

Abstract
Progesterone receptors (PR) are critical mediators of mammary gland development and contribute to breast cancer progression. Progestin-induced rapid activation of cytoplasmic protein kinases leads to selective regulation of growth-promoting genes by phospho-PR species. Herein, we show that phosphorylation of PR Ser81 is ck2 dependent and progestin regulated in intact cells but also occurs in the absence of PR ligands when cells enter the G1/S phase of the cell cycle. T47D breast cancer cells stably expressing a PR-B mutant receptor that cannot be phosphorylated at Ser79/81 (S79/81A) formed fewer soft agar colonies. Regulation of selected genes by PR-B, but not PR-A, also required Ser79/81 phosphorylation for basal and/or progestin-regulated (BIRC3, HSD11β2, and HbEGF) expression. Additionally, wild-type (wt) PR-B, but not S79/81A mutant PR, was robustly recruited to a progesterone response element (PRE)-containing transcriptional enhancer region of BIRC3; abundant ck2 also associated with this region in cells expressing wt but not S79/81A PR. We conclude that phospho-Ser81 PR provides a platform for ck2 recruitment and regulation of selected PR-B target genes. Understanding how ligand-independent PRs function in the context of high levels of kinase activities characteristic of breast cancer is critical to understanding the basis of tumor-specific changes in gene expression and will speed the development of highly selective treatments.
INTRODUCTION
The ovarian steroid hormone progesterone acts by binding to and activating progesterone receptor (PR) A, B, and C isoforms expressed in target tissues. In the normal breast, PR-A and PR-B are typically expressed in a minority population (7 to 10%) of luminal epithelial cells. PR-B is required for mammary gland development during puberty and pregnancy and acts by contributing to lobulo-alveolar proliferation and ductal side branching (8, 46). Studies from PR-knockout mice show that these mice have significant defects in mammary gland morphology (primarily PR-B dependent) and reproductive abnormalities (primarily PR-A driven) (46, 54). Additionally, the presence of PR was shown to be required for the formation of mammary tumors in a carcinogen-induced mouse model of breast cancer (47). Finally, recent clinical data have shown that women taking hormone replacement therapy (HRT) whose regimens included both estrogen and a progestin, but not estrogen alone, experienced increased breast tumor numbers and sizes (1, 5, 12). Interestingly, the effect of combined HRT on breast cancer risk was reversible (5, 13), suggestive of epigenetic events.
In the absence of progesterone, PR molecules rapidly shuttle between the cytoplasm and the nucleus; cytoplasmic PRs contain membrane-associated species capable of direct binding and signaling to mitogenic protein kinases (c-Src, MAPK, PI3K) (3, 7, 25, 50). Following ligand binding, PRs dissociate from heat shock protein-containing chaperone complexes, undergo dimerization, and are largely retained in the nucleus. Nuclear receptors activate transcription of PR target genes, either directly through binding to progesterone response elements (PREs) or indirectly through tethering interactions with other transcription factors (AP1, SP1, STATs) (14, 61, 70). Notably, PR is highly posttranslationally modified, primarily on serine (phosphorylation) and lysine (acetylation, ubiquitination, and sumoylation) residues located in the N-terminal region (16, 17, 43, 76). These modifications are frequently ligand dependent but can also occur independently of progestin binding and significantly alter receptor stability, localization, tethering interactions, transcriptional activity, and promoter selectivity (18, 75). For example, MAPK and cdk2 have previously been shown to phosphorylate and modulate the activity of both liganded and unliganded PR (43, 62, 79).
The serine-threonine protein kinase ck2 (formerly casein kinase II) is ubiquitously expressed with over 300 substrates, many of which are involved in proliferation, cell survival, and gene expression (49). Moreover, ck2 has been shown to be overexpressed in many different types of cancer, including breast cancer (31). ck2, a holoenzyme composed of two catalytic subunits (α and α′) and two regulatory subunits (β), is a unique kinase in that it is constitutively active and does not require modifications or signaling inputs to modulate its kinase activity. In contrast, one mode of ck2 regulation likely occurs via altered subcellular localization of ck2 and/or its respective substrates (27). ck2 localization appears to be altered in a cell cycle-dependent manner, with nuclear accumulation occurring primarily in G1/S (51, 78). However, subcellular sequestration is not the only proposed mechanism for ck2 regulation. Others include regulated assembly of the ck2 holoenzyme, protein complex formation with substrates, autophosphorylation, and small molecule interactions (59); little is known about this topic.
Understanding how a cancer-associated kinase, like ck2, modulates PR function may provide insight into how PR promotes breast cancer cell proliferation (a PR-B-dependent action) and tumor progression (31). ck2 has previously been shown in vitro to phosphorylate human PR at Ser81, a residue located in the N-terminal region of PR unique to PR-B, termed the B-upstream segment (BUS) (80). Subsequent in silico analysis revealed 11 potential ck2 phosphorylation sites in PR (80). Mass spectrometry studies and in vitro kinase assays revealed that Ser81 was the primary site for ck2 phosphorylation; these studies failed to detect phosphorylation on any of the other ck2 consensus sites in PR (80). However, these studies were done using solely in vitro model systems; regulated phosphorylation at this site has not been studied in intact cells. Herein, we sought to understand the functional significance of ck2 regulation of PR-B Ser81 in breast cancer models.
MATERIALS AND METHODS
Cell lines.
The estrogen-independent ER/PR positive T47Dco (T47D) variant cell line has been previously described (35). T47D-Y (PR negative), T47D-YB (stably expressing wild-type [wt] PR-B), and T47D-YA (stably expressing wt PR-A) cells were characterized by Sartorius et al. (66). HeLa-PR cells have been previously described (62). T47D-S79/81A PR cells were created by stable expression of pSG5-S79/81A PR and pSV-neo in T47D-Y cells using FuGene-HD (Roche). Individual colonies were selected in 500 μg/ml G418 and maintained in 200 μg/ml G418 after initial selection. The pSG5-S79/81A PR plasmid (containing serine-to-alanine mutations at Ser79 and Ser81) was generated by GenScript Corporation. T47D-Y and HeLa cells were maintained at 37°C in 5% CO2 in minimum essential media (MEM) (CellGro) supplemented with 5% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1% nonessential amino acids, and 6 ng/ml insulin (cMEM). T47D-YB, T47D-YA, T47D-S79/81A PR, and HeLa-PR cells were maintained under the same conditions, with the addition of 200 μg/ml G418.
T47D cells containing an inducible PR expression system were created as follows using the ARGENT regulated transcription retrovirus kit (ARIAD Pharmaceuticals, Inc.). T47D-Y cells were first stably retrovirally transduced with the transcription factor vector pL2N2-RHS3H/ZF3 (necessary for activating subsequent transcription from the target gene vector). A clone from this cell line was stably retrovirally transduced with the target gene vector (pLH-Z12I-PL) containing wt PR-B (iPR-B) or with the empty vector (iEV). Upon addition of a chemical dimerizer (AP21967; 10−9 M), PR-B protein expression occurs within 24 to 48 h (as measured by Western blotting). These cells are maintained in cMEM supplemented with 200 μg/ml G418 and hygromycin B (CalBioChem).
Transient-transfection experiments were performed as follows: 24 h after cell plating, HeLa cells were transfected with pSG5-vector, pSG5-wt PR or pSG5-S79/81A PR using FuGene6 (Roche). At 24 h following transfection, cells were starved for 18 h in serum-free iMEM (modified improved MEM). Following starvation, cells were treated as noted in the respective figure legend and total cell lysates were isolated as described below.
Immunoblotting.
For most of the immunoblotting presented here (exceptions noted in figure legends), cells were starved for 18 h in serum-free iMEM. Following 18 h starvation, cells were treated, if applicable. Whole-cell lysates were isolated using a modified radioimmune precipitation assay (RIPA) buffer (0.15 M NaCl, 6 mM Na2HPO4, 4 mM NaH2PO4, 2 mM EDTA, 1% Triton-X, 0.1 M NaF; in H2O) supplemented with protease and phosphatase inhibitors. Lysates containing equal protein levels (between 25 and 30 μg protein was loaded per lane on each gel) were separated by SDS-PAGE and transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore) for subsequent immunoblotting analysis. Membranes were probed with primary antibodies recognizing total PR (number MS-298-P; ThermoScientific), phospho-Ser294 (MS-1332; Lab Vision Corp.), Erk1/2 (9102; Cell Signaling), phospho-Erk1/2 (9101; Cell Signaling), ck2α (sc-12738; Santa Cruz Biotechnology), and ck2β (sc-12739; Santa Cruz Biotechnology). The phospho-Ser81 (p-S81) PR antibody was a custom antibody commissioned from Invitrogen designed to recognize the following phospho-specific peptide sequence (PR-B amino acids 76 to 85): DQQSL-pS-DVEG. Mouse and rabbit horseradish peroxidase-conjugated secondary antibodies were obtained from Bio-Rad, and chemiluminescence was visualized using SuperSignal West Pico chemiluminescent substrate (Pierce Chemical Company). All Western blotting experiments were performed at a minimum in triplicate, and representative experiments are shown in each respective figure.
Luciferase transcription assays.
Luciferase assays were performed as previously described (25) using the dual luciferase reporter assay (Promega). Relative luciferase units (RLU) were normalized to the mean result ± standard deviation (SD) for Renilla luciferase.
Reagents.
Cells were treated with the following reagents (when applicable): R5020 (10 nM; Sigma), RU486 (100 nM; Sigma), EGF (30 ng/ml; Sigma), TBB (1 to 100 μM; CalBioChem), DMAT (1 to 100 μM; CalBioChem), PP2 (10 μM; CalBioChem), roscovitine (100 μM; CalBioChem), U0126 (10 μM; CalBioChem), and AP21967 (1 nM; ARIAD Pharmaceuticals, Inc.).
Cell cycle analysis/flow cytometry.
A total of 1.5 × 105 T47D-YB cells were plated in 10-cm2 dishes in cMEM (day 0). Synchronized cells were treated on day 1 with cMEM containing 2.5 μg/ml thymidine (Sigma) for 18 h. Cells were then washed with phosphate-buffered saline (PBS) and fresh iMEM-5% dextran-coated charcoal (DCC)-treated serum was added for 7 h. Synchronized cells were then treated for 18 h with iMEM-5% DCC-50 μg/ml mimosine. Following the 18-h mimosine treatment (and, if applicable, 60 min treatment with vehicle or TBB), cells were harvested in RIPA for Western blotting (as above) or trypsinized and fixed for flow cytometry. For flow cytometry analysis, media and wash (2 ml PBS) were collected. Trypsinized cells and collected media/wash were combined and pelleted by centrifugation. Cells were resuspended in 300 μl PBS-10% FBS, following which 4 ml ice cold 80% ethanol was added dropwise to fix samples. Samples were stored at −20°C until analyzed for cell cycle phase. Fixed cells were pelleted and washed three times with 5 ml cold PBS. Samples were resuspended in 100 to 400 μl staining buffer: 1× PBS with 10% RNase A (10 mg/ml Sigma), 5% FBS, 0.5 mM EDTA, 0.1% TX-100, and 200 μg/ml propidium iodide (Sigma). Propidium iodide staining was detected using a FACSCalibur (BD Biosciences). Cells were gated for cell cycle phase using FlowJo (Tree Star Inc.).
Soft agar anchorage-independent growth assays.
Soft agar assays were performed as previously described (16). Briefly, cells were suspended in 0.48% SeaPlaque GTG agarose (Lonza) in iMEM supplemented with 5% DCC serum containing either ethanol (EtOH) or 10 nM R5020. Cells were plated in triplicate/condition at 9.6 × 103/well over a bottom layer of 0.8% agarose/iMEM with 5% DCC serum. Cells were incubated under normal growth conditions for 21 days, following which colonies were counted in 15 fields/treatment group. The data are represented as an average number of colonies per field ± standard error of the mean (SEM). Soft agar experiments were performed in triplicate.
qPCR.
Cells were plated at 5 × 105 cells/well in triplicate wells of a 6-well plate. Following 18 h starvation in serum-free iMEM, cells were treated for 1 to 18 h with 10 nM R5020 or EtOH (if applicable; see relevant figure legend). Total RNA was isolated using Trizol (Invitrogen); cDNA was created using the Transcriptor cDNA first-strand cDNA synthesis kit (Roche) by following the manufacturer's recommendations. Real-time quantitative PCR (qPCR) was performed on equal amounts of cDNA using the Light Cycler 480 SYBR Green1 master mix on a Roche 480 light cycler. Results in triplicate for each gene of interest were normalized to those for either β-actin, 18S, or GAPDH (as indicated in each respective graph) ± SD.
For qPCR experiments on G1/S synchronized cells, cells were plated at 2.5 × 105/well in triplicate wells of a 6-well plate. Cells were synchronized as described above, and RNA/cDNA was created and analyzed as described above.
ChIP assays.
ChIP and ReChIP assays were performed using the ChIP-IT express or Re-ChIP-IT kit (Active Motif), according to the manufacturer's instructions using sonication as the method for chromatin shearing. Lysates were immunoprecipitated (IP) overnight (18 h) with the following antibodies: PR (number MS-298-P; ThermoScientific), ck2α (number sc-12738; Santa Cruz), or an equal amount of mouse or rabbit IgG. Resulting DNA was analyzed using qPCR as described above, and data are represented as a percentage of input DNA. In silico analysis using MatInspector (Genomatix) identified potential PRE-binding sites using the following consensus sequence: RGNACANRNTGTNCY. Primer sets used for qPCR analysis of ChIP data are as follows: BIRC3 PRE1-F (5′-AAAACAATAGTGCCAGTTCAATGAC-3′), BIRC3 PRE1-R (5′-ATGTTCTCTTTGATTCCCTGACAC-3′), BIRC3 (neg control 1)-F (5′-TTATGCTGAGCTGGAAGTTAAATAAAAAG-3′), BIRC3 (neg control 1)-R (5′-TTGGCCACTGGTCTCAAACTC-3′), BIRC3 (neg control 2)-F (5′-TGGGAAAAGTGCAGTATTTGG-3′), BIRC3 (neg control 2)-R (5′-GTTCATCTAATTGGGACTGGTTG-3′), TF PRE2-F (5′-TCATTTTAAGACGTCAGCTATTTCAC-3′), TF PRE2-R (5′-ATATTCTCCAGTCAGCATTTCAAAG-3′), TF (neg control 1)-F (5′-CTGAGAATCTATTGGTATTGCTTGG-3′), TF (neg control 1)-R (5′-CCCTTACGTGAGAAAGTCATTTTG-3′), TF (neg control 2)-F (5′-CTAGATGTGGATGAAATGAGTTGG-3′), and TF (neg control 2)-R (5′-TTCTGAAAGAAAACTAAGCCAAAAC-3′).
Statistics.
Statistical significance for all experiments was determined using an unpaired Student's t test.
RESULTS
Hormone- and ck2-dependent regulation of PR Ser81 phosphorylation.
Previous studies have shown that PR is phosphorylated on Ser81 in vitro (80). However, regulation of this site in vivo has yet to be defined. Using custom-made polyclonal antibodies created to recognize PR phospho-Ser81, we measured progestin-induced phosphorylation of this site in T47Dco human breast cancer cells (Fig. 1A). T47Dco cells are unmodified breast cancer cells that naturally constitutively express both PR-A and PR-B, without the requirement of estrogen treatment to induce PR expression (35). We detected weak basal PR-B Ser81 phosphorylation that substantially increased in response to treatment with the synthetic progesterone R5020 (Fig. 1A). PR-A does not contain Ser81, located within the BUS domain of PR-B. As expected, our phospho-Ser81-specific antibodies detected PR-B but not PR-A.
Fig. 1.
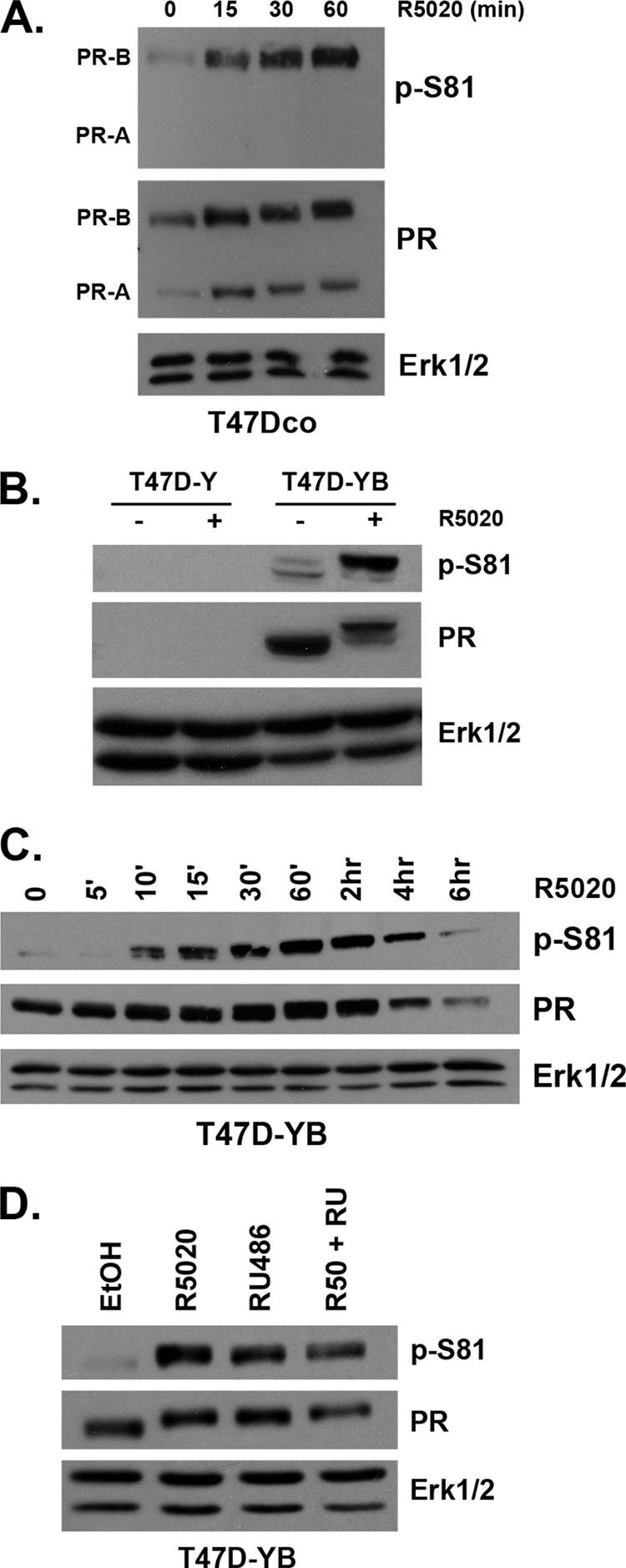
In vivo phosphorylation of PR Ser81. (A) T47Dco cells were starved for 18 h in serum-free media followed by treatment with 10 nM R5020 or ethanol (vehicle) for 0 to 60 min. Lysates were analyzed by Western blotting using antibodies against total Erk1/2 (loading control), total PR, and a custom-designed antibody that specifically recognizes phosphorylated Ser81 PR (p-S81). (B) Cells lacking PR (T47D-Y) and cells stably expressing PR-B (T47D-YB) were serum starved for 18 h and then treated with 10 nM R5020 or EtOH for 60 min. Lysates were analyzed by Western blotting as described for panel A. (C) Following 18 h serum starvation, T47D-YB cells were treated with a time course of 10 nM R5020 for 0 min to 6 h. Lysates were analyzed by Western blotting as described for panel A. (D) Following 18 h serum starvation, T47D-YB cells were treated with 10 nM R5020, 100 nM RU486, both, or vehicle control (EtOH). Lysates were analyzed by Western blotting as described for panel A.
In most steroid hormone receptor-positive breast cancer cell models, the levels of PR are primarily upregulated by estradiol, making experimental isolation of PR action (i.e., as studied independently of estrogen) very difficult (34). A naturally occurring PR-negative variant of the T47Dco human breast cancer cell line, termed T47D-Y, was first described by Sartorius and coworkers (66). This parental cell line was used to create stable cell lines constitutively expressing either wild-type (wt) PR-B (T47D-YB) or PR-A (T47D-YA) (66). As observed in unmodified T47Dco cells (Fig. 1A), we also detected low basal levels of Ser81 phosphorylation in T47D-YB cells (Fig. 1B). Again (as in T47Dco cells), the level of PR Ser81 phosphorylation increased significantly in response to R5020 (Fig. 1B). Control cells not expressing PR (T47D-Y) failed to exhibit any nonspecific bands with phospho-S81 or total PR antibodies, indicating a high degree of specificity.
T47D and HeLa cells (stably or transiently expressing PR isoforms) are routinely used as model systems for studying PR action; these cell lines behave similarly with regard to the regulation of posttranslational PR modifications and subsequent changes in receptor function (19, 24, 62). To determine the kinetics of PR Ser81 phosphorylation, we analyzed T47D and HeLa cells stably expressing PR-B. Following a time course of 10 nM R5020 treatment (0 min to 6 h), we observed increased Ser81 phosphorylation beginning at 10 min (T47D-YB) (Fig. 1C) to 15 min (HeLa-PR) (data available on request). This reached a maximum level in both cell lines at 30 to 60 min (Fig. 1C and data available on request). PR Ser81 phosphorylation preceded the ligand-dependent PR upshift primarily mediated by phosphorylation events on one or more unidentified residues (71). Note that ligand-dependent downregulation of PR was observed after at least 4 h of R5020 treatment in both cell lines (58).
PR phosphorylation on Ser294, Ser345, and Ser400 occurs in response to either progestins (i.e., R5020) or mitogenic inputs to MAPKs and/or cdk2 (i.e., EGF, serum) (24, 62, 79). To determine the potential for mitogenic inputs to regulate Ser81 phosphorylation, we performed a time course of EGF treatment in HeLa-PR cells (data available on request). PR Ser81 phosphorylation was not affected by this mitogen, following up to 60 min of EGF treatment, despite significant activation of Erk1/2 over the same time course. To test a broader spectrum of mitogens, we used fetal bovine serum (FBS; 20%) as a rich source of multiple growth factors. HeLa-PR cells were grown overnight either in serum-free medium, medium supplemented with 5% DCC (charcoal-stripped steroid-free medium), or full growth medium (5% FBS), followed by treatment with either R5020 (positive control for Ser81 phosphorylation; 60 min) or 20% FBS (15 or 60 min). Only R5020 treatment induced robust PR Ser81 phosphorylation (data available on request); no phosphorylation was detected following any of the serum treatments. MAPK (Erk1/2) phosphorylation served as a positive control for serum/mitogenic treatment. Finally, we used the synthetic PR antagonist/partial agonist, RU486, to demonstrate the specificity of PR ligand induction of Ser81 phosphorylation. T47D-YB (Fig. 1D) and HeLa-PR (data available on request) cells were treated with R5020, RU486, or a combination of both. Both ligands induced potent PR Ser81 phosphorylation, while the combination of R5020 plus RU486 was neither additive nor inhibitory. Cumulatively, these data suggest that PR Ser81 phosphorylation occurs primarily in response to progestins, although we frequently observed a low level of basal phosphorylation at this site (see Fig. 1; addressed below).
In vitro kinase assays suggest that ck2, a ubiquitously expressed Ser/Thr protein kinase, directly phosphorylates PR on Ser81 (80). We probed the requirement for ck2 kinase activity in intact cells using two different synthetic, highly specific ck2 kinase inhibitors, TBB and DMAT (23). HeLa-PR and T47D-YB cells were pretreated with increasing concentrations of either TBB or DMAT (or dimethyl sulfoxide [DMSO] vehicle alone) for 30 min, followed by 30 min of R5020. Again, PR Ser81 was potently phosphorylated in response to treatment of cells with R5020 alone (30 min). However, hormone-induced PR Ser81 phosphorylation was completely blocked with either of the ck2 inhibitors in both HeLa-PR (Fig. 2A) and T47D-YB (Fig. 2B) cells. We observed a loss of PR protein at high doses of TBB, the more potent of the two ck2 inhibitors. This is likely due to increased PR degradation, as ck2 is a key regulator of the PR chaperone molecule, hsp90; ck2-mediated phosphorylation of hsp90 is essential for its chaperone activity (52). These data suggest that ck2 kinase activity is required for ligand-dependent PR Ser81 phosphorylation. To determine the specificity of this phosphorylation event in vivo, we examined Ser81 phosphorylation in the presence of a broad spectrum of inhibitors for kinases known to affect PR phosphorylation at other N-terminal serine residues, including PP2 (c-Src; Ser345), Roscovitine (cdk2; Ser400), and U0126 (MEK1-MAPK; Ser294) (24, 62, 68). HeLa-PR cells were pretreated with each kinase inhibitor, followed by R5020 for 30 min. Again, Ser81 was robustly phosphorylated in response to R5020. While DMSO alone (the vehicle for each kinase inhibitor) somewhat reduced R5020-induced PR Ser81 phosphorylation (Fig. 2C, compare lane 2 to lane 8), this ligand-regulated phosphorylation event was completely inhibited (compare lane 8 to lanes 3 and 4) only in the presence of the ck2 inhibitors. Together, these data suggest that in the presence of progestin, PR is phosphorylated on Ser81 specifically by (endogenous) ck2.
Fig. 2.

PR Ser81 is phosphorylated by endogenous ck2. (A and B) HeLa-PR (A) and T47D-YB (B) cells were serum starved for 18 h. Cells were then pretreated with increasing doses of TBB (1 to 100 μM), DMAT (1 to 100 μM), or DMSO (vehicle) for 30 min, followed by 10 nM R5020 for 30 min. Alternatively, cells were treated with R5020 for 30 min or vehicle (EtOH) with no pretreatment. Lysates were analyzed by Western blotting using p-S81, PR, and Erk1/2 antibodies. (C) HeLa-PR cells were starved for 18 h in serum-free medium. Cells were then pretreated (30 min) with TBB (10 μM), DMAT (10 μM), PP2 (10 μM), Roscovitine (100 μM), U0126 (10 μM), or vehicle (DMSO) or left untreated. Following kinase inhibitor pretreatments, cells were treated with 10 nM R5020 or vehicle (EtOH) for 30 min. Lysates were analyzed by Western blotting as described for panel A. (D) Left: T47D-YB cells were serum starved for 18 h and treated with EtOH or 10 nM R5020 for 60 min (left two lanes). Alternatively, cells were treated sequentially as follows: 18 h with thymidine (2.5 μg/ml) or vehicle (PBS), iMEM plus 5% DCC for 7 h, iMEM-5% DCC-mimosine (50 μg/ml; G1/S Sync.) or vehicle (EtOH; Unsync.) for 18 h. Following synchronization (confirmed by flow cytometry; data not shown), protein was analyzed via Western blotting with antibodies for p-S81, phospho-Ser294 (p-S294), or PR. Right: T47D-YB cells were synchronized as just described (or treated with vehicle; Unsync). Following synchronization, cells were treated for 60 min with vehicle (DMSO) or TBB (10 μM). Protein was analyzed via Western blotting with antibodies for p-Ser81, PR, or Erk1/2 (loading control).
ck2 has been shown to be regulated in part by cell cycle-dependent localization to the nucleus (51, 78). Steroid receptors rapidly shuttle between the cytoplasm and nucleus; in the presence of progestins, PRs are primarily nuclear. To further address the potential for ck2-mediated regulation of PR Ser81 in the absence of progestins (i.e., basal phosphorylation levels observed above), we tested the cell cycle dependence of this event. For these studies, T47D-YB cells were synchronized at the G1/S transition using mimosine, a chemical inhibitor of DNA replication; synchronization of control (vehicle) and mimosine-treated T47D-YB cultures was confirmed by flow cytometry (data not shown). In G1/S-synchronized T47D-YB cells, but not vehicle controls, we observed robust PR Ser81 phosphorylation in the complete absence of ligand (Fig. 2D, left), but it was comparable in magnitude to levels induced following progestin (R5020 or RU486) treatment of unsynchronized cells (Fig. 1D and Fig. 2D, left). Ser294, a MAPK site primarily regulated only in PR-B, was unaffected by mimosine-induced synchronization (Fig. 2D, left). To confirm the ck2 dependence of PR Ser81 phosphorylation in G1/S phase cells, we treated synchronized populations of cells with or without the ck2 inhibitor, TBB. As in progestin-treated cells above (Fig. 2A to C), ligand-independent PR Ser81 phosphorylation in G1/S phase cells was completely blocked by addition of the ck2 inhibitor (Fig. 2D, right). Cumulatively, these data suggest that phosphorylation of PR Ser81 occurs independently of ligand when breast cancer cells are passing through the G1/S phase of the cell cycle, a period when ck2 is primarily nuclear (51, 78). Notably, ck2 is both cytoplasmic and nuclear in untreated T47D cells. Upon progestin-induced nuclear localization of PR, we observed only subtle increases in nuclear relative to cytoplasmic ck2 (data not shown).
PR Ser81-dependent transcriptional activity and promoter selectivity.
To investigate the functional consequences of PR Ser81 phosphorylation by ck2, we created a phospho-mutant receptor. Point mutation of phosphorylated residues within phospho-proteins can shift specificity to adjacent or very nearby phospho-acceptor sites that are not detected using mass spectrometry of the wt protein (63). Thus, both PR residues (Ser79 and Ser81) were mutated to ensure that nearby Ser79 is not weakly targeted by highly active kinases (in vivo) when Ser81 is mutated. Phospho-Ser81 PR antibody specificity was verified using the double phospho-mutant receptor (S79/81A PR). Western blotting showed that when transiently transfected into HeLa cells, wt PR and S79/81A PR-B were expressed at equal levels; following treatment with R5020, Ser81 phosphorylation was detected only in cells transfected with wt PR (Fig. 3A). Notably, wt and S79/81A receptors were similarly phosphorylated on all other PR phosphorylation sites tested (Ser190, Ser294, Ser345, and Ser400; data not shown), suggesting that mutant receptors fold properly and bind ligand. To determine if phospho-mutant S79/81A PR was capable of binding DNA and subsequently activating transcription, we analyzed wt and mutant PRs using PRE-luciferase reporter gene assays. In transiently transfected HeLa cells treated with vehicle or R5020, wt and S79/81A PRs behaved similarly (Fig. 3B); each receptor activated PRE-luciferase transcription to similar levels (∼15- to 20-fold) in the presence of progestin (Fig. 3B). Additional characterization of the S79/81A PR mutant using confocal microscopy showed no apparent differences in subcellular localization of S79/81A PR relative to wt PR, in both the presence and absence of ligand (data not shown). Single mutant receptors (S79A and S81A) behaved similarly to the double mutant (not shown).
Fig. 3.
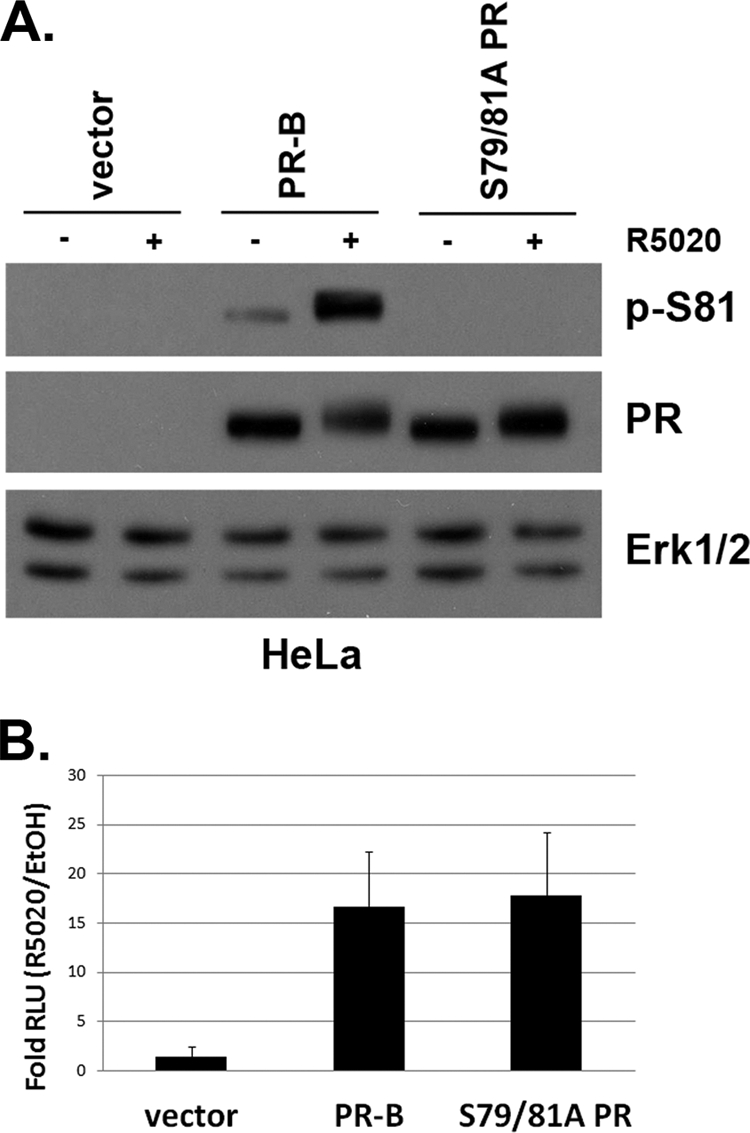
S79/81A PR phospho-mutant is transcriptionally active. (A) HeLa cells were transiently transfected with wt PR-B, S79/81A PR, or empty vector. At 24 h following transfection, cells were starved for 18 h in serum-free medium and then treated with 10 nM R5020 for 60 min. Lysates were analyzed via Western blotting using p-S81, PR, and Erk1/2 antibodies. (B) HeLa cells were transiently transfected with plasmids expressing wt PR-B, S79/81A PR, or vector only, as well as a firefly PRE-luciferase reporter construct and Renilla expression control. At 24 h following transfection, cells were starved for 18 h in serum-free medium, followed by an 18-h 10 nM R5020 (or vehicle) treatment. Fold relative luciferase units (RLU; PRE-luciferase over Renilla luciferase controls) of R5020-treated cells over EtOH-treated cells is plotted. Error bars represent means ± standard deviations (SD) of results from three independent experiments.
We then created multiple clones of stable T47D-Y cell lines expressing S79/81A mutant PR (T47D-S79/81A). Cells expressing wt PR (T47D-YB) in the same parental cell line background served as controls. Western blotting demonstrated that S79/81A PR-B is expressed at similar levels relative to wt PR-B in this model system (Fig. 4A). Again, upon progestin treatment, we detected robust Ser81 phosphorylation in wt, but not S79/81A, PR-B-expressing cells. Additionally, ligand-dependent receptor downregulation, which has been shown to be augmented by MAPK-dependent PR phosphorylation (i.e., at Ser294) (58), followed a similar time course in cell lines expressing either wt or phospho-mutant S79/81A PR. To verify that ck2 expression levels remained equal among the clonal cell lines, we analyzed ck2α and β protein levels via Western blotting (Fig. 4B). T47D-Y cells stably expressing wt PR-B, mutant S79/81A PR, or PR-null exhibited equal levels of both ck2 subunits; neither subunit appeared to be affected by treatment with R5020.
Fig. 4.

Stable S79/81A PR cell lines have impaired anchorage-independent survival in soft agar. (A) T47D-Y cells stably expressing wt PR-B (T47D-YB) or S79/81A PR (T47D-S79/81A) were serum starved for 18 h and then treated with 10 nM R5020 for 0 to 18 h or vehicle (EtOH; 18 h). Lysates were analyzed by Western blotting using p-S81, PR, and Erk1/2 antibodies. (B) T47D-Y cells stably expressing wt PR-B (T47D-YB) or S79/81A PR (T47D-S79/81A) or unmodified were serum starved for 18 h and then treated with 10 nM R5020 or EtOH for 60 min. Lysates were analyzed via Western blotting using antibodies against ck2α, ck2β, PR, and Erk1/2 (loading control). (C) T47D-Y cells (PR-null) or T47D cells stably expressing PR-B or S79/81A PR were plated in soft agar containing 5% DCC medium and either EtOH or 10 nM R5020 for 21 days. Colonies were counted in 15 fields/treatment group, and error bars represent the standard errors of the means (SEM) of these measurements. Soft agar assays were performed in triplicate with similar results. Asterisks indicate statistical significance (P < 0.05; determined using an unpaired Student's t test) compared to the respective treatment group (EtOH or R5020) in control cells (PR-null).
In soft agar assays performed in vitro, the proliferative and survival effects of progestins are mediated by PR-B but not PR-A (25). We therefore assayed the ability of S79/81A mutant PR to induce breast cancer cell growth in anchorage-independent soft agar assays. Stable T47D cell lines expressing either wt PR or S79/81A PR-B or PR-null were plated for soft agar colony formation assays in the presence of either vehicle or R5020 (10 nM). Following 21 days, established colonies were counted. Cells stably expressing S79/81A PR retained their ability to form colonies in response to R5020; total numbers of R5020-induced colonies were similar between cells expressing wt or S79/81A PR by the end of the 21-day assay, while PR-null cells failed to grow well in either condition (Fig. 4C; data from additional clones are available on request). Interestingly, however, cells expressing S79/81A PR formed significantly fewer colonies in the ligand-independent condition than cells expressing wt PR-B; S79/81A PR cells resembled PR-null cells in this regard (Fig. 4C). These data suggest that in the absence of exogenously added progestin, phospho-Ser81 PR may regulate genes that primarily contribute to cell survival and/or proliferation. Ligand binding is able to overcome this deficit, perhaps because the same set of genes are also highly responsive to hormone (addressed below).
Although our PRE-luciferase reporter gene analysis (Fig. 3B) indicated that S79/81A PR behaved similarly to wt PR, transcriptional activity on endogenous PR target genes offers a more sensitive and relevant readout of PR genomic action (i.e., PR-dependent regulation of complex promoters/distant enhancer elements arrayed in chromatin). Additionally, we have shown that PR phosphorylation by rapidly activated cytoplasmic protein kinases provides a mechanism for altered PR target gene selectivity, recruiting differentially phosphorylated PR species to specific gene subsets (reviewed in reference 18). Using our stable T47D cell line models, we surveyed mRNA expression of known PR target genes in the absence and presence of progestin (R5020; 0 to 18 h) by quantitative real-time PCR (qPCR). While many progestin-regulated genes were similarly expressed in cells containing either wt PR or S79/81A PR-B, others were differentially regulated (see below, Fig. 5; data from additional clones are available on request). These included the previously identified progestin-regulated genes BIRC3 (64), HSD11β2 (2), and HbEGF (4, 20, 81).
Fig. 5.

Endogenous PR target gene expression is attenuated in cells containing S79/81A PR relative to wt PR. (A, B, and C) Top: T47D-Y cells stably expressing either wt PR-B or S79/81A PR, or unmodified (PR-null) cells, were starved for 18 h in serum-free medium, followed by treatment with 10 nM R5020 or EtOH for 6 h. BIRC3 (A), HSD11β2 (B), HbEGF (C), or β-actin (internal control) mRNA levels were analyzed by qPCR. Middle: T47D-Y cells stably expressing wt PR-A, PR-B, or S79/81A PR were serum starved for 18 h, followed by treatment with 10 nM R5020 or EtOH for 6 h. BIRC3 (A), HSD11β2 (B), HbEGF (C), or 18S (internal control) mRNA levels were analyzed by qPCR. Asterisks indicate statistical significance (P < 0.05; determined using an unpaired Student's t test) compared to the respective treatment group (EtOH or R5020) in control cells (PR-null or PR-A). Bottom (C): T47D-YB cells were starved for 18 h in serum-free medium. Cells were then pretreated with TBB (10 μM) or DMSO (vehicle) for 30 min, followed by 60 min of 10 nM R5020. HbEGF and β-actin (internal control) mRNA expression was analyzed using qPCR. Error bars represent means ± SD of triplicate measurements.
Notably, in the absence of progestin, BIRC3 (baculovirus inhibitor of apoptosis repeat 3), an antiapoptosis gene recently identified as a PR target gene (64), exhibited decreased levels of basal transcription in cells stably expressing S79/81A mutant PR relative to cells stably expressing wt PR-B (Fig. 5A, top). Unliganded PR appears to contribute to basal BIRC3 expression, as PR-null cells (T47D-Y) also contain lower levels of BIRC3 mRNA relative to cells expressing wt PR-B (T47D-YB). Thus, mutation of the Ser81 phosphorylation site in PR appears to have abrogated basal expression of this gene. Additionally, although mutant S79/81A PR was able to weakly induce BIRC3 mRNA in response to ligand, levels of this transcript never reached those observed in R5020-treated cells containing wt PR-B. Finally, T47D cells stably expressing PR-A (T47D-YA), and thus lacking the BUS region containing Ser81, displayed significantly lower basal expression of BIRC3 and failed to respond to progestin relative to cells expressing wt PR-B (Fig. 5A, bottom), indicating that the structural requirements for regulation of this gene (basal and ligand dependent) are localized to the segment of PR unique to the B isoform, which includes the Ser81 phosphorylation site. Together, these data indicate that phosphorylation at PR-B Ser81 significantly contributes to the basal expression of BIRC3 and is also required for robust responses to ligand.
HSD11β2 (11β-hydroxysteroid dehydrogenase type 2), a dehydrogenase enzyme that mediates tissue-specific metabolism of glucocorticoids (9), has previously been identified as a cancer-associated proliferative protein (40) and a progestin-responsive gene (2, 21). HSD11β2 behaved similarly to BIRC3 in that basal mRNA levels were significantly decreased in cells containing mutant S79/81A PR, as well as in PR-null cells, relative to wt PR-B-expressing cells, again strongly suggesting that wt PR Ser81 phosphorylation is responsible for the maintenance of basal transcription of this gene (Fig. 5B, top). Similar to the regulation of BIRC3, cells containing S79/81A PR further enhanced HSD11β2 mRNA expression in response to ligand, while overall transcript levels remained significantly lower relative to those induced in cells expressing wt PR-B. Finally, cells stably expressing PR-A contained HSD11β2 mRNA levels similar to those seen in S79/81A PR cells (both basally and in response to ligand), again suggesting that regulation of this gene is linked to PR-B-specific phosphorylation of Ser81 (Fig. 5B, bottom). These data indicate that PR-B Ser81 phosphorylation primarily regulates the basal expression of these genes (BIRC3, HSD11β2) but can also alter the magnitude of their response to hormone. Taken together with the above effects on soft agar colony formation (Fig. 4C), our data suggest that phospho-Ser81 PR contributes to gene regulation and breast cancer cell survival, even when progestins are absent or limiting.
HbEGF (heparin-binding epidermal growth factor-like growth factor) is a well-characterized phosphorylation-sensitive PR target gene shown to be important for the growth of mammary epithelial cells (4, 16, 81). In cells expressing wt PR-B, HbEGF mRNA levels were responsive to ligand (Fig. 5C, top). In contrast, cells expressing mutant S79/81A PR failed to induce HbEGF mRNA in response to R5020. Interestingly, in contrast to the previously discussed genes (Fig. 5A and B), basal HbEGF transcript levels remained comparable in the absence of ligand in cells expressing either wt PR-A or PR-B, mutant S79/81A PR, or no PR, suggesting that PR does not influence basal transcription of this gene. Cells expressing PR-A and treated with progestin failed to induce HbEGF, again implicating the Ser81-containing region unique to PR-B in the progestin-dependent regulation of this gene (Fig. 5C, middle). Finally, cells treated with the ck2 inhibitor, TBB, also failed to induce HbEGF mRNA in response to ligand (Fig. 5C, bottom). Together, these data implicate the kinase activity of ck2, presumably through direct phosphorylation of PR Ser81, in progestin-induced upregulation of HbEGF mRNA expression.
To verify that the transcriptional differences described above (BIRC3, HSD11β2, and HbEGF) between cells expressing wt PR and S79/81A PR indeed reflect a functional requirement for phosphorylation of PR Ser81 in gene activation, rather than altered kinetics of gene activation, we analyzed mRNA isolated from cells following a time course of R5020 treatment (0 to 18 h) (Fig. 6). Impaired transcription observed in S79/81A PR-B-expressing cells relative to cells containing wt PR-B remained significant throughout this time course. Absolute mRNA levels (HbEGF and HSD11β2) became equal only after the peak of transcriptional activation, when mRNA levels began to decline. These data support the conclusion that PR Ser81 is required for absolute regulation of selected PR target genes over an extended time course.
Fig. 6.
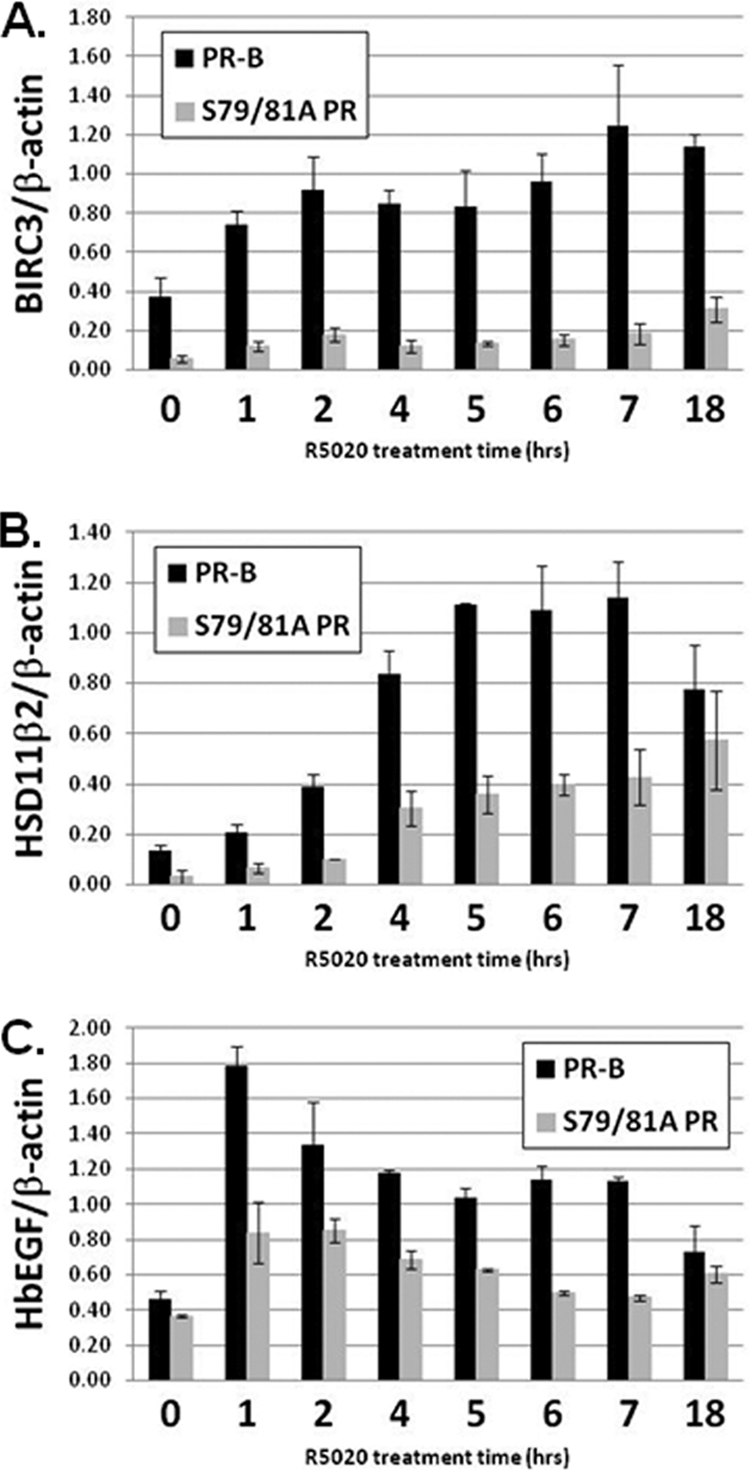
Time course of endogenous gene expression in wt and S79/81A PR-expressing cells. T47D-Y cells stably expressing either wt PR-B or S79/81A PR were starved for 18 h in serum-free medium, followed by treatment with 10 nM R5020 for 0 to 18 h. BIRC3 (A), HSD11β2 (B), HbEGF (C), or β-actin (internal control) mRNA levels were analyzed by qPCR. Statistical significance (P < 0.05; determined using an unpaired Student's t test) was achieved for all time points when comparing wt PR-B- and S79/81A PR-expressing cells with the following exceptions: HSD11β2 (18 h) and HbEGF (0 and 18 h). Error bars represent means ± SD of triplicate measurements.
Notably, the expression of well-characterized PR target genes, including c-Fos, tissue factor (TF), and EGFR (epidermal growth factor receptor) (38, 55, 56) was not differentially affected either basally or in response to ligand in cells expressing mutant S79/81A PR compared to expression of those expressing wt PR (data not shown). These genes represent a diverse spectrum of progestin-responsive promoters that display a variety of transcriptional kinetics (i.e., peak mRNA expression) following ligand treatment at 1 h (c-Fos), 6 h (TF), and 18 h (EGFR). These data suggest that mutation of the Ser81 phosphorylation site has not disrupted the ability of PR to activate endogenous target genes via general mechanisms (i.e., that may alter all PR transcriptional complexes or effect PR localization), indicating that the genes discussed above are uniquely regulated by phospho-PR Ser81. Results repeated in multiple clones of T47D cells stably expressing wt and phospho-mutant PRs (data available on request).
There are few reports of ligand-independent PR action. Surprisingly, both BIRC3 and HSD11β2 exhibited basal upregulation in cells expressing wt but not phospho-mutant PR-B (Fig. 5A and B and data available on request). To confirm that these genes are regulated by phospho-PRs independently of progestin, we employed an isogenic model of inducible PR expression. T47D-iEV (empty vector) and T47D-iPR-B (inducible wt PR-B) cells were treated with a small molecule inducer (AP21967; AP) or vehicle (EtOH) for 48 h; Western blotting confirmed PR-B expression (Fig. 7A, inset). In the absence of progestin, mRNA isolated from these cells showed significant increases in both BIRC3 (Fig. 7A, left) and HSD11β2 (Fig. 7A, right) transcripts only when PR-B was expressed. In contrast, transcription of two control genes, HbEGF, a ligand-dependent PR Ser81-regulated gene that is not basally regulated by wt PR (Fig. 5C), and TF, a gene that is not responsive to PR Ser81 phosphorylation, were not significantly affected by PR expression (data not shown). These data confirm that basal transcription of these phospho-Ser81-regulated genes is indeed PR dependent, but independent of exogenously added progestins.
Fig. 7.

Basal transcriptional regulation of phospho-Ser81-dependent genes. (A) T47D-iEV and T47D-iPR-B cells were treated for 48 h with 1 nM AP21967 (+AP and −AP) or vehicle (EtOH) to induce PR-B expression (inset). BIRC3 (left), HSD11β2 (right), or GAPDH (internal control) mRNA levels were analyzed by qPCR. Asterisks (*) indicate statistical significance (P < 0.05; determined using an unpaired Student's t test) compared to the respective treatment group (+AP or −AP) in control cells (iEV), as well as in response to treatment (+AP or −AP) within each cell line. (B) T47D-Y cells stably expressing wt PR-B (PR-B.3), S79/81A PR (S79/81A PR.3 and S79/81A PR.4), or PR-null (PR-null.2) were G1/S synchronized as described in the legend to Fig. 2D. BIRC3 (left), TF (right), or β-actin (internal control) mRNA levels were analyzed by qPCR. Asterisks indicate statistical significance (P < 0.05; determined using an unpaired Student's t test) compared to the respective treatment group (Unsync. or Sync.) in control cells (PR-null.2). Error bars represent means ± SD of triplicate measurements.
Ligand-independent regulation of selected PR target genes provides a mechanism for PR coupling to cell cycle regulation in rapidly dividing cells. To link ck2-induced (ligand-independent) PR Ser81 phosphorylation (occurring in G1/S phase; Fig. 2D) to functional changes in gene expression, we examined BIRC3 mRNA levels during the G1/S phase of the cell cycle in synchronized populations of T47D cells either lacking PR or stably expressing wt or S79/81A PR-B. Upon G1/S phase synchronization of PR-null cells, we observed PR-independent (G1/S-dependent) increased BIRC3 mRNA expression (Fig. 7B, left). However, cells containing wt PR-B, but not phospho-mutant S79/81A PR, exhibited a further significant increase in BIRC3 mRNA levels (relative to PR-null cells). TF mRNA levels were similar among G1/S-synchronized cells, independent of their PR status (Fig. 7B, right). These data indicate that BIRC3, a gene regulated basally in response to Ser81 PR phosphorylation, is transcriptionally activated during G1/S phase, a period when ck2-dependent PR Ser81 phosphorylation occurs in the absence of progestins.
Recruitment of phospho-Ser81 PR and ck2 to target gene promoters.
To confirm direct regulation of PR target genes by phospho-Ser81 PR-B, we performed chromatin immunoprecipitation (ChIP) assays. In silico analysis of promoter and enhancer regions of the BIRC3 gene revealed several putative full-length PRE binding sites, including sites located just after the transcriptional start site (Fig. 8A). ChIP analysis was performed on lysates from EtOH- or R5020-treated cells stably expressing wt or S79/81A PR, or from PR-null cells, using PR-specific antibodies. In the presence of ligand, we detected robust recruitment (∼70-fold) of wt PR to a full-length PRE (PRE1) located within 4 kb (downstream) of the BIRC3 transcriptional start site (Fig. 8B). This is in contrast to much-decreased S79/81A PR recruitment (∼10-fold) to the same area observed in side-by-side assays performed from R5020-treated cells (Fig. 8B). PR-B recruitment to PRE1 appeared to be highly specific, as other areas tested within the proximal and distal promoter regions were negative for PR binding (data not shown). In the presence of progestin, wt and S79/81A PR-B were equally recruited to a PRE located in the TF promoter region (data available on request), a gene shown earlier not to be regulated by Ser81 phosphorylation. These data indicate that decreased recruitment of S79/81A PR to the PRE1-containing region of BIRC3 is specific to this phospho-Ser81-responsive gene and does not represent a general defect in DNA-binding and/or tethering to general transcription factors by mutant S79/81A PR. Interestingly, although we observed significant differences in the basal levels of BIRC3 mRNA expression between cells containing wt and S79/81A PR (Fig. 5 and 6), we did not detect appreciable recruitment of PR to PRE1 in the absence of progestin. It is possible that PRE1 primarily regulates the ligand-activated transcriptional response of this gene, whereas another PRE(s) in the region may regulate basal activities and would, therefore, not be detected in our ChIP analyses (focused on PRE1).
Fig. 8.

Decreased recruitment of S79/81A PR and ck2α to a PRE-containing BIRC3 enhancer region. (A) Schematic of PRE1 location in BIRC3 gene. PRE1 is located 3.4 kb downstream of the transcriptional start site (denoted with the arrow). The sequence of PRE1 is shown. (B and C) T47D-Y cells stably expressing either wt PR-B or S79/81A PR or unmodified cells (PR-null) were serum starved for 18 h. Cells were then treated with EtOH or 10 nM R5020 for 60 min. Fixed lysates were subjected to ChIP with antibodies against PR-B (B) or ck2α (C), and qPCR was performed on the isolated DNA using primers designed to amplify PRE1. Fold recruitment of PR or ck2α in R5020 conditions over EtOH is shown. Error bars represent means ± SEM of results from triplicate experiments. (D) T47D-Y cells stably expressing wt PR-B were serum starved for 18 h. Cells were then treated with EtOH or 10 nM R5020 for 60 min. Fixed lysates were subjected to ChIP with antibodies against PR-B (left), followed by ck2α (right), and qPCR was performed on the isolated DNA using primers designed to amplify PRE1 in BIRC3. Species-specific IgG antibodies were used as controls (IgG). ChIP-reChIP experiments were performed in duplicate, and a representative experiment is shown.
To determine if ck2, the kinase responsible for PR Ser81 phosphorylation and, therefore, functional activation of PR-B at Ser81-dependent target genes, was also present at this site, we repeated our ChIP assays using antibodies directed against ck2α, one of the active subunits comprising the ck2 holoenzyme. Interestingly, ck2α was also strongly recruited to PRE1 in cells containing wt PR-B (∼8-fold), but not in those containing S79/81A PR (∼1-fold) (Fig. 8C). These data indicate that in the presence of progestin, both wt PR-B and its activating kinase, ck2, are recruited to PR-binding sites within transcriptional regulatory regions of BIRC3. Moreover, surprisingly, mutation of PR Ser81 greatly diminished not only PR-B recruitment to this PRE but recruitment of ck2 as well, suggesting that phosphorylation of this residue is important for the formation of stable protein complexes that are associated with direct regulation of this gene.
To determine if PR and ck2α were corecruited to this site in the BIRC3 enhancer, we performed ChIP-reChIP analysis (Fig. 8D). In cells expressing wt PR-B, sequential immuno-precipitations using PR antibodies (Fig. 8D, left) followed by ck2α antibodies (Fig. 8D, right) showed that the two proteins were present together at PRE1. This interaction was detected only in cells following treatment with R5020. Reversing the order of the antibodies for the ChIP-reChIP experiment yielded similar results (data not shown). We conclude that phospho-Ser81 PR-B provides a platform for the early recruitment of ck2-containing transcriptional complexes that direct promoter-specific PR target gene regulation.
DISCUSSION
Our studies reveal novel hormone and cell cycle-dependent regulation of PR Ser81 by ck2, a protein kinase tightly associated with prosurvival and uncontrolled proliferative phenotypes that characterize human malignancy. We show that progestin induces robust ck2-dependent phosphorylation of PR Ser81. Interestingly, this ck2-dependent event also occurs in the absence of added PR ligands, during the G1/S transition point of the cell cycle (Fig. 2). This result highlights the important linkage that exists between PR and cell cycle regulation (22). Notably, hormone-dependent PR Ser81 phosphorylation is a relatively rapid event, occurring as early as 10 min following treatment with PR ligands (R5020, RU486; Fig. 1). Other potent mitogenic stimuli, including EGF and serum, failed to appreciably induce phosphorylation at this site (data available on request). Protein kinase inhibitor studies confirmed that ck2 is the kinase primarily responsible for PR Ser81 phosphorylation in vivo (Fig. 2). Mutational analysis revealed that phospho-mutant S79/81A PR, while equally transcriptionally active as wt PR in PRE-luciferase reporter gene assays (i.e., a minimal artificial promoter), exhibited dramatically impaired recruitment and transcriptional responses relative to wt PR on selected endogenous PR target genes (Fig. 5 to 8). PR Ser81 phosphorylation is required for efficient PR and ck2 recruitment to PRE1, located within the BIRC3 downstream enhancer region (Fig. 8). Taken together, these data indicate that PR/ck2 complexes may regulate a distinct subset of phospho-Ser81-specific PR-B target genes in both the presence and the absence of ligand (i.e., in proliferating/cycling cells). Our findings provide novel insight into how PR-B may contribute to breast cancer prosurvival and tumor progression, even when hormone concentrations are limiting.
Role of PR phosphorylation events in breast cancer models.
Phosphorylation can impact diverse properties of the respective substrate. Direct phosphorylation of PR at specific amino-terminal Ser residues has been shown to alter receptor stability, localization, protein complex formation, dimerization, transcriptional activity, and promoter selectivity (18, 75). Data presented here indicate that tightly regulated (i.e., in response to hormone-binding and/or during G1/S transition) Ser81 phosphorylation directs target gene specificity; we identified at least three PR target genes that are differentially regulated by phosphorylation at this site. One class of genes is altered in both the presence and the absence of progestin (BIRC3 and HSD11β2), while HbEGF is an example of a gene whose expression is primarily ligand and ck2 dependent (i.e., induced via hormone-regulated PR Ser81 phosphorylation), lacking regulation in the absence of ligand. The precise mechanism(s) through which Ser81 phosphorylation alters PR-B target gene specificity is not clear, but such phosphorylation might occur via complex mechanisms that may include altered formation of transcriptional complexes and/or recognition/binding affinity for PRE elements and associated regulatory elements, thus altering early events in promoter recruitment (Fig. 8 and further discussed below).
Related to this finding, phosphorylation on Ser81 contributes in part to PR isoform specificity (Fig. 5). The two predominant PR isoforms, PR-B and PR-A, have overlapping but distinct transcriptional profiles (64) and have tissue-specific effects on growth (54), presumably through activation of different subsets of target genes. These receptors are generally expressed at a 1:1 ratio (i.e., equal levels) in normal mammary epithelial cells, but the ratio of expression is often altered in breast cancers (53). The full-length receptor, PR-B, contains an N-terminal region (the BUS) unique to PR-B where Ser81 is located. Data presented here showing that PR-B-activated gene transcription is lost on selected genes following mutation of the Ser81 phosphorylation site, and that mutant S79/81A PR-B mimics PR-A in this regard, suggest that Ser81 may be critical for PR-B versus PR-A target gene specificity. Related to this concept, we have begun to explore the possibilities of altered PR-A/B protein-protein interactions with associated transcriptional coactivators, corepressors, and other cofactors. Changes in further posttranslational modifications of PR (sumoylation, acetylation, ubiquitination, subsequent multisite phosphorylation events) may also be isoform specific and dictated in part by early phosphorylation events (16) and/or sequential events (15) but are outside the scope of the present study.
Transcriptional mechanisms are highly ordered and dynamic processes, characterized by waves of interactions between DNA and dozens of regulatory molecules. Given this enormous complexity, the precise role of ck2-dependent PR Ser81 phosphorylation may remain elusive. Notably, preliminary cell fractionation and confocal experiments suggested identical subcellular localization of wt PR and S79/81A PR, independent of ligand (data not shown). Additionally, the rate of ligand-dependent downregulation/receptor turnover appeared to be unaltered by Ser79/81 mutation (Fig. 4). Effects on PR dimerization are unlikely, as S79/81A PR was able to activate PRE-luciferase transcription (Fig. 3) as well as regulate other endogenous PR target genes to levels equal to that of wt PR (c-Fos, TF, EGFR). These data indicate that mutant S79/81A PR is a fully functional transcription factor for some promoters but not others (i.e., promoter selectivity is primarily altered). Interestingly, much less phospho-mutant PR protein appeared to be recruited to a PRE located in the BIRC3 enhancer region relative to wt PR-B (Fig. 8), while recruitment to other Ser79/81-independent genes (TF; data available on request and Garabedian) was unaffected. This finding suggests a block at some early event required for efficient PR/DNA recognition and/or interaction. Recent work from Blind and Garabedian. (6) also suggests that phospho-specific steroid receptor isoforms are differentially recruited to the promoters of specific genes based on their phosphorylation status. Using ChIP analysis, the authors showed that phosphorylation patterns on the glucocorticoid receptor (GR) dictate which gene promoters those phospho-GRs were recruited to, the kinetics of that respective recruitment, and, therefore, which GR target genes were subsequently activated (6). Our data showing decreased recruitment of mutant S79/81A PR to select PR target genes (Fig. 8) are in concordance with this finding and suggest that this mechanism of transcriptional regulation may be a characteristic shared by many steroid receptors.
In addition to PR recruitment to the BIRC3 enhancer region, data presented here also show that ck2α, the kinase responsible for Ser81 phosphorylation of PR, is similarly recruited to the same region in the presence of progestin (Fig. 8). ChIP-reChIP experiments demonstrated that wt PR and ck2α reside together in the same DNA-bound protein complexes. Surprisingly, less ck2α is recruited to the BIRC3 enhancer region in cells expressing mutant S79/81A PR. These data suggest that PR Ser81 phosphorylation mediates the formation of stable transcriptional complexes that may contain multiple proteins/phospho-proteins. Other factors (not assayed herein), functioning similarly to estrogen receptor (ER) or AR-associated pioneer factors (45), may require ck2-dependent PR Ser81 phosphorylation for assembly and/or stable association (i.e., that can be detected upon cross-linking); no obvious sequences that could serve as binding sites for additional PR- or ck2-associated factors were noted in the BIRC3 or HSD11β2 gene regulatory regions. Notably, Narayanan et al. (57) showed that cyclin A and PR are recruited to PRE regions within the MMTV promoter (stably incorporated into the T47D cell genome). In these studies, the interaction between cyclin A and active cdk2 was necessary to stimulate PR transcriptional activity, primarily via phosphorylation of SRC-1 coactivator molecules (57). These findings using an exogenous MMTV promoter system, and our data presented herein, performed on endogenous PR target genes expressed in breast cancer cells, suggest that phosphorylation events and subsequent transcriptional activation of PR are tightly linked at selected promoters and that the protein kinases responsible for these modifications (of PR and/or coregulators) are an integral part of PR-containing transcriptional complexes. ER was recently shown to associate with ERK2 and CREB at selected estrogen-responsive genes important for breast cancer cell proliferation, although the required substrate(s) in transcriptional complexes that are phosphorylated by ERK2 activity (i.e., possibly CREB) has yet to be defined (48).
Notably, weak PR Ser81 phosphorylation occurred in the absence of progestins (Fig. 1 to 4). However, this site was potently phosphorylated in cells entering the G1/S boundary (Fig. 2D), as in response to progestin. Ligand binding to PR sets up an exquisite program of cell cycle synchronization wherein cells enter S phase following precisely timed regulation of cell cycle mediators (reviewed in reference 22). Indeed, PR target genes include cyclins (D, E, and A) and cdk inhibitors (p21 and p27), and progestin-treated breast cancer cells are known to pause or accumulate at the G1/S boundary (30). Given the tight coupling of PR to cell cycle control, it is perhaps not surprising that selected PR target genes depend upon PR Ser81 phosphorylation for regulation both in the presence (HbEGF) and absence (BIRC3 or HSD11β2) of ligand. Ligand-independent PR gene regulation may provide important clues to how ck2 is regulated during cell cycle traverse. Protein complex formation involving Ser81-phosphorylated PR and ck2 is the topic of future studies.
Functional significance of ck2 and PR Ser81 target gene regulation in breast cancer.
The Ser/Thr protein kinase ck2 is upregulated in every cancer studied thus far (72). Although ck2 itself does not appear to be an oncogene, it is thought that ck2 works in an oncogenic fashion by potentiating the activity of other oncogenes and progrowth signaling molecules that function as its major substrates (reviewed in reference 74). For example, numerous studies have shown that ck2 overexpression promotes tumorigenesis in existing transgenic mouse models of cancer (11, 39, 41, 42). In the context of breast cancer, where progestins have been implicated as a risk factor for tumor development and early progression (1, 5, 12), overexpressed ck2 could further enhance the oncogenic potential of PR through inappropriate phosphorylation (on Ser81). Notably, the genes that are transcriptionally regulated by PR Ser81 phosphorylation have been shown to be important in cell growth and have each been identified in various types of cancer, including breast cancer. BIRC3 is an anti apoptosis protein belonging to the inhibitor of apoptosis (IAP) family of proteins (65). IAPs bind to and inhibit other pro-death-associated proteins, such as caspases, thereby preventing apoptosis (44). BIRC3, a mammalian-specific IAP also known as cellular IAP2 (cIAP2), is overexpressed, along with other closely related IAP family members, in breast cancer (28). HSD11β2 is a dehydrogenase enzyme that is responsible for the tissue-specific metabolism of glucocorticoids (reviewed in reference 9). Specifically, HSD11β2 expression has proliferative effects, especially in tumors, through inactivation of the anti proliferative effects of GR (36). Of note, HSD11β2 is upregulated in many different cancers, including breast, whereas the corresponding normal nonneoplastic tissue normally lacks HSD11β2 expression (36, 40). As a PR target gene, HSD11β2 may be an important mediator of progestin action. Finally, HbEGF, a gene shown here to be regulated by ligand-induced PR Ser81 phosphorylation, has been shown to contribute to mammary cell proliferation and breast cancer cell growth (4, 20). Moreover, ck2 is frequently upregulated in breast cancer. This fact, coupled with our findings that phospho-Ser81 PR can drive the expression of genes that clearly contribute to breast cancer biology, suggests a scenario for ck2-high breast tumors, in which PR may be inappropriately or persistently phosphorylated on Ser81 (i.e., either basally or in response to ligand) and thereby contribute to a hyperproliferative state. Indeed, we observed increased ligand-independent soft agar colony formation in cells expressing wt PR-B relative to cells expressing S79/81A PR and PR-null cells. Thus, the basal level of anchorage-independent growth was abrogated in cells expressing phospho-mutant S79/81A PR (Fig. 4C); cells expressing PR-A also fail to grow in soft agar (25). Related to this finding, we suspect that many additional prosurvival and/or proliferative genes are regulated by phospho-Ser81 PR. The identification of a more complete Ser81-regulated gene signature in breast cancer cells awaits detailed gene array analyses. Additionally, the presence of phospho-PR Ser81 in breast tumors may provide a marker of activated PRs in S-phase cells (in progress).
Due to the diverse nature and subcellular distribution of the >300 substrates of ck2, it is not surprising that ck2 has been localized to nearly every cellular compartment, including, but not limited to, the nucleus, cytoplasm, plasma membrane, and mitochondria (reviewed in reference 26). Conflicting reports exist regarding a correlation between ck2 localization and cell cycle; this discrepancy is likely due to cell type-specific differences in ck2 distribution. Reports indicate that ck2 localization (either the holoenzyme or specific subunits) shifts to predominantly nuclear during the G1 phase of the cell cycle and at the G1/S border (51, 78); we have also detected a similar shift in PR localization in G1/S synchronized cells (data not shown). Phosphorylation of PR Ser81 in the absence of ligand (observed in cells arrested at the G1/S transition; Fig. 2D) may be regulated as a consequence of increased nuclear accumulation of ck2 and PR observed at this stage of the cell cycle. In addition, work from the Ahmed lab (reviewed in reference 32) showed that in response to androgenic or growth factor signals in prostate cancer cells, ck2 localization was strongly nuclear and specifically associated with the nuclear matrix and chromatin, areas of high transcriptional activity (33). Progestins may work similarly to their androgenic counterparts and direct PR to the ck2-containing nuclear compartment, subsequently inducing prolonged phosphorylation of PR Ser81. Interestingly, PR nuclear entry appears to precede Ser81 phosphorylation (data not shown), similar to the pattern recently described for PR phosphorylation on Ser294 and Ser400 (17).
Significantly, nearly 70% of breast cancers express both ER and PR at the time of diagnosis, in contrast to PR/ER expression in just 7 to 10% of normal breast luminal epithelium (67). As steroid hormone receptor (SR)-positive tumors progress, they frequently become hormone independent while retaining receptor expression, indicating an early switch to autocrine or paracrine growth factor signaling (60). In addition, many breast cancers have upregulated protein kinases, such as MAPK, c-Src, cdk2, and ck2, which can modify and hyperactivate PR (29, 69, 72, 77). Recently, progesterone was shown to mediate mammary stem cell self-renewal via paracrine mechanisms in which secreted factors (Wnt, RANKL) derived from PR-positive cells influence the PR-null stem cell niche (37). In PR-positive breast cancer cells, PR action drives proliferation, prosurvival signaling, and early invasion primarily by autocrine mechanisms (10, 25, 61). In an environment where steroid hormones are no longer required to drive cellular proliferation (i.e., during SR-positive tumor progression), the increased expression and constitutive activation of PR-activating protein kinases may promote increased cell survival and uncontrolled growth (i.e., in the face of endocrine therapies primarily directed against ER). Understanding how mitogenic protein kinases, such as ck2, alter PR phosphorylation and function is critical to fully understanding breast tumor etiology and developing better targeted therapies. Due to the ubiquitous nature of ck2 and its prevalence in many different types of cancer, there has been much interest in the development of ck2 inhibitors as anti cancer agents (73). Clinical ck2 inhibitors, in combination with more specific anti-progestins (new classes of selective progesterone receptor modulators or SPRMs), could provide an effective combination of targeted therapy for breast cancer treatment.
ACKNOWLEDGMENTS
We thank Khalil Ahmed (University of Minnesota) for providing test aliquots of ck2 inhibitors. We thank Andrea R. Daniel (Lange lab) for helpful comments on the manuscript and Todd Knutson (Lange lab) for supplying cDNA isolated from the inducible control and PR-B isogenic cell system.
This work was supported by NIH/NCI grant number R01 CA123763 (C.A.L.), Department of Defense Post-Doctoral Fellowship number USDOD ARMY/W81XWH-09-1-0639 PK0001 (C.R.H.), and NIH Institutional Training Grant number T32 CA009138 (C.R.H. and G.E.D.).
Footnotes
Published ahead of print on 25 April 2011.
REFERENCES
- 1. Anderson G. L., et al. 2004. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA 291: 1701–1712 [DOI] [PubMed] [Google Scholar]
- 2. Arcuri F., et al. 2000. Progestin regulation of 11beta-hydroxysteroid dehydrogenase expression in T-47D human breast cancer cells. J. Steroid Biochem. Mol. Biol. 72: 239–247 [DOI] [PubMed] [Google Scholar]
- 3. Bagowski C. P., Myers J. W., Ferrell J. E. 2001. The classical progesterone receptor associates with p42 MAPK and is involved in PI3-K signaling in Xenopus oocytes. J. Biol. Chem. 276: 37708–37714 [DOI] [PubMed] [Google Scholar]
- 4. Beerli R. R., Hynes N. E. 1996. Epidermal growth factor-related peptides activate distinct subsets of ErbB receptors and differ in their biological activities. J. Biol. Chem. 271: 6071–6076 [DOI] [PubMed] [Google Scholar]
- 5. Beral V. 2003. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362: 419–427 [DOI] [PubMed] [Google Scholar]
- 6. Blind R. D., Garabedian M. J. 2008. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J. Steroid Biochem. Mol. Biol. 109: 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boonyaratanakornkit V., et al. 2001. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol. Cell 8: 269–280 [DOI] [PubMed] [Google Scholar]
- 8. Brisken C., et al. 1998. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc. Natl. Acad. Sci. U. S. A. 95: 5076–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bush I. E., Hunter S. A., Meigs R. A. 1968. Metabolism of 11-oxygenated steroids. Metabolism in vitro by preparations of liver. Biochem. J. 107: 239–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carvajal A., et al. 2005. Progesterone pre-treatment potentiates EGF pathway signaling in the breast cancer cell line ZR-75. Breast Cancer Res. Treat. 94: 171–183 [DOI] [PubMed] [Google Scholar]
- 11. Channavajhala P., Seldin D. C. 2002. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene 21: 5280–5288 [DOI] [PubMed] [Google Scholar]
- 12. Chlebowski R. T., et al. 2003. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women's Health Initiative randomized trial. JAMA 289: 3243–3253 [DOI] [PubMed] [Google Scholar]
- 13. Chlebowski R. T., et al. 2009. Breast cancer after use of estrogen plus progestin in postmenopausal women. N. Engl. J. Med. 360: 573–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cicatiello L., et al. 2004. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol. Cell. Biol. 24: 7260–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clemm D. L., et al. 2000. Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol. Endocrinol. 14: 52–65 [DOI] [PubMed] [Google Scholar]
- 16. Daniel A. R., Faivre E. J., Lange C. A. 2007. Phosphorylation-dependent antagonism of sumoylation derepresses progesterone receptor action in breast cancer cells. Mol. Endocrinol. 21: 2890–2906 [DOI] [PubMed] [Google Scholar]
- 17. Daniel A. R., et al. 2010. The progesterone receptor hinge region regulates the kinetics of transcriptional responses through acetylation, phosphorylation, and nuclear retention. Mol. Endocrinol. 24: 2126–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daniel A. R., Knutson T. P., Lange C. A. 2009. Signaling inputs to progesterone receptor gene regulation and promoter selectivity. Mol. Cell Endocrinol. 308: 47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Daniel A. R., Lange C. A. 2009. Protein kinases mediate ligand-independent derepression of sumoylated progesterone receptors in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 106: 14287–14292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daniel A. R., et al. 2007. Linkage of progestin and epidermal growth factor signaling: phosphorylation of progesterone receptors mediates transcriptional hypersensitivity and increased ligand-independent breast cancer cell growth. Steroids 72: 188–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Darnel A. D., Archer T. K., Yang K. 1999. Regulation of 11beta-hydroxysteroid dehydrogenase type 2 by steroid hormones and epidermal growth factor in the Ishikawa human endometrial cell line. J. Steroid Biochem. Mol. Biol. 70: 203–210 [DOI] [PubMed] [Google Scholar]
- 22. Dressing G. E., Lange C. A. 2009. Integrated actions of progesterone receptor and cell cycle machinery regulate breast cancer cell proliferation. Steroids 74: 573–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duncan J. S., et al. 2008. An unbiased evaluation of CK2 inhibitors by chemoproteomics: characterization of inhibitor effects on CK2 and identification of novel inhibitor targets. Mol. Cell. Proteomics 7: 1077–1088 [DOI] [PubMed] [Google Scholar]
- 24. Faivre E. J., Daniel A. R., Hillard C. J., Lange C. A. 2008. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol. Endocrinol. 22: 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Faivre E. J., Lange C. A. 2007. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol. Cell. Biol. 27: 466–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faust M., Montenarh M. 2000. Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res. 301: 329–340 [DOI] [PubMed] [Google Scholar]
- 27. Filhol O., Cochet C. 2009. Protein kinase CK2 in health and disease: cellular functions of protein kinase CK2: a dynamic affair. Cell. Mol. Life Sci. 66: 1830–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foster F. M., et al. 2009. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 11: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gregory C. W., et al. 2004. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J. Biol. Chem. 279: 7119–7130 [DOI] [PubMed] [Google Scholar]
- 30. Groshong S. D., et al. 1997. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1). Mol. Endocrinol. 11: 1593–1607 [DOI] [PubMed] [Google Scholar]
- 31. Guerra B., Issinger O. G. 2008. Protein kinase CK2 in human diseases. Curr. Med. Chem. 15: 1870–1886 [DOI] [PubMed] [Google Scholar]
- 32. Guo C., Davis A. T., Yu S., Tawfic S., Ahmed K. 1999. Role of protein kinase CK2 in phosphorylation nucleosomal proteins in relation to transcriptional activity. Mol. Cell. Biochem. 191: 135–142 [PubMed] [Google Scholar]
- 33. Guo C., Yu S., Davis A. T., Ahmed K. 1999. Nuclear matrix targeting of the protein kinase CK2 signal as a common downstream response to androgen or growth factor stimulation of prostate cancer cells. Cancer Res. 59: 1146–1151 [PubMed] [Google Scholar]
- 34. Horwitz K. B., Koseki Y., McGuire W. L. 1978. Estrogen control of progesterone receptor in human breast cancer: role of estradiol and antiestrogen. Endocrinology 103: 1742–1751 [DOI] [PubMed] [Google Scholar]
- 35. Horwitz K. B., Mockus M. B., Lessey B. A. 1982. Variant T47D human breast cancer cells with high progesterone-receptor levels despite estrogen and antiestrogen resistance. Cell 28: 633–642 [DOI] [PubMed] [Google Scholar]
- 36. Hundertmark S., Buhler H., Rudolf M., Weitzel H. K., Ragosch V. 1997. Inhibition of 11 beta-hydroxysteroid dehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF-7 and ZR-75-1 breast cancer cells. J. Endocrinol. 155: 171–180 [DOI] [PubMed] [Google Scholar]
- 37. Joshi P. A., Jackson H. W., Beristain A. G., Di Grappa M. A., Mote P. A., Clarke C. L., Stingl J., Waterhouse P. D., Khokha R. 2010. Progesterone induces adult mammary stem cell expansion. Nature 465: 803–807 [DOI] [PubMed] [Google Scholar]
- 38. Kato S., et al. 2005. Progesterone increases tissue factor gene expression, procoagulant activity, and invasion in the breast cancer cell line ZR-75-1. J. Clin. Endocrinol. Metab. 90: 1181–1188 [DOI] [PubMed] [Google Scholar]
- 39. Kelliher M. A., Seldin D. C., Leder P. 1996. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J. 15: 5160–5166 [PMC free article] [PubMed] [Google Scholar]
- 40. Koyama K., Myles K., Smith R., Krozowski Z. 2001. Expression of the 11beta-hydroxysteroid dehydrogenase type II enzyme in breast tumors and modulation of activity and cell growth in PMC42 cells. J. Steroid Biochem. Mol. Biol. 76: 153–159 [DOI] [PubMed] [Google Scholar]
- 41. Landesman-Bollag E., Channavajhala P. L., Cardiff R. D., Seldin D. C. 1998. p53 deficiency and misexpression of protein kinase CK2alpha collaborate in the development of thymic lymphomas in mice. Oncogene 16: 2965–2974 [DOI] [PubMed] [Google Scholar]
- 42. Landesman-Bollag E., et al. 2001. Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol. Cell. Biochem. 227: 153–165 [PubMed] [Google Scholar]
- 43. Lange C. A., Shen T., Horwitz K. B. 2000. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc. Natl. Acad. Sci. U. S. A. 97: 1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liston P., et al. 1996. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379: 349–353 [DOI] [PubMed] [Google Scholar]
- 45. Lupien M., Brown M. 2009. Cistromics of hormone-dependent cancer. Endocr. Relat. Cancer 16: 381–389 [DOI] [PubMed] [Google Scholar]
- 46. Lydon J. P., et al. 1995. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9: 2266–2278 [DOI] [PubMed] [Google Scholar]
- 47. Lydon J. P., Ge G., Kittrell F. S., Medina D., O'Malley B. W. 1999. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Res. 59: 4276–4284 [PubMed] [Google Scholar]
- 48. Madak-Erdogan Z., Lupien M., Stossi F., Brown M., Katzenellenbogen B. S. 2011. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol. Cell. Biol. 31: 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meggio F., Pinna L. A. 2003. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17: 349–368 [DOI] [PubMed] [Google Scholar]
- 50. Migliaccio A., et al. 1998. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 17: 2008–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miro F. A., et al. 2002. Persistent nuclear accumulation of protein kinase CK2 during the G1-phase of the cell cycle does not depend on the ERK1/2 pathway but requires active protein synthesis. Arch. Biochem. Biophys. 406: 165–172 [DOI] [PubMed] [Google Scholar]
- 52. Miyata Y. 2009. Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell. Mol. Life Sci. 66: 1840–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mote P. A., Bartow S., Tran N., Clarke C. L. 2002. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res. Treat. 72: 163–172 [DOI] [PubMed] [Google Scholar]
- 54. Mulac-Jericevic B., Lydon J. P., DeMayo F. J., Conneely O. M. 2003. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. U. S. A. 100: 9744–9749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murphy L. C., Murphy L. J., Shiu R. P. 1988. Progestin regulation of EGF-receptor mRNA accumulation in T-47D human breast cancer cells. Biochem. Biophys. Res. Commun. 150: 192–196 [DOI] [PubMed] [Google Scholar]
- 56. Musgrove E. A., Lee C. S., Sutherland R. L. 1991. Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor alpha, epidermal growth factor receptor, c-fos, and c-myc genes. Mol. Cell. Biol. 11: 5032–5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Narayanan R., Adigun A. A., Edwards D. P., Weigel N. L. 2005. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol. Cell. Biol. 25: 264–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nardulli A. M., Katzenellenbogen B. S. 1988. Progesterone receptor regulation in T47D human breast cancer cells: analysis by density labeling of progesterone receptor synthesis and degradation and their modulation by progestin. Endocrinology 122: 1532–1540 [DOI] [PubMed] [Google Scholar]
- 59. Olsten M. E., Litchfield D. W. 2004. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem. Cell Biol. 82: 681–693 [DOI] [PubMed] [Google Scholar]
- 60. Osborne C. K., Schiff R., Arpino G., Lee A. S., Hilsenbeck V. G. 2005. Endocrine responsiveness: understanding how progesterone receptor can be used to select endocrine therapy. Breast 14: 458–465 [DOI] [PubMed] [Google Scholar]
- 61. Owen G. I., Richer J. K., Tung L., Takimoto G., Horwitz K. B. 1998. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J. Biol. Chem. 273: 10696–10701 [DOI] [PubMed] [Google Scholar]
- 62. Pierson-Mullany L. K., Lange C. A. 2004. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol. Cell. Biol. 24: 10542–10557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Resing K. A., et al. 1995. Determination of v-Mos-catalyzed phosphorylation sites and autophosphorylation sites on MAP kinase kinase by ESI/MS. Biochemistry 34: 2610–2620 [DOI] [PubMed] [Google Scholar]
- 64. Richer J. K., et al. 2002. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J. Biol. Chem. 277: 5209–5218 [DOI] [PubMed] [Google Scholar]
- 65. Rothe M., Pan M. G., Henzel W. J., Ayres T. M., Goeddel D. V. 1995. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83: 1243–1252 [DOI] [PubMed] [Google Scholar]
- 66. Sartorius C. A., et al. 1994. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors; only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 54: 3868–3877 [PubMed] [Google Scholar]
- 67. Seagroves T. N., Lydon J. P., Hovey R. C., Vonderhaar B. K., Rosen J. M. 2000. C/EBPbeta (CCAAT/enhancer binding protein) controls cell fate determination during mammary gland development. Mol. Endocrinol. 14: 359–368 [DOI] [PubMed] [Google Scholar]
- 68. Shen T., Horwitz K. B., Lange C. A. 2001. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol. Cell. Biol. 21: 6122–6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Steeg P. S., Zhou Q. 1998. Cyclins and breast cancer. Breast Cancer Res. Treat. 52: 17–28 [DOI] [PubMed] [Google Scholar]
- 70. Stoecklin E., Wissler M., Schaetzle D., Pfitzner E., Groner B. 1999. Interactions in the transcriptional regulation exerted by Stat5 and by members of the steroid hormone receptor family. J. Steroid Biochem. Mol. Biol. 69: 195–204 [DOI] [PubMed] [Google Scholar]
- 71. Takimoto G. S., et al. 1996. Role of phosphorylation on DNA binding and transcriptional functions of human progesterone receptors. J. Biol. Chem. 271: 13308–13316 [DOI] [PubMed] [Google Scholar]
- 72. Tawfic S., et al. 2001. Protein kinase CK2 signal in neoplasia. Histol. Histopathol. 16: 573–582 [DOI] [PubMed] [Google Scholar]
- 73. Trembley J. H., Chen Z., Unger G., Slaton J., Kren B. T., Van Waes C., Ahmed K. 2010. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 36: 187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Trembley J. H., Wang G., Unger G., Slaton J., Ahmed K. 2009. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell. Mol. Life Sci. 66: 1858–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ward R. D., Weigel N. L. 2009. Steroid receptor phosphorylation: assigning function to site-specific phosphorylation. Biofactors 35: 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weigel N. L., et al. 1995. Phosphorylation and progesterone receptor function. J. Steroid Biochem. Mol. Biol. 53: 509–514 [DOI] [PubMed] [Google Scholar]
- 77. Wilson G. R., et al. 2006. Activated c-SRC in ductal carcinoma in situ correlates with high tumour grade, high proliferation and HER2 positivity. Br. J. Cancer 95: 1410–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yu I. J., Spector D. L., Bae Y. S., Marshak D. R. 1991. Immunocytochemical localization of casein kinase II during interphase and mitosis. J. Cell Biol. 114: 1217–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang Y., et al. 1997. Phosphorylation of human progesterone receptor by cyclin-dependent kinase 2 on three sites that are authentic basal phosphorylation sites in vivo. Mol. Endocrinol. 11: 823–832 [DOI] [PubMed] [Google Scholar]
- 80. Zhang Y., Beck C. A., Poletti A., Edwards D. P., Weigel N. L. 1994. Identification of phosphorylation sites unique to the B form of human progesterone receptor. In vitro phosphorylation by casein kinase II. J. Biol. Chem. 269: 31034–31040 [PubMed] [Google Scholar]
- 81. Zhang Z., Funk C., Roy D., Glasser S., Mulholland J. 1994. Heparin-binding epidermal growth factor-like growth factor is differentially regulated by progesterone and estradiol in rat uterine epithelial and stromal cells. Endocrinology 134: 1089–1094 [DOI] [PubMed] [Google Scholar]