antibodies (Fig. 2B). These included antibodies against phospho-eIF4G(Ser1148), a C-terminal (Ct) eIF4G epitope (raised against a region surrounding Ala881), and an N-terminal (Nt) epitope (raised against a region surrounding Gly188). Antibodies detecting C-terminal signatures of eIF4G revealed the characteristic, uniform appearance of eIF4G and related degradation products (Fig. 2B). This pattern was similar to the signal obtained with p-substrate (PKC) antibodies, suggesting that PKCα may phosphorylate a site in the C-terminal part of eIF4G. There was enhanced signal with antibodies recognizing phosphorylated epitopes in the C-terminal portion of eIF4G [p-substrate (PKC) and p-eIF4G(Ser1148)] compared to eIF4G(Ct), detecting a nonphosphorylated epitope in the same region. eIF4G degradation has been shown to be associated with preferential phosphorylation of C-terminal truncation fragments in vivo, explaining this finding (5).
Fig. 2.
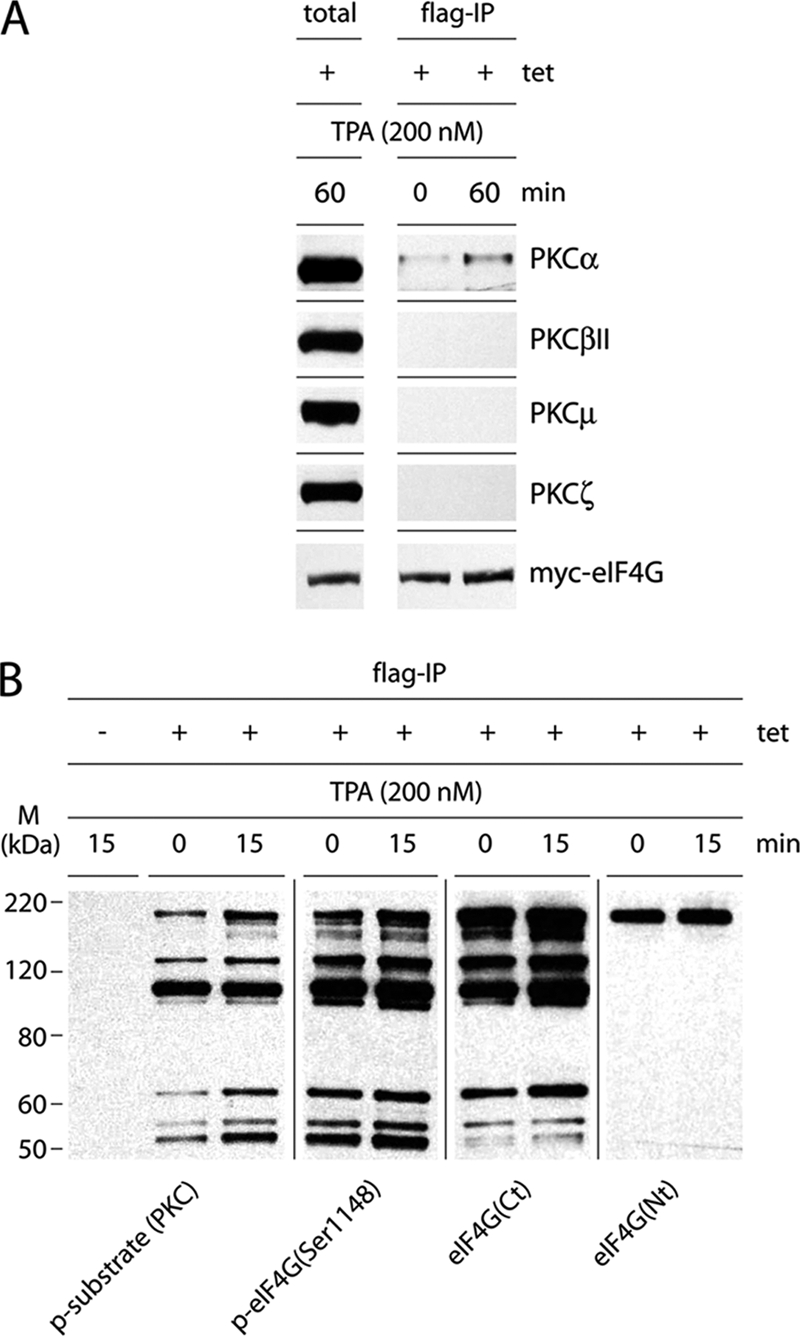
eIF4G is a substrate for PKCα. (A) Co-IP of PKC isoforms with eIF4G from Tet-induced, TPA-stimulated HEK293eIF4G-b cells. Serum-starved HEK293eIF4G-b cells were Tet induced for 24 h and treated for 60 min with DMSO (0) or 200 nM TPA. Cell lysates were subjected to Flag-IP followed by immunoblotting with the indicated antibodies. The experiment was repeated four times with similar results. A representative assay is shown. (B) Immunoblot of eIF4G and its cleavage products with site-specific antibodies. Serum-starved HEK293eIF4G-b cells were Tet or mock induced for 24 h and treated for 15 min with DMSO (0) or 200 nM TPA. The experiment was repeated twice with similar results. A representative assay is shown. M, molecular mass.
For further mapping of a PKC phosphorylation site in eIF4G, we conducted Flag-IP analyses from Tet-induced, TPA-stimulated HEK293eIF4G-b cells or HEK293 cells transfected with a series of tagged C-terminal eIF4G truncation products (Fig. 3A). First, we confirmed that C-terminal eIF4G fragments respond to TPA in an authentic manner. CTMC (encompassing aa 1085 to 1600) showed similar responses to TPA as full-length eIF4G (Fig. 3B). Phosphorylation of Ser1148 was modestly enhanced in intact eIF4G, but phosphorylation of Ser1232 was strongly stimulated in either protein (Fig. 3B). Co-IP after TPA stimulation showed induced PKCα binding to CTMC similar to that of eIF4G-b (Fig. 3B).
Second, for mapping of the PKCα phosphorylation site, we conducted Flag-IP analyses from TPA-stimulated HEK293 cells transfected with the indicated C-terminal eIF4G fragments. The p-substrate (PKC) antibody detected TPA-dependent phosphorylation of CTMC (aa 1085 to 1600), CTM (aa 1085 to 1412), but not CTC (aa 1245 to 1600) (Fig. 3C, top panel). This correlated with co-IP of PKCα, which was successful with CTMC and CTM only (Fig. 3C, middle panel). Since CTM lacks the Mnk1 binding site in eIF4G (aa 1585 to 1600), this excludes Mnk1 as a possible kinase for the site recognized by p-substrate (PKC) antibodies. To confirm this finding directly, we constructed CTMCΔMnk, carrying a 15-aa C-terminal deletion that abolishes Mnk1 binding (28). As expected, co-IP of Mnk1 revealed the lack of binding to CTM and CTMCΔMnk (Fig. 3C). The p-substrate (PKC) antibodies readily detected TPA-mediated phosphorylation of CTMCΔMnk and CTMC alike, demonstrating that Mnk1 is not directly involved in phosphorylation of this site.
As reported for full-length eIF4G (35), binding of Mnk1 to CTMC or CTC was induced by TPA. Interestingly, Mnk1 binding to CTC was significantly enhanced both in the uninduced and TPA-induced states, suggesting that this eIF4G fragment lacks a conformational arrangement that inherently restricts the association with Mnk1. The levels of exogenous myc-eIF4G fragments were comparable in all samples and assays (Fig. 3C, bottom panel).
Ser1186 in eIF4G is phosphorylated by PKCα and modulates binding to Mnk1.
To determine the precise site for PKCα phosphorylation in eIF4G, we surveyed the sequence of aa 1085 to 1245 to identify a possible consensus motif(s) or sequence similar to known natural substrates. Similarities with the optimal consensus and the phosphorylation site in the confirmed PKCα substrate P-glycoprotein of MDR1 (37) yielded Ser1186, 2 aa upstream of the previously identified, serum-responsive Ser1188 site (Fig. 4A) (29). To confirm this prediction, we constructed variants of CTMC with Ser1186 and/or Ser1188 to Ala mutations. Flag-IP of CTMC-transfected HEK293 cells after TPA stimulation revealed efficient detection by p-substrate (PKC) antibodies of wild-type (wt) CTMC and the single Ser1188Ala substitution but not the Ser1186Ala mutants (Fig. 4B). Flag-IP of CTN, a fragment encompassing aa 683 to 1133 of eIF4G (Fig. 3A), failed to react with p-substrate (PKC) antibodies, as expected (Fig. 4B). The Ser1186Ala substitution did not affect binding of PKCα to CTMC but prevented reactivity with p-substrate (PKC) antibodies, suggesting that PKCα phosphorylates Ser1186 in eIF4G (Fig. 4B). Similarly, CTMC(Ser1188Ala) retained TPA-induced binding of PKCα with eIF4G. Upon TPA treatment, this variant was readily detected by p-substrate (PKC) antibodies, but the signal was lower than for wt CTMC (Fig. 4B). We speculate that this may be due to reduced affinity of the p-substrate (PKC) antibodies to the mutant site. Also, Ser1188 is a serum-responsive phosphorylation site in eIF4G (29) which may influence the efficiency of Ser1186 phosphorylation. Since the kinase or signaling pathways involved in Ser1188 phosphorylation are unknown, we cannot currently explain the effect of the Ser1188Ala substitution on Ser1186 phosphorylation.
Fig. 4.

eIF4G(Ser1186) is phosphorylated by PKCα. (A) Optimal substrate consensus for PKCα, sequences of a confirmed substrate (P-glycoprotein of MDR1) (37) and of the proposed phosphorylation site in eIF4G (aa 1179 to 1190). Values in parentheses indicate the relative preference for the amino acids shown (bold letters represent amino acids with values of >1.5). Bold letters in the eIF4G sequence emphasize positions that are equal to the consensus or to the P-glycoprotein phosphorylation site. (B) Identification of the PKCα phosphorylation site in eIF4G. HEK293 cells were transfected for 16 h with CTMC variants or CTN fragments, serum starved for 24 h, and treated for 60 min with DMSO (0) or 200 nM TPA. Cell lysates were subjected to Flag-IP followed by immunoblotting. This assay was repeated three times; a representative experiment is shown. ND, not done.
The consensus substrate sequence for PKCα specifies a positively charged Arg residue at the +2 position. In eIF4G it is Ser1188. After phosphorylation by an as of yet unknown kinase, p-Ser1188 would become negatively charged, which should decrease the efficiency of Ser1186 phosphorylation by PKCα. The amino acid selectivity index (0.1 for Glu; 3.4 for Arg) (Fig. 4A) supports this assumption. To prevent the unpredictable effects of Ser1188 phosphorylation in our assays, we decided to use the double mutant Ser1186/1188Ala for further experiments.
TPA activation of Mnk1 stimulates binding to eIF4G, presumably via conformational changes to a structural arrangement of Mnk1 which prevents eIF4G binding in the uninduced state (35). Our data on Mnk1 binding to the C-terminal eIF4G fragments CTMC and CTC (Fig. 3C) suggest that Mnk1-eIF4G binding also faces inherent restraints due to eIF4G structure. It was thus compelling to investigate whether PKCα-mediated phosphorylation of eIF4G participates in coordinating Mnk1 binding. Co-IP of Mnk1 with CTMC revealed a pattern paralleling Ser1186 phosphorylation (Fig. 4B). Ser1186Ala, but not Ser1188Ala, prevented TPA-induced binding of Mnk1 to CTMC (Fig. 4B). The CTN fragment lacks the Mnk1 binding site (Fig. 3A) and, thus, fails to bind Mnk1 (Fig. 4B). These findings indicated a role for Ser1186 phosphorylation in coordinating Mnk1 interactions with eIF4G. Our observations also suggest that Mnk1 does not phosphorylate Ser1232 in eIF4G since Ser1232 phosphorylation occurs despite absent TPA-induced Mnk1 binding to the CTMC(Ser1186Ala) or CTMC(Ser1186/1188Ala) variants (Fig. 4B).
The kinetics of PKCα-specific signaling to eIF4F.
To correctly identify effects of PKCα activation on translation machinery, it is imperative to use specific stimuli and avoid unintended activation of confounding signaling pathways. The PKC Ser/Thr kinase family is divided into three subfamilies based on sequence similarities and modes of activation. Classic PKCs are activated by Ca+2, phosphatidylserine (PS), and diacyl glycerol (DAG); novel PKCs require only PS and DAG; and atypical PKCs (ξ, ι, and λ) respond to PS alone (25, 38, 42). TPA mimics DAG and broadly activates classic and novel PKCs (19). Therefore, TPA treatment of cells may elicit signaling events that affect eIF4G function independent of the pathways studied in our investigation.
Co-IP of PKCα with eIF4G (Fig. 2A, 3C, and 4B) and inhibition of eIF4G phosphorylation after TPA stimulation by Bis1 support involvement of the α-isoform in eIF4G phosphorylation. Yet Bis1 specificity for classic PKCs is not absolute (1). Therefore, to unambiguously document phosphorylation of Ser1186 in eIF4G by PKCα, we used an isoform-specific activator of PKC (42), sapintoxin D (STD), at a concentration of 0.5 nM. Previous studies showed that STD binds a different PKC phorbol binding site than TPA (36) and activates only PKCα without concomitantly stimulating other PKC family members (11). Treatment of HEK293 cells with 0.5 nM STD induced PKCα and Erk1/2 phosphorylation, accompanied by phosphorylation of eIF4G(Ser1232) (Fig. 5A, bottom panel), which was similar to results with TPA (Fig. 5A, top panel). Also, we detected phosphorylation of Ser235/236 in ribosomal protein S6, substrates of Rsk1/Rsk2, which are activated by Erk1/2 (Fig. 5B, left panel) (31, 33). Flag-IP revealed that 0.5 nM STD elicited eIF4G phosphorylation detected with p-substrate (PKC) antibodies, similar to treatment with TPA (Fig. 5B, right panel). STD stimulation increased phosphorylation of eIF4E(Ser209) and S6(Ser235/6), but we did not observe changes in association of eIF4G-b with eIF4E, eIF4A, or S6 upon STD treatment (Fig. 5B, right panel).
Fig. 5.

Isoform-specific activation of PKCα by STD produces sequential phosphorylation of translation factors. (A) Time course of phosphorylation of PKCα, Erk1/2, and eIF4G(Ser1232) upon TPA or STD activation. Serum-starved, Tet-induced HEK293eIF4G-e cells were treated for the intervals shown with 200 nM TPA or 0.5 nM STD. Total cell lysates were subjected to SDS-PAGE followed by immunoblotting. (B) Time course of STD-stimulated activation of the PKCα/Raf/Erk pathway and phosphorylation of eIF4G, eIF4E, and S6. Serum-starved, Tet-induced HEK293eIF4G-e cells were treated for the intervals shown with 0.5 nm STD. Extracts were probed by immunoblotting (left panel) or subjected to Flag-IP and immunoblotting (right panel). (C) Lysates from cells treated as described in panel B were processed for immunoblotting or Flag-IP/immunoblotting and quantitative chemiluminescence measurements. The interval with maximum signal was set at 100%, and the signal intensities at all other time points were calculated in relation to this. This experiment was repeated four times with consistent results; average percent phosphorylation values are shown. Standard deviation values were ≤3% at 2 and 5 min, ≤5% at 15 min, and ≤7% at all other time points.
To establish the context of eIF4G phosphorylation by PKCα within a broader signaling network converging on translation machinery, we quantitatively studied the time course of signaling events upon STD stimulation of cells (Fig. 5C). Activation of PKCα was rapid (τ1/2 = 60 to 90 s), peaked around 15 min, and then markedly declined since PKC phosphorylation triggers proteasomal degradation of the protein (17). Phosphorylation of eIF4G(Ser1186) (τ1/2 = 8 to 9 min) closely tracked the kinetics of PKCα phosphorylation, reaching its maximum after 15 min before declining by 45 to 90 min (Fig. 5C). Phosphorylation of Erk1/2 (τ1/2 = 3 to 4 min) followed that of PKCα and preceded phosphorylation of Ser209 in eIF4E (τ1/2 = 15 min), in line with the projected signaling pathways implied by our findings. Phosphorylation of eIF4G(Ser1232) (τ1/2 = 35 min) and of Ser235/236 in S6 (τ1/2 of ≥ 45 min) lagged behind all other signaling events.
Phospho-proteomic analysis of in vitro phosphorylated, recombinant eIF4G (GST-Ct) by recombinant PKCα.
To confirm that PKCα phosphorylates eIF4G at Ser1186 directly, we expressed a recombinant C-terminal eIF4G fragment encompassing aa 683 to 1600 (GST-Ct) in E. coli (18) and investigated its phosphorylation by purified recombinant PKCα in vitro. Recombinant PKCα phosphorylated GST-Ct in a time- and dose-dependent manner (Fig. 6A), as evident from the increasing signal with p-substrate (PKC) antibody upon incubation of GST-Ct with recombinant PKCα. Untreated GST-Ct and in vitro phosphorylated product were subjected to phospho-proteomic analysis. For this, the material was digested with trypsin, and the resulting fragments were affinity purified using phosphopeptide enrichment via a TiO2-column and subjected to LC-MS/MS analysis. Several sites in eIF4G-Ct were phosphorylated by PKCα in vitro, but phosphorylation of Ser1186 was preponderant (Fig. 6B). Incubation of GST-Ct with recombinant PKCα increased phosphorylation of Ser1186 ∼900-fold (Fig. 6C). Phosphorylation of Ser1145/1146 or Thr1120 occurred at much reduced levels (Fig. 6C). The p-substrate (PKC)-specific antibody failed to recognize CTMC (aa 1085 to 1600) after mutation of Ser1186 (Fig. 4B). This indicates that minor sites in eIF4G, which are phosphorylated by PKCα in vitro, fail to respond to STD stimulation in vivo or are not recognized by the p-substrate (PKC)-specific antibody.
Fig. 6.

Identification of eIF4G phosphorylation sites by PKCα in vitro. (A) Time course of phosphorylation of recombinant GST-Ct by purified, recombinant GST-PKCα. This experiment was repeated four times; a representative assay is shown. (B) Sequences of phosphorylated eIF4G peptides determined from tandem mass spectrometry. Bold pS or pT denotes a phosphorylated Ser or Thr residue, respectively. (C) The relative intensities of phosphorylation of corresponding residues in eIF4G were determined by quantitative phospho-proteomic analysis of in vitro phosphorylated, recombinant GST-Ct by purified, recombinant GST-PKCα.
Phosphorylation of Ser1186 in eIF4G modulates Mnk1 binding.
Tests in TPA-stimulated cells expressing the C-terminal eIF4G fragment CTMC suggested that PKCα signaling to eIF4G may affect Mnk1 binding (Fig. 4B). However, intact eIF4G may react differently to PKCα activation. We therefore studied Mnk1-eIF4G interactions in Tet-inducible HEK293eIF4G-e cell lines expressing wt protein or the Ser1186/1188Ala or Ser1186/1188Glu variant. These cells were serum starved, Tet induced, and treated with STD (0.5 nM) for the periods indicated in Fig. 7A. Flag-IP revealed approximately even recovery of Flag-eIF4G and co-IP of eIF4E in all samples (Fig. 7A). Mnk1 co-IPs, however, differed substantially among eIF4G variants (Fig. 7A).
Fig. 7.

Kinetic analysis of STD-induced Mnk1 binding to wt eIF4G or its Ser1186/1188 double mutants. (A) Serum-starved, Tet-induced HEK293eIF4G-e cells or the corresponding eIF4G(Ser1186/1188Ala) and eIF4G(Ser1186/1188Glu) mutants were pretreated for 1 h with 10 μM CGP57380 and stimulated with 0.5 nM STD as indicated. Cell lysates were processed for co-IP of Mnk1 with Flag-IP of exogenous eIF4G. IP of Flag-eIF4G and co-IP of eIF4E are included as controls. This experiment was repeated three times; a representative assay is shown. (B) Chemiluminescence quantification of the kinetics of STD-induced Mnk1-eIF4G binding shown in panel A. The quantification of the kinetics of STD-stimulated eIF4G(Ser1186) phosphorylation reflects data shown in Fig. 5B. The interval with maximum signal was set at 100%, and the signal intensities at all other time points were calculated in relation to this. The data represent average percent phosphorylation values from three independent experiments. Standard deviation values were ≤4% at 0, 2, and 5 min, ≤6% or less at 15 and 90 min, and ≤7% at 45 min. (C) Phosphorylation of Ser1232 in eIF4G occurs upon STD stimulation of cells independent of Ser1186/1188 phosphorylation. IP of Flag-eIF4G-e in serum-starved, Tet-induced HEK293 cells expressing wt eIF4G-e or eIF4G-e(Ser1186/8A). (D) Time course of STD-stimulated Mnk1 co-IP in serum-starved, Tet-induced HEK293 cells expressing the eIF4G-e variants shown. The cells were pretreated for 1 h with 10 μM CGP57380 and stimulated with 0.5 nM STD as indicated. The experiment was repeated twice with similar results. A representative assay is shown.
Catalytic activity of Mnk1 itself is a factor in regulating binding to eIF4G (35). To eliminate this confounding factor and focus on the role of eIF4G phosphorylation in controlling Mnk1 binding, we conducted our investigations in cells treated with the Mnk1 inhibitor CGP57380 (10 μM). We reported that CGP57380 inhibition of Mnk1 enhanced eIF4G binding upon phorbol ester stimulation, most likely because inhibiting Mnk1 catalytic activity prevents Mnk1 dissociation and effectively locks the binding complex (35). The kinetics of STD stimulation of Mnk1 binding to wt eIF4G was highly reproducible. The Ser1186/1188Ala mutant displayed delayed and significantly impaired Mnk1 interactions (Fig. 7A). In contrast, the phospho-mimic mutant exhibited enhanced Mnk1 binding kinetics upon STD stimulation compared to wt. Elevated Mnk1 binding of the phospho-mimic form at 0 and 2 min after STD treatment (Fig. 7A) indicates higher affinity in the absence of PKCα activation. However, Mnk1 binding remained strongly responsive to phorbol ester stimulation, reflecting the contributing role of MAPK-mediated Mnk1 activation in eIF4G binding (35). Quantitative chemiluminescent tracking of the kinetics of Mnk1 binding to eIF4G confirmed these observations and corroborate a role for eIF4G phosphorylation by PKCα in Mnk1 binding (Fig. 7B).
Since PKCα activation invariably leads to downstream Erk1/2 activation, the phosphorylation of Ser1232 in eIF4G may be influenced by preceding phosphorylation of Ser1186. To examine this possibility, we performed Flag-IP of eIF4G in serum-starved, Tet-induced HEK293 cells expressing wt eIF4G-e or the Ser1186/1188Ala variant (Fig. 7C). The Ser1186/1188Ala substitutions did not alter the kinetics of Ser1232 phosphorylation upon STD treatment, indicating that STD-mediated phosphorylation of Ser1232 occurs independent of PKCα-mediated phosphorylation of Ser1186 (Fig. 7C).
To investigate if Ser1232 phosphorylation may affect the kinetics of STD-stimulated Mnk1-eIF4G binding, we performed Mnk1 co-IP from serum-starved, Tet-induced HEK293eIF4G-e cells expressing wt protein or a eIF4G(Ser1232Ala) variant. This yielded similar kinetics of STD-induced Mnk1-eIF4G binding (Fig. 7D). These results suggest that Ser1232 phosphorylation is not involved in modulating phorbol ester-induced Mnk1 binding to eIF4G.
Using CGP57380 in our assay may skew results because this compound has a relatively broad inhibitory spectrum (1) that may affect the kinetics of eIF4G binding to Mnk1 independent of Ser1186. For example, in Mnk1/2 double-knockout cells, CGP57380 inhibited protein synthesis (at concentrations of >10 μM), suggesting effects on signal transduction to protein synthesis machinery unrelated to Mnk1/2 (34). To rule out this possibility, we limited the dose of CGP57380 at 10 μM and investigated the effect of CGP57380 in our assay (Fig. 8). Immunoblot studies of serum-starved, Tet-induced HEK293eIF4G-e cells expressing wt protein or the Ser1186/1188 variants revealed that addition of CGP57380 (10 μM) did not change the efficiency of phosphorylation of PKCα, eIF4G(Ser1186), or Erk1/2 upon STD treatment of cells (Fig. 8A). As anticipated, CGP57380 inhibited phosphorylation of eIF4E(Ser209) upon STD stimulation (Fig. 8A).
Fig. 8.

Inhibition of Mnk1 with CGP57380 does not affect the activation of PKCα, Erk1/2, and phosphorylation of eIF4G(Ser1186). (A) Inhibition of eIF4E(Ser209) phosphorylation by CGP57380. Lysates from serum-starved, Tet-induced HEK293eIF4G-e cells expressing the indicated forms of eIF4G-e were subjected to immunoblotting. The effects of pretreatment with 10 μM CGP57380 and stimulation with 0.5 nM STD are shown. (B) Treatments with CGP57380 and STD-mediated stimulation enhance Mnk1 binding to eIF4G. Serum-starved, Tet-induced HEK293eIF4G-e cells or the corresponding eIF4G(Ser1186/1188Ala) or eIF4G(Ser1186/1188Glu) mutants were pretreated for 1 h with 10 μM CGP57380 and stimulated with 0.5 nM STD as indicated. Cell lysates were subjected to Flag-IP followed by immunoblotting. Bands were quantified as described in Materials and Methods, and the Mnk1 signal density was adjusted for the signal of the loading control (eIF4E). The Mnk1 signal in mock-induced, untreated cells was set at 1. Comparison of exogenous eIF4G-e variants reveals a potent increase in Mnk1 binding upon STD stimulation in cells pretreated with CGP57380.
Evaluation of Mnk1 binding to eIF4G or its Ser1186/1188 variants by Flag-IP confirmed the previously observed diminished binding upon Ser1186/1188Ala mutation when cells were treated with CGP57380 (Fig. 8B). A higher-exposure film also revealed the previously observed increase of eIF4G-Mnk1 binding with the Ser1186/1188Glu variant both in the unstimulated and the STD-stimulated state and in the presence or absence of CGP57380. In untreated cells, the amount of Mnk1 coimmunoprecipitated with the Ser1186/1188Glu mutant was 3.7-fold higher than with wt eIF4G. Treatment of cells with CGP57380 and STD increased Mnk1 binding 22-fold for the Ser1186/1188Ala mutant, 66-fold for wt eIF4G, and 87-fold for the phospho-mimic variant, relative to wt eIF4G in untreated cells. These results corroborate our hypothesis that phosphorylation of Ser1186 in eIF4G enhances Mnk1 binding.
DISCUSSION
In this report, we describe an important link between the PKC-Ras-Erk signal transduction pathway and translation machinery. We found that phorbol esters activate eIF4G phosphorylation at Ser1186 by PKCα. Furthermore, Ser1232, a serum-responsive phosphorylation site in eIF4G (29), may be a target of MAPK signal transduction. Our studies indicate that PKCα activation and resulting eIF4G phosphorylation modulate interactions with the eIF4E kinase, Mnk1.
Classic PKCs (e.g., PKCα) are distinct from other subfamilies in that they prefer substrates with basic residues at positions +2/+3 (25). The context of eIF4G(Ser1186) is similar but not identical to the PKCα consensus substrate. The main difference is a polar Ser1188 residue at position +2 with a neutral selectivity index (25). This residue is a target for an unknown serum-stimulated kinase (29), which converts it to the negatively charged phospho-Ser. The presence of a negatively charged Glu residue at the +2 position in the consensus strongly decreases the selectivity index, suggesting that phosphorylation of Ser1188 may negatively regulate Ser1186 phosphorylation by PKCα. Unraveling the functional relationship of these adjacent substrate sites requires identification of the kinase targeting Ser1188.
Time course studies revealed a series of coordinated events converging on eIF4F upon phorbol ester stimulation. Phosphorylation of eIF4G(Ser1186) (τ1/2 = 8 to 9 min) closely tracked activation of PKCα (τ1/2 = 1 to 1.5 min) and Erk1/2 (τ1/2 = 3 to 4 min) and preceded phosphorylation of eIF4E(Ser209) (τ1/2 = 15 min), eIF4G(Ser1232) (τ1/2 = ∼35 min) and S6(Ser235/6) (τ1/2 = ∼45 min). Phorbol ester-stimulated binding of Mnk1 to eIF4G was diminished upon introducing a Ser1186Ala mutation but not Ser1186Glu, suggesting that Ser1186 phosphorylation by PKCα modulates the association with Mnk1. Our studies indicate that phosphorylation of eIF4G(Ser1232) does not participate in this function although our results do not exclude a role of other phosphorylation sites in eIF4G.
X-ray structural analyses of eIF4G revealed three α-helical HEAT domains (21, 22). HEAT1 and HEAT2 jointly bind eIF4A, stimulating its helicase activity (7, 22). The 155-aa interdomain linker connecting the HEAT1 and -2 domains appeared to be unstructured (22) and contains the three initially reported serum-responsive phosphorylation sites in eIF4G (Ser1148/1188/1232 [29]) in addition to Ser1186 identified here (Fig. 9A and B). The most conserved region in the C-terminal HEAT3 domain contains two tandem AA (aromatic and acidic residues) boxes, which commonly mediate specific binding to positively charged motifs of partner proteins (Fig. 9A). In the case of eIF4G, this motif is involved in binding Mnk1 at an N-terminal stretch of eight basic amino acids (RK box) (2). The crystal structure of the first (proximal) AA box of eIF4G (aa 1543 to 1564) has been investigated in detail (2). With the exception of Tyr1551 and Trp1564, all aromatic/hydrophobic residues contribute to a hydrophobic core, while the conserved acidic residues are solvent exposed, creating an acidic cluster that easily interacts with positively charged motives (Fig. 9C). In contrast, the five C-terminal acidic residues in the (distal) AA box 2 (aa 1586 to 1598) do not form a rigid structure (2). This flexible loop of the second AA box appears prevalent for binding to Mnk1 because deleting the C-terminal 15 aa abolishes Mnk1 binding to eIF4G (24).
Fig. 9.

Proposed schematic model of eIF4G binding to Mnk1 upon PKCα-dependent Ser1186 phosphorylation (22). Linear (A) and three-dimensional (B) domain/motif organization of eIF4G. Mnk1 binding to AA box 2 (gray) and the position of Ser1186 in the interdomain linker of eIF4G are indicated. The three-dimensional structure depicts the mutual orientation of eIF4G/eIF4A and eIF4H structured domains; the interdomain linker is reconstructed based on topology analyses of the helicase complex with eIF4G (22). AA boxes 1 and 2 are represented as filled black and gray circles, respectively. The position of Ser1186 in the interdomain linker suggests a possible role in controlling access to AA box 2 and the Mnk1 binding site in eIF4G. (C) Structure-based sequence alignment of AA box 1 with the eIF4G interdomain linker in the vicinity of the PKCα phosphorylation site (Ser1186; asterisk). Solvent-exposed acidic residues in AA box 1 (gray boxes) and positively charged amino acids in the interdomain linker (open boxes) are indicated. Solvent-exposed aromatic residues Tyr1551 and Trp1564 in AA box 1 (2) are shown in black boxes.
The interdomain linker region surrounding the PKCα phosphorylation site may exert inhibitory effects on binding Mnk1, which may be alleviated upon Ser1186 phosphorylation (Fig. 9B). Based on ultrastructural data on eIF4G (22), we propose that the PKCα phosphorylation site region may interact with the first AA box (Fig. 9B). The conserved Phe1187 of the interdomain linker may be stacked between Tyr1551 and Trp1564 of the first AA box, and positively charged amino acid residues in the vicinity of Ser1186 may interact with the negatively charged cluster of the first AA box (Fig. 9C). Ser1186 phosphorylation would generate a negatively charged phospho-Ser and cause electrostatic repulsion with the negatively charged residues of the first AA box. The resulting displacement of the interdomain linker may free access for Mnk1 binding to the HEAT3 domain (Fig. 9B).
mTORC1 activation promotes interactions of eIF4G with its ligands eIF4E (13, 20), eIF3 (14), or S6 (31). Signaling cascades implicated in these events modulate eIF4G binding by targeting 4E-BPs, eIF3, or S6. The role of mTORC1-responsive signaling to eIF4G itself, however, is unclear. The phorbol esters TPA and STD do not induce phosphorylation of mTORC1(Ser2448). Accordingly, while the phosphorylation status of eIF4E and S6 changed upon PKCα activation in our assays, the levels of their association with eIF4G were unaltered. Our studies suggest that PKC-Ras-Erk signaling to eIF4F modulates the association of eIF4G with Mnk1. Binding to eIF4G approximates Mnk1 to its substrate eIF4E (28). Mnk1-eIF4G binding is stimulated upon MAPK activation (35) and is required for efficient eIF4E phosphorylation. Recent evidence demonstrated a prominent role for Mnk1-catalyzed eIF4E phosphorylation in malignant transformation (9, 40, 43). Although this strongly suggests altered protein synthesis rates upon Mnk1 activation, the precise effects of Ser209 phosphorylation in eIF4E on translation control are controversial. Our studies suggest that stimuli which produce eIF4E phosphorylation may exert their influence on translation control through concerted action on eIF4E and its binding partner eIF4G. This view is supported by kinetic analyses that show phorbol ester-induced eIF4E phosphorylation coincident with at least two phosphorylation events affecting eIF4G. Structural changes to the interdomain linker elicited by phosphorylation of target sites may produce far-reaching effects on eIF4G's function in translation beyond modulation of Mnk1 binding. Flexible structural arrangements of the interdomain linker in response to distinct signals may portend an enormous potential for translation adjustment via modulation of eIF4G function.
ACKNOWLEDGMENTS
We thank C. Kaiser for providing recombinant GST-Ct. We are grateful to Erik Soderblom and other members of the Proteomics Core Facility at Duke University for phospho-proteomic analyses.
This work was supported by PHS grants CA124756 and CA140510 (M.G.).
Footnotes
Published ahead of print on 16 May 2011.
REFERENCES
- 1. Bain J., et al. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408:297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bellsolell L., Cho-Park P. F., Poulin F., Sonenberg N., Burley S. K. 2006. Two structurally atypical HEAT domains in the C-terminal portion of human eIF4G support binding to eIF4A and Mnk1. Structure 14:913–923 [DOI] [PubMed] [Google Scholar]
- 3. Brunn G. J., et al. 1997. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277:99–101 [DOI] [PubMed] [Google Scholar]
- 4. Byrd M. P., Zamora M., Lloyd R. E. 2002. Generation of multiple isoforms of eukaryotic translation initiation factor 4GI by use of alternate translation initiation codons. Mol. Cell. Biol. 22:4499–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeGracia D. J., Rafols J. A., Morley S. J., Kayali F. 2006. Immunohistochemical mapping of total and phosphorylated eukaryotic initiation factor 4G in rat hippocampus following global brain ischemia and reperfusion. Neuroscience 139:1235–1248 [DOI] [PubMed] [Google Scholar]
- 6. Dephoure N., et al. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 105:10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujita Y., et al. 2009. Domain-dependent interaction of eukaryotic initiation factor eIF4A for binding to middle and C-terminal domains of eIF4G. J. Biochem. 146:359–368 [DOI] [PubMed] [Google Scholar]
- 8. Fukunaga R., Hunter T. 1997. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 16:1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Furic L., et al. 2010. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. U. S. A. 107:14134–14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gauci S., et al. 2009. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 81:4493–4501 [DOI] [PubMed] [Google Scholar]
- 11. Geiges D., et al. 1997. Activation of protein kinase C subtypes alpha, gamma, delta, epsilon, zeta, and eta by tumor-promoting and nontumor-promoting agents. Biochem. Pharmacol. 53:865–875 [DOI] [PubMed] [Google Scholar]
- 12. Gingras A. C., Raught B., Sonenberg N. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68:913–963 [DOI] [PubMed] [Google Scholar]
- 13. Haghighat A., Mader S., Pause A., Sonenberg N. 1995. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14:5701–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harris T. E., et al. 2006. mTOR-dependent stimulation of the association of eIF4G and eIF3 by insulin. EMBO J. 25:1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herbert T. P., Kilhams G. R., Batty I. H., Proud C. G. 2000. Distinct signalling pathways mediate insulin and phorbol ester-stimulated eukaryotic initiation factor 4F assembly and protein synthesis in HEK 293 cells. J. Biol. Chem. 275:11249–11256 [DOI] [PubMed] [Google Scholar]
- 16. Hershey J. W., Merrick W. C. 2000. Pathway and mechanism of initiation of protein synthesis, p. 637–654 In Sonenberg N., Hershey J. W., Mathews M. B. (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 17. Huang F. L., Yoshida Y., Cunha-Melo J. R., Beaven M. A., Huang K. P. 1989. Differential down-regulation of protein kinase C isozymes. J. Biol. Chem. 264:4238–4243 [PubMed] [Google Scholar]
- 18. Kaiser C., et al. 2008. Activation of cap-independent translation by variant eukaryotic initiation factor 4G in vivo. RNA. 14:2170–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mackay H. J., Twelves C. J. 2007. Targeting the protein kinase C family: are we there yet? Nat. Rev. Cancer. 7:554–562 [DOI] [PubMed] [Google Scholar]
- 20. Marcotrigiano J., Gingras A. C., Sonenberg N., Burley S. K. 1999. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 3:707–716 [DOI] [PubMed] [Google Scholar]
- 21. Marcotrigiano J., et al. 2001. A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol. Cell 7:193–203 [DOI] [PubMed] [Google Scholar]
- 22. Marintchev A., et al. 2009. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 136:447–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marintchev A., Wagner G. 2005. eIF4G and CBP80 share a common origin and similar domain organization: implications for the structure and function of eIF4G. Biochemistry 44:12265–12272 [DOI] [PubMed] [Google Scholar]
- 24. Morino S., Imataka H., Svitkin Y. V., Pestova T. V., Sonenberg N. 2000. Eukaryotic translation initiation factor 4E (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation, and the C-terminal one-third functions as a modulatory region. Mol. Cell. Biol. 20:468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nishikawa K., Toker A., Johannes F. J., Songyang Z., Cantley L. C. 1997. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272:952–960 [DOI] [PubMed] [Google Scholar]
- 26. Olsen J. V., et al. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648 [DOI] [PubMed] [Google Scholar]
- 27. Pause A., et al. 1994. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371:762–767 [DOI] [PubMed] [Google Scholar]
- 28. Pyronnet S., et al. 1999. Human eukaryotic translation initiation factor 4G (eIF4G) recruits Mnk1 to phosphorylate eIF4E. EMBO J. 18:270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raught B., et al. 2000. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 19:434–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roux P. P., Ballif B. A., Anjum R., Gygi S. P., Blenis J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. U. S. A. 101:13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roux P. P., et al. 2007. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 282:14056–14064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rush J., et al. 2005. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23:94–101 [DOI] [PubMed] [Google Scholar]
- 33. Shahbazian D., et al. 2006. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25:2781–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shenberger J. S., et al. 2007. Roles of mitogen-activated protein kinase signal-integrating kinases 1 and 2 in oxidant-mediated eIF4E phosphorylation. Int. J. Biochem. Cell Biol. 39:1828–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shveygert M., Kaiser C., Bradrick S. S., Gromeier M. 2010. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol. Cell. Biol. 30:5160–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slater S. J., Ho C., Stubbs C. D. 2002. The use of fluorescent phorbol esters in studies of protein kinase C-membrane interactions. Chem. Phys. Lipids 116:75–91 [DOI] [PubMed] [Google Scholar]
- 37. Songyang Z., et al. 1996. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 16:6486–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steinberg S. F. 2008. Structural basis of protein kinase C isoform function. Physiol. Rev. 88:1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ubersax J. A., Ferrell J. E., Jr 2007. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8:530–541 [DOI] [PubMed] [Google Scholar]
- 40. Ueda T., et al. 2010. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc. Natl. Acad. Sci. U. S. A. 107:13984–13990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waskiewicz A. J., Flynn A., Proud C. G., Cooper J. A. 1997. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16:1909–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Way K. J., Chou E., King G. L. 2000. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 21:181–187 [DOI] [PubMed] [Google Scholar]
- 43. Wendel H. G., et al. 2007. Dissecting eIF4E action in tumorigenesis. Genes Dev. 21:3232–3237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang H., et al. 2002. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem. 277:39379–39387 [DOI] [PubMed] [Google Scholar]