

Abstract
Magnesium, the second most abundant cellular cation after potassium, is essential to regulate numerous cellular functions and enzymes, including ion channels, metabolic cycles, and signaling pathways, as attested by more than 1000 entries in the literature. Despite significant recent progress, however, our understanding of how cells regulate Mg2+ homeostasis and transport still remains incomplete. For example, the occurrence of major fluxes of Mg2+ in either direction across the plasma membrane of mammalian cells following metabolic or hormonal stimuli has been extensively documented. Yet, the mechanisms ultimately responsible for magnesium extrusion across the cell membrane have not been cloned. Even less is known about the regulation in cellular organelles. The present review is aimed at providing the reader with a comprehensive and up-to-date understanding of the mechanisms enacted by eukaryotic cells to regulate cellular Mg2+ homeostasis and how these mechanisms are altered under specific pathological conditions.
Keywords: Mg2+, Mg2+ transport, channel, exchanger, cellular organelles, signaling
1. Introduction
Magnesium is the second most abundant cellular cation after potassium. High concentrations of total and free magnesium ion (Mg2+) have been measured within mammalian cells through a variety of techniques [1]. These concentrations are essential to regulate numerous cellular functions and enzymes, including ion channels, metabolic cycles, and signaling pathways as attested by the large number of observations gathered in the last twenty years. Yet, despite significant progress, our understanding of how cells regulate Mg2+ homeostasis still remains incomplete. There are both conceptual and methodological reasons for this limitation. The relative slow turn-over of Mg2+ across the plasma membrane or other biological membranes in the absence of metabolic and hormonal stimuli, the absolute abundance of total and free Mg2+ within the cell, and the limited occurrence of significant changes in free [Mg2+] have all contributed for a long time to the assumption that cellular Mg2+ concentration does not change significantly over time as it is consistently at a level more than adequate for the role of co-factor for various cellular enzymes and proteins. This assumption has consequently hindered the interest to develop techniques and methodologies able to rapidly and accurately measure changes in cellular Mg2+ content. In the last two decades, however, an increasing number of experimental and clinical observations have challenged this assumption. More than 1000 entries in the literature support a regulatory role of Mg2+ for various cellular functions, and indicate the occurrence of major fluxes of Mg2+ in either direction across the plasma membrane of mammalian cells following metabolic or hormonal stimuli. In turn, these fluxes have resulted in appreciable changes in cytosolic free [Mg2+] and total Mg2+ content within the cell and cellular organelles. Genetic and electrophysiological approaches in prokaryotes and eukaryotes have identified several Mg2+ entry mechanisms that operate in the cell membrane or in the membrane of cellular organelles (e.g. mitochondria and Golgi). The increased interest in elucidating the mechanism(s) by which Mg2+ regulates biological functions has promoted the development of new methodologies able to better detect and measure variations in cellular Mg2+ content, and to relate variations in Mg2+ homeostasis with specific pathologies and complications in human patients.
The present review will attempt to provide the reader with a comprehensive and up-to-date understanding of the mechanisms enacted by eukaryotic cells to regulate cellular Mg2+ homeostasis and how these mechanisms are altered under specific pathological conditions.
2. Cellular Mg2+ Distribution
Determinations of total and free Mg2+ concentrations by electron probe X-rays microanalysis (EPXMA), 31P-NMR, selective Mg2+-electrode, 13C-NMR citrate/isocitrate ratio or fluorescent indicators [Table I in ref. 1, and 3] consistently indicate total Mg2+ concentration ranging between 17 to 20mM in the majority of mammalian cell types examined [1, 2], with equivalent levels of Mg2+ localized within mitochondria, nucleus, and endo-(sarco)-plasmic reticulum. The presence of 15 to 18mM total Mg2+ content within these organelles has been attributed to Mg2+ binding to phospholipids, proteins, nucleic acids, chromatin and nucleotides depending on the particular organelle considered. As a result, only a fraction varying from 15% to 22% of such a large Mg2+ content is free in the lumen of these structures. Concentrations of 0.8 to 1.2 mM free [Mg2+] have in fact been measured in the matrix of cardiac and liver mitochondria [4,5], i.e. a value not too dissimilar to what reported to be in the cytoplasm of these cells, or in the extracellular space [1–3]. Similar free [Mg2+] concentrations are envisioned to be present in the nucleus and the endo-(sarco)-plasmic reticulum although no direct determinations have been carried out due to technical limitations. For the nucleus, this assumption is merely based upon the porous structure of the nuclear envelope, which makes reasonable to envision a lack of a gradient between cytoplasm and intranuclear environment. On the other hand, the free [Mg2+] within the lumen of the endoplasmic or sarcoplasmic reticulum cannot be reliably determined because of the elevated millimolar concentration of Ca2+ inside the organelle [6], and its high affinity (~50μM) as compared to a Mg2+ affinity of ~1.5 mM for fluorescent dyes such as Mag-Fura or Mag-Indo [7]. Cytoplasmic Mg2+ represents the last large and well detectable pool of Mg2+ within the cell. The majority of this Mg2+ pool (~4–5mM) is present in the form of a complex with ATP, phosphonucleotides in general, and phosphometabolites [8]. Because of its abundance (more than 5mM) and Mg2+ binding affinity (Kd ~78μM), ATP constitutes the largest metabolic pool able to bind Mg2+ within the cytoplasm and the mitochondrial matrix as well [9]. The binding/buffering capacity of ATP, phosphonucleotides and phosphometabolites, and perhaps proteins, maintains cytosolic free [Mg2+] between 0.5–1mM, or less than 5% of total cellular Mg2+ content (Table I in ref. 1) as attested by measurements obtained using fluorescent dyes, 31P-NMR and citrate/isocitrate ratio [1]. Overall, these results support the presence of a very limited chemical Mg2+ gradient across the cell membrane, and across the membrane of cellular organelles. In cells lacking intracellular compartmentation such as erythrocytes, Mg2+ buffering depends mostly on ATP, phosphonucleotides and phosphometabolites, proteins, and metabolic pools. Flatman and Lew [10] have observed three kinetically distinct binding pools for Mg2+ within erythrocytes. A low capacity, high affinity pool is represented by cell proteins, whereas the other two pools correspond reasonably well to ATP and 2, 3-diphosphoglycerate (2, 3-DPG) content, respectively [11]. This distribution has been further refined by Raftos et al. [12] to take into account Mg2+ binding to hemoglobin under oxygenated and not oxygenated conditions.
Little is known about the ability of cellular proteins to bind Mg2+. Aside from the aforementioned hemoglobin [12] in red blood cells, consensus sequences for Mg2+ binding have been reported for calmodulin [13], troponin C [14], parvalbumin [15], and S100 protein [16]. No indication is available as to whether other cytosolic or intra-organelle proteins can bind substantial amount of Mg2+ under physiological conditions and contribute to the elevated total Mg2+ concentrations measured within mitochondria and discrete regions of the endoplasmic or sarcoplasmic reticulum. A report by Bogucka et al. [17] more than forty years ago has suggested the presence of two proteins able to bind Mg2+ with high affinity/low capacity and high capacity/low affinity, respectively, in the intermembraneous space of the mitochondrion. However, the presence of these proteins has not been confirmed by subsequent studies, nor have the proteins been identified. The presence of Mg2+ binding sites has been reported for several other cellular proteins, but no information is available as to which extent these proteins bind Mg2+ under basal conditions and whether the binding changes significantly following hormonal or metabolic stimuli, or under pathological conditions. Moreover, the potential physiological relevance of Mg2+ binding by any of the mentioned proteins has been questioned by the observation that parvalbumin null mice do not exhibit hypomagnesaemia or any detectable changes in tissue Mg2+ handling and homeostasis [18].
Lastly, Mg2+ concentration in plasma and extracellular fluid is approximately 1.2–1.4 mM, one-third of which is bound by extracellular proteins (e.g. albumin) or other biochemical moieties [19]. Comparing this concentration to those available for cellular Mg2+ distribution, it becomes evident that chemical free [Mg2+] concentration across the cell membrane or the biomembranes of cellular organelles (e.g. mitochondria) is at or near zero trans condition in the majority of mammalian cells. Because the electrochemical equilibrium potential for cellular free [Mg2+] is ~50mM in most eukaryotes under resting conditions [20], it is evident that mechanisms must operate in the cell membrane to maintain cytosolic free Mg2+ and total cellular Mg2+ content within the measured levels. Figure 1 summarizes the main regulatory mechanisms involved in controlling cellular Mg2+ homeostasis.
Figure 1. Regulation of cellular Mg2+ homeostasis.
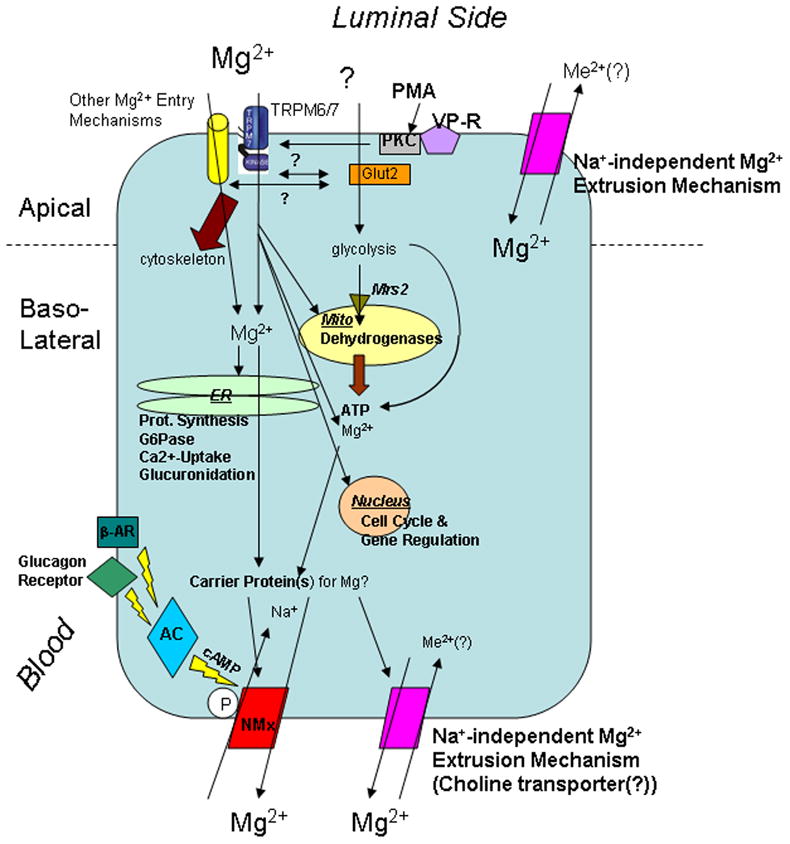
The cartoon summarizes the principal mechanisms controlling cellular Mg2+ homeostasis, compartmentation, and transport in and out of mammalian cell, as well as the main cellular functions regulated by changes in Mg2+ content within different compartments. For mere practical purpose, the entry mechanisms have been assigned to the apical domain of the cell. Abbreviation used in the figure: ER = endoplasmic reticulum; G6Pase = glucose 6 phosphatase; Mito = mitochondria; β-AR = β-adrenergic receptor; AC = adenylyl cyclase; NMx = Na+/Mg2+ exchanger; Me2+ = divalent cations; VP-R = vasopressin receptor; PMA = Phorbol-Myristate Acetate; PKC = Protein Kinase C.
3. Mg2+ Transport Mechanisms
Many mammalian cells maintained in culture in the presence of very low or even virtual zero [Mg2+]o do not shown a significant decrease in cellular Mg2+ content despite the relatively large gradient present across the cell membrane [2,21]. Different turn-over rates ranging from 1 hour in adipocytes to several days in lymphocytes, however, can be observed as a result of structural and functional differences in specific tissues and cells [21]. Furthermore, discrepancies can be observed in the same cell types based upon the experimental conditions or modality of isolation (e.g. cells in situ versus freshly isolated cells versus cells in culture). For example, cardiac ventricular myocytes attain 28Mg equilibrium within 3 hours in the whole animal but require 72–80 hours as dispersed cells incubated at 37°C, or even a longer period of time when incubated at 20°C [22–24]. Similar differences in the amplitude of Mg2+ transport have been observed in freshly isolated [25] vs. cultured [26] lymphocytes.
For a long time, the slow Mg2+ turn-over observed in various cells has contributed to the erroneous idea that cellular Mg2+ content does not change, or changes at such a slow pace that it lacks physiological relevance. In the last twenty-five years, this notion has been completely reversed by a large body of experimental evidence documenting the occurrence of large fluxes of Mg2+ across the plasma membrane of eukaryotic cells within minutes from the application of metabolic or hormonal stimuli [21, 27–29]. Lymphocytes [25,30], erythrocytes [31], cardiac myocytes [32,33] and liver cells [34–36] are just a few examples of the mammalian cells extruding 10% to 20% of their total cellular Mg2+ content in less than 10min from the application of adrenergic stimuli. The amplitude and rapidity of these fluxes suggest the presence and operation of powerful Mg2+ transport mechanisms at the cell membrane level [see ref. 29 for a list of experimental models and conditions]. Under the majority of conditions examined, however, these large fluxes have resulted in relatively small changes in free [Mg2+]i, suggesting that the operation of these Mg2+ entry mechanisms is tightly coupled with the ability of the cell to rapidly and effectively buffering the magnesium ions lost or accumulated [37,38].
As reported for other cations, Mg2+ is transported across the cell membrane or the membrane of cellular organelles through channels (Mg2+ accumulation) and exchanger mechanisms (Mg2+ extrusion). As illustrated in Figure 1, and reported in Table 1, the majority of the recently identified Mg2+ entry mechanisms operate at the cell membrane level with two notable exceptions that favor Mg2+ transport across the membrane of mitochondria and Golgi system, respectively. For the most part, these entry mechanisms present a modest selectivity for Mg2+ over other divalent cations. No structural information is currently available about the nature of the mechanisms that promote Mg2+ entry in the endoplasmic or sarcoplasmic reticulum, or favor Mg2+ extrusion across the cell membrane or the membrane of the cellular organelles.
Table 1.
Mg2+ Transporters in Mammalian Cells
| Family | Members | Apparent Km | Type of Transporter | Ref. | |
|---|---|---|---|---|---|
| Entry Mechanisms | |||||
| Cell Membrane | TRPM | TRPM6 | ~0.7 Mm | Channel | 43 |
| TRPM7 | ~0.7 mM | Channel | 42 | ||
| Claudins | claudin 16 (PCLN-1) | ~0.7mM | Channel | 92 | |
| claudin-19 | ~0.7mM | Channel | 100 | ||
| MagT1 | MagT1 | 0.2 mM | Channel | 101,103 | |
| SLC41 | SLC41A1 | 0.7 – 3 mM | Carrier(?)* | 147 | |
| SLC41A2 | 0.7 – 3 mM | Carrier(?) | 150,151 | ||
| ACDP | ACDP1 | ~0.7 mM | Carrier | 152 | |
| ACDP2 | ~0.5 mM | Carrier | 153 | ||
| NIPA | NIPA1 (SPG6) | 0.7 mM | Carrier | 157 | |
| NIPA2 | 0.7 mM | Carrier | 158 | ||
| Huntingtin | Huntingtin1 (HIP14) | 0.87 mM | Carrier | 160 | |
| HIP14L | 0.74 mM | Carrier | 160 | ||
| Mitochondria | Mrs2 | Mrs2/AtMrs2, Lpe10 | ~1.5 mM | Channel | 106 |
| Golgi | MMgt | MMgT1 | 1.5 mM | Channel | 112 |
| MMgT2 | 0.6 mM | Channel | 112 | ||
| Exit Mechanisms | |||||
| Cell Membrane | Na+/Mg2+ exchanger | ND | 15–20 mM | antiport | 131,133 |
| Na+-Independent | ND (choline?) | ~20mM | exchanger(?) | 137 | |
| SLC41 | SLC41A1 | ~0.7 mM | Carrier(?)* | 149 | |
| H+/Mg2+ exchanger | AtMHX# | ~15mM | exchanger | 141 | |
Identified only in plants and yeast and not in mammalian/human cells.
3.1 Channels
Channels favoring Mg2+ entry into the cell were originally described in prokaryotes [39, 40] and protozoan [41]. Recently, several Mg2+ entry mechanisms with channels or channels-like features have been identified in eukaryotic cells. Some of these mechanisms exhibit a relatively high specificity for Mg2+ although they can permeate other divalent cations as well. The majority of these channels is located in the cell membrane or perhaps translocate between early endosomal vesicles and the cell membrane whereas two of them are located in the mitochondrial membrane or in the Golgi cysternae (Table 1). As the characterization of these Mg2+ channels is far from complete, information relative to their regulation is still fragmentary. Furthermore, the perceived abundance of Mg2+ entry mechanisms raises the question as to which extent the different mechanisms contribute to regulate Mg2+ entry, or rather exert an absolute (or relative predominance) in specific cells under well defined conditions.
3.1.1 TRPM Channels
TRPM7 [42] and TRPM6 [43] were the first Mg2+ channels identified in mammalian cells by different approaches. Whereas TRPM7 is ubiquitous and therefore more in control of Mg2+ homeostasis in individual cells, TRMP6 is specifically localized in the colon and the distal convolute tubule of the nephron, a distribution that strongly emphasizes the role of this channel in controlling whole body Mg2+ homeostasis via intestinal absorption and renal re-sorption.
The original observations have originated a flurry of studies aimed at better understanding role, regulation, and interaction of these channels with other cellular components possibly involved in Mg2+ homeostasis. Presently, about 350 publications relative to TRPM7 and TRPM6 can be found in the literature. These publications clearly illustrate how these two channels share several similarities in terms of structure and operation and yet differ in various aspects ranging from location to hormonal modulation.
3.1.1.1 TRPM7
Fleig’s group first reported a key role of TRPM7 in promoting Mg2+ accumulation and cell growth [42]. At the time, the group identified the channel as LTRPC7, or long TRP channel 7, owing to the presence of a long extension outside the channel component [44]. This protein was already known as CHAK1 (channel kinase 1) [45] due to the presence of an alpha-kinase domain at its C-terminus [45], and its functional homology to eEF2-kinase [46]. Shortly after the initial observation of Nadler et al. [42], Runnels et al., [47] evidenced how TRPM7 combines a channel structure with an alpha-kinase domain at the C-terminus. Investigated for its possible role in Ca2+ signaling in lymphocyte, it soon became clear that the channel would carry preferentially Mg2+ and Ca2+ [42] as well as trace amounts of other divalent cations including Ni2+ and Zn2+ [48,49].
Located at the locus 15q21 of the human chromosome 15, TRPM7 is formed by 1865 amino acids arranged in 10 trans-membrane domains with both the C- and N- termini internalized. Ubiquitously expressed, the functional structure of the protein is supposed to be a tetramer. Yet, it is unclear whether the channel is a homotetramer or is formed by a varying stoichiometry combination of TRPM7 and TRPM6 monomers, perhaps in different portions of the cell membrane, or in different cells. Voets and colleagues [50] reported the functional expression of TRPM6 channels in HEK-293 cells with electrophysiological properties similar to those of TRPM7. On the other hand, Chubanov et al. [51] reported no electrical conductance through TRPM6 when this channel is expressed by itself in HEK-293 cells or in X. Laevis oocytes, and suggested that TRPM7 needed to be co-expressed with TRPM6 for the latter to be incorporated into channel complexes in the cell membrane. The association of TRPM6 and TRPM7 channel proteins to obtain a functional structure was subsequently confirmed by Schmitz et al. [52]. The functional characterization of TRPM6/TRPM7 chimeras remained controversial [53] until Yue’s group demonstrate that TRPM6 and TRPM7 do form a heterotetramer, and that pure TRPM6, pure TRPM7, and TRPM6/TRPM7 chimeras constitute three distinct ion channels with different divalent cation permeability, pH sensitivity, and unique single channel conductance [54,55]. In addition, this group reported that the activities of TRPM6, TRPM7, and TRPM6/TRPM7 can be differentiated by using 2-2-aminoethoxydiphenyl-borate (2-APB), which markedly increases Mg2+ and Ca2+ entry through TRPM6 [54]. These results support the notion that TRPM6 can form functional homotetrameric channels as well as heterotetrameric channels with TRPM7 [56]. Thus, it is conceivable that homotetrameric TRPM6, homotetrameric TRPM7, and heterotetrameric TRPM6/TRPM7 channels may play different roles in different tissues under various physiological conditions and pathological scenarios. As we still lack and accurate mapping of the distribution of TRPM6, TRPM7 and heterotetrameric TRPM6/TRPM7 channels in the various tissues, however, the relative role of these channels remains largely undefined.
Recently, some progress has been registered in understanding how TRPM7 is regulated. At the channel level, TRPM7 inward current is markedly enhanced by protons, which compete with Ca2+ and Mg2+ for binding sites, most likely at the level of the channel pore. As a result, the blockade of divalent cations on inward monovalent currents is released [48, 49]. High concentrations of extracellular protons, in fact, decrease the apparent affinity of TRPM7 for Ca2+ or Mg2+ even under conditions in which external Ca2+ or Mg2+ concentrations are increased. Hence, at physiological pH, Ca2+ or Mg2+ bind to TRPM7 and inhibit monovalent cation currents. At higher H+ concentrations, instead, the protons decrease the affinity of TRPM7 for Ca2+ and Mg2+, allowing monovalent cations to permeate the channel [57]. Another level of regulation is provided by PIP2. This regulation was initially reported by Clapham and collaborators [58], but was not subsequently confirmed by Fleig’s group, which reported a regulatory role by cAMP [59]. Recent report from Langeslag et al. [60] and from Mubagwa’s group [61, 62], however, have confirmed that the depletion in PIP2 level following PLC-activation counteracts TRPM7 activation. In particular, Mubagwa’s group has reported that inhibition of phospholipase C (PLC), or addition of exogenous PIP2 decreases TRPM7 run-down whereas the extracellular addition of phenylephrine, which activates phospholipase C, accelerates it [61]. Also, this group has observed that both ATP [62] and non-hydrolysable GTP analogs [61] modulate the channel activity, most likely by forming Mg*ATP [62] and by accelerating the channel run-down via PLC activation, respectively [61]. The regulatory role of PIP2 is further emphasized by the experimental evidence that bradykinin or angiotensin-II, which activates PLC coupled receptors via Gq signaling [60,63] in a manner similar to phenylephrine [61] can modulate TRPM7 activity via PIP2 metabolism. Activation of TRPM7, however, only occurs in the presence of physiological [Mg2+]i, within the cell. Reducing this concentration below its physiological level by EDTA-AM it results in PLC-mediated inactivation of TRPM7 activity, most likely via PIP2 depletion [60]. All together these results suggest a PLC/PIP2-centered TRPM7 regulation whereby PLC-activation accelerates TRPM7 ‘rundown’ via PIP2 depletion. Alternatively, PIP2 depletion would play a feed-back regulatory role on the channel activation by PLC [60].
That phosphatidyl-inositol metabolites play a significant role in modulating TRPM7 activity is further supported by the recent evidence provided by the Scharenberg’s group that a functional TRPM7 is required for a sustained phosphoinositide-3-kinase (PI3K)-mediated signaling in lymphocytes [64]. Irrespective of the presence of physiological [Mg2+]o, TRPM7-deficient cells rapidly down-regulate their rate of growth as a result of signaling deactivation downstream PI3-Kinase [64]. The cells, however, can be rescued by supplementing the culture medium with Mg2+ [64]. This group has further expanded their investigation investigating the role of TRPM7 in regulating the transition of lymphocytes from quiescent to proliferative metabolic state [65]. The results these authors reported indicate that modulation of TRPM7 channel function in B-lymphocytes promotes the transition from quiescent to proliferative state. In contrast, TRPM7 deficient cells exit cell cycle and enter quiescence [65]. This transition is regulated by p27, which becomes up-regulated in TRPM7-deficient cells undergoing quiescence [65]. Because TRPM7 is widely expressed in the immune system but also outside it, the results of this group suggest that TRPM7 may play an essential role in regulating rapid cell proliferation and possibly malignancy development.
As indicated previously, TRPM7 was originally known based on the alpha-kinase activity present at its C-terminus [45]. At variance of other better known kinases, which phosphorylate residues in a beta-sheet, this kinase domain phosphorylates serine and threonine residues within an alpha-helix [66]. Initially, this kinase domain was considered to be essential to modulate TRPM7 activity and gating [58]. This initial claim, however, was not supported by subsequent studies, which indicated that TRPM7 channels lacking the kinase domain could still be activated by internal Mg2+ depletion [67]. Despite our progress in understanding the regulation of TRPM7 by cations and phosphatidyl-inositol metabolites, and the role of the TRPM7 kinase domain in regulating the channel activity, little has been uncovered about the molecular mechanisms that activate the channel in toto within the cell, and the alpha-kinase domain in particular. Data by Clark et al. [68] suggests that kinase autophosphorylation plays a significant role in target recognition by this domain. Phosphomapping by mass-spectrometry has confirmed the occurrence of massive autophosphorylation of TRPM7 kinase domain, which – in turn – increases the rate of substrate phosphorylation. Phosphomapping indicates the 37 autophosphorylation sites out of 46 total take place in a region rich in serine and threonine residues located immediately upstream the kinase catalytic domain [70]. Deletion of this region does not affect the intrinsic catalytic activity of the kinase but prevents substrate phosphorylation, confirming the essential role of this region in substrate recognition [70]. Although this Ser/Thr region is poorly conserved in the TRPM6 in terms of amino acid sequence, the kinase domain of this channel appears to require a similar massive autophosphorylation of its Ser/Thr residues for proper recognition and efficient phosphorylation of its substrate [70].
The identification of TRPM7 targets is still incomplete. Presently, only annexin I [71], myosin IIA heavy chain [70,72], and calpain [73] have been clearly identified as substrates phosphorylated by TRPM7 kinase domain. Thus, it would appear that at the cellular level TRPM7 plays a double role by regulating Mg2+ homeostasis as well as cell adhesion, contractility or inflammation, based upon the cell type. In the particular case of smooth muscle cells, this double role of TRPM7 is clearly supported by recent reports from Touyz and collaborators [74]. Results form this group, in fact, indicate that aortic segments from mice exhibiting low intracellular Mg2+ levels present increased medial cross-section and TRPM7 expression, and decreased annexin-I expression. Because annexin-I has an important antiinflammatory role within cells [75], these results suggest that TRPM7 and cellular Mg2+ are essential to regulate vascular structure, integrity, and inflammation. This is further corroborated by the observation that a decrease in TRPM7 expression and activity results in a decrease in cellular Mg2+ content, which – in turn - promote the differentiation of endothelial cells from a quiescent to a calcification-prone phenotype [76].
One consequence of generating TRPM7 lacking the kinase domain is clearly the inability to properly phosphorylate and consequently activate downstream cellular components. Two recent publications by the groups of Ryazanov and Fleig [69,77] support this notion. Ryazanov’s group generated TRPM7–deficient mice carrying the deletion of the kinase domain [69]. Homozygous TRPM7ΔKinase mice presented early embryonic lethality. The heterozygous mice were viable but presented signs of hypomagnesaemia due to a defect in intestinal Mg2+ absorption. Cells derived from these heterozygous mice presented reduced TRPM7 currents with an increased sensitivity to inhibition by Mg2+. Embryonic stem cells lacking TRPM7 kinase domain showed an arrest in proliferation that could be rescues by Mg2+ supplementation, validating results reported by Scharenberg’s group [65]. The relevance of the kinase domain in mediating several of TRPM7 intracellular signals and functions is further confirmed by a recent publication by Perraud et al. [77] reporting a role of TRPM7 in regulating the rate of protein synthesis based upon Mg2+ availability. These authors observed that the phosphorylation of Thr56 residue of eEF2, which is inhibitory on this protein activity, is increased under hypomagnesaemic conditions and that the regulation by Mg2+ requires TRPM7 with an active and viable kinase. The regulation of eEF2 by TRPM7 is indirect, occurring through eEF2 cognate kinase (eEF2-k), which becomes phosphorylated by TRPM7 kinase on Ser77 [77].
As our understanding of TRPM7 expression and regulation has advanced, evidence has mounted about a significant role of this channel in modulating specific functions within different cell types.
For examples, in neurons TRPM7 regulates neuronal function and survival under hypoxia or ischemia-reperfusion conditions. Because it can transport either Ca2+ or Mg2+, TRPM7 exhibits an ambivalent role based upon the permeating cation. Following activation by reactive oxygen/nitrogen species and prolonged oxygen and glucose deprivation, TRPM7 favors Ca2+ fluxes that result in a toxic event for neurons [78]. In contrast, Mg2+ permeation enhances anti-apoptotic and cell-survival mechanisms, preventing anoxic death of neurons [68]. The key role of TRPM7 in detecting extracellular divalent cations is supported by a recent report by Wei et al. [79] indicating that TRPM7 activation by low extracellular divalent cations concentrations is lethal to the cell. At the same time, Jiang et al. [80] have reported that 1 hour occlusion of middle cerebral artery enhances TRPM7 expression in ipsilateral hippocampus, with deleterious consequences for the neurons. Pre-treatment of neurons with nerve growth factor counteracted both the increase in TRPM7 expression and its harmful consequences by activating TrkA pathway [80]. More recently, Runnels and collaborators reported that 5-lipoxygenase inhibitors can prevent cell death by blocking TRPM7 current without affecting protein expression and cell membrane concentration [81]. In addition, TRPM7 facilitates the fusion of cholinergic vesicles with the pre-synaptic membrane of parasympathetic fibers without affecting large dense core vesicle secretion, thus promoting the specific secretion of acetylcholine at the synaptic level [82].
The effect of TRPM7 on proliferation and differentiation is not restricted to endothelial cells [76] but it extends to osteoblasts as well [83,84]. Expression of TRPM7, in fact, increased during osteoblast differentiation, suggesting an essential role of cellular Mg2+ homeostasis, perhaps in conjunction with Ca2+ homeostasis on cell differentiation. Culturing osteoblasts in low extracellular Mg2+ or Ca2+ significantly reduced their differentiation as based upon alkaline phosphatase activity and osteocalcin expression [83]. Matrix mineralization was also reduced under these conditions whereas expression of collagen type I, which predominates in the extracellular matrix, was increased, indicating that dysfunction in matrix protein production cannot explain the reduced mineralization observed [83,84]. Osteblastic differentiation and extracellular matrix mineralization were affected to a comparable extent by TRPM7 silencing during the differentiation stage, further connecting cellular Mg2+ homeostasis with TRPM7 expression and activity. Expression of the osteoblastic transcription factor Runx2 was also reduced by culturing the cells in the presence of low extracellular Mg2+ level, or by TRPM7 silencing [83]. Hence, these results indicate that cellular Mg2+ and Ca2+ homeostasis via TRPM7 are important for osteoblastic differentiation. It still remains to be properly determined to which extent Mg deficiency, which is relative common in the population, is associated with altered osteoblastic differentiation and ultimately with inadequate bone formation and osteoporosis development.
Despite the mounting evidence for a preferential role of TRPM7 as a Mg2+ entry mechanism, data by the Clapham’s group have casted some doubts about the effective role of this channel in regulating Mg2+ homeostasis [85]. Having developed a TRPM7 null mouse, this group observed altered embryonic development and tissue specific deletion of the channel in T cell lineage. The latter condition resulted in disrupted thymopoiesis and progressive depletion of thymic medullary cells. Deleting TRPM7, however, did not affect acute Mg2+ accumulation or total Mg2+ content in T cells but significantly dysregulated the synthesis of several growth factors thus altering thymic epithelial cells differentiation [85]. Based upon these results, the group is proposing that TRPM7 is the first TRP channel with a non-redundant but actually essential role in embryogenesis and thymopoiesis. Whether the observed defects are the result on an altered Ca2+ rather than Mg2+ homeostasis it is presently undefined. It is also unclear how removal of this protein alters T cells differentiation.
3.1.1.2 TRPM6
At variance of TRPM7 isoform, TRPM6 channel is uniquely localized in the colon and the renal distal convolute tubule, two epithelia highly impermeable to salt re-absorption. This specific localization supports the specific role of this channel in controlling whole-body Mg2+ homeostasis by regulating intestinal Mg2+ absorption and renal Mg2+ reabsorption.
The TRPM6 gene was originally identified by genetic analysis as the site of various mutations responsible for Hypomagnesaemia with Secondary Hypocalcaemia (HSH, OMIM 602014). A rare autosomal recessive disease, HSH is characterized by Mg2+ and Ca2+ wasting and by symptoms that cannot be ameliorated by massive intravenous Mg2+ administration and oral Mg2+ supplementation [43]. More specifically, while hypocalcaemia is completely alleviated by this treatment, serum Mg2+ level in these patients remains around 0.5–0.6 mmol/L, or half the physiological level [43]. Because the primary defect is at the level of the TRPM6 channels expressed in the intestine [43], the excess Mg2+ supplementation is rapidly filtered at the glomerular level and increases passive renal absorption via paracellin-1 (see Section 3.1.2). Trans-cellular absorption via apical TRPM6 channels in the renal epithelium, however, remains depressed and unable to restore physiological serum Mg2+ level [43].
As indicated for TRPM7, experimental evidence suggests that the channel forms a functional tetramer at the plasma membrane level. As reported earlier, it is unclear as to whether the channel forms a homotetramer, or a heterotetramer with TRPM7 with a varying stoichiometry. Irrespective of the possibilities, several point mutations within the TRPM6 amino acid sequence have been identified [86], which result in the expression of a truncated and non-functional channel [86]. The missense mutation S141L, for example, occurs at the N-terminus of the channel and prevents its proper assembly as a homotetramer, or a heterotetramer with TRPM7 [86]. Another missense identified in humans is the P1017R mutation [86], which occurs in a region putatively identified as the pore region of the channel. Yet, this mutation affects negatively and more significantly TRPM7 function when this protein is co-expressed with TRPM6 [86]. More recently, TRPM6 null mice have been developed by Sheffield and his collaborators [87]. Aside for a modest low plasma Mg2+ level (~0.67 vs. 0.75) the heterozygous Trpm6+/− mice present normal electrolyte levels [87]. The majority of the homozygous Trpm6−/− mice, instead, die by embryonic day 12.5. Of the few animals that survive to term, the majority presents significant neural tube defects such as exencephaly and spina bifida occulta. Offspring survival to weaning can be obtained by administering high Mg diet to dams [87]. Recent data by Woudenberg-Vrenken and colleagues [88] confirmed that homozygous TRPM6 deletion is embryonic lethal whereas heterozygous TRPM6 deletion is associated with a mild hypomagnesaemia. This group, however, reported that Mg2+ -enriched diet could not compensate for the embryonic lethality and hypomagnesaemia caused by TRPM6 deficiency [88]. In fact, 30% of the offsprings on either regular (0.19% wt/wt) or high Mg2+ diet (0.48% wt/wt) were TRPM6 wild type (+/+), 70% were TRPM6 heterozygous (+/−), and none were TRPM6 (−/−). Furthermore, TRPM6 heterozygous (+/−) presented low serum Mg2+ levels and reduced renal and colon TRPM6 mRNA expression irrespective of the diet administered.
Like its homologous TRPM7, TRPM6 also presents an alpha-kinase domain at its C-terminus with functional homology to eEF2-kinase, and consequently was originally termed CHAK2 (channel kinase 2) [46]. As mentioned for the TRPM7, this kinase domain phosphorylates serine and threonine residues located within an alpha-helix instead of a beta-sheet [45,46,66]. Currently, owing to their dual function as a channel and a kinase, TRPM6 and TRPM7 are referred to as chan-zymes. Also in the case of TRPM6, removal of the kinase domain does not abolish the channel activity but modulates the extent to which the channel is regulated by intracellular free Mg2+ or Mg*ATP complex [51–54,89], and affect the ability of the chan-zyme to phosphorylate its downstream targets. At variance of what reported for the TRPM7 (see previous Section), no substrate phosphorylated by TRPM6 kinase has been properly identified, with the exception of TRPM7 itself. Ryazanov and his group [52] have provided evidence that TRPM6 kinase domain can phosphorylate residues on TRPM7 channel within a heterotetramer structure whereas TRPM7 kinase does not appears to phosphorylate residues on TRPM6 [52]. Hence, it is presently undefined as to whether the kinase domains associated with TRPM7 and TRMP6 phosphorylate similar or different substrates within the tissues in which the chanzymes are expressed.
How expression and activity of TRPM6 channel are modulated in vivo is slowly being elucidated. TRPM6 but not TRPM7 appears to be extremely sensitive to changes in estrogen level and dietary Mg2+ intake. Estrogens (17β-estradiol) selectively up-regulate TRPM6 mRNA in both colon and kidney while having no effect on TRPM7 mRNA in other tissues [90,91]. In the absence of estrogen, the repressor of estrogen receptor activity (REA) binds to the 6th, 7th and 8th beta-sheets of TRPM6 kinase domain in a phosphorylation-dependent manner and inhibits its activity [90]. REA binding is rapidly dissociated by estrogen administration, resulting in increased TRPM6 activity [90]. Dietary Mg2+ restriction also up-regulates TRPM6 mRNA in both colon and kidney but has not effect on TRPM7 mRNA [91,92]. In contrast, exposure to Mg2+ enriched diet up-regulates TRPM6 mRNA only in the colon, in keeping with an increased intestinal absorption [91]. Mice selected for the their low erythrocyte and plasma Mg2+ status exhibit hypomagnesaemia and hypomagnesuria, and increased TRPM6 expression in kidney and intestine when fed a severely Mg2+-deficient diet [92]. In contrast, feeding these mice an Mg2+ adequate diet results in hypomagnesaemia and hypermagnesuria, and reduced intestinal and renal TRPM6 expression [92]. These changes in TRPM6 expression and Mg2+ level in blood and urine do not occur in mice exhibiting normal or high erythrocyte and plasma Mg2+ levels [92]. Therefore, it is becoming progressively apparent that genetic factors control TRPM6 expression and activity, and that dietary Mg2+ restriction promotes a compensatory increase in Mg2+ absorption and reabsorption by enhancing TRPM6 expression in intestine and kidney, respectively [91,92].
As observed for TRPM7, cellular ATP decreased TRPM6 current [51–54]. The site of inhibition resides in the conserved ATP-binding motif GxG(A)xxG within the alpha-kinase domain [89]. Full deletion of the kinase domain and point mutations within the ATP-binding motif (G1955D) completely abolish the inhibitory effect of ATP. The effect of ATP, however, does not depend on alpha-kinase autophosphorylation activity [89].
The activity of TRPM6 channels is also modulated by cellular signaling molecules. Bindels and collaborators [93] have reported that over-expression of RACK1 (receptor for activated protein kinase C) results in the direct binding of this protein to the alpha-kinase domain of TRPM6, and possibly TRPM7 due to the high homology (>84%) between the two kinase domains. The RACK1 binding site on TRPM6 is located to the region between 1857 to 1885 amino acid residues, which corresponds to the 6th, 7th and 8th β sheets, the same sheets involved in REA regulation [90] Whether any interplay occurs between REA and RACK1 in modulating the channel activity is still unrevealed. Accessibility analysis of the RACK1 binding site on TRPM6 suggests that 18 of the indicated 28 amino acid residues in this site are localized at the surface of the alpha-kinase domain [93]. Following the interaction of RACK1 with the kinase domain of TRPM6, and TRPM7, the activities of the channels are inhibited. As expected, co-expression of RACK1with alpha-kinase deleted TRPM6 mutant fails to suppress channel activity. The inhibitory effect of RACK1 depends on threonine 1851 (T1851) autophosphorylation within the kinase domain. This residue is localized at the end of the 4th alpha-helix adjacent to the RACK1 binding site. Mutating T1851 to alanine (T1851A) or to aspartate (T1851D) it decreases TRPM6 autophosphorylation but does not affect RACK1 binding. The inhibitory effect of RACK1 on channel activity, however, is only abolished following T1851A mutation, persisting virtually unaltered in the case of T1851D mutation [93]. It has to be noted that following T1851D mutation, autophosphorylation of the kinase is directly proportional to the Mg2+ concentration, steadily increasing in the 0.1 to 1mM range. The T1851A mutant, instead, is less sensitive to intracellular Mg2+ concentrations as compared to the wild type (IC50 ~0.7 vs. 0.5mM, respectively). Pre-treatment of the cells with phorbol-myristate acetate (PMA), which activates protein kinase C (PKC), completely prevents the inhibitory effect of RACK1 on TRPM6 channel activity [93]. The inhibition, however, can be restored by pre-treating the cells with the PKC inhibitor chelerythrine [93], suggesting a competing effect of PKC for RACK1.
Clinical and experimental evidence gathered in recent time is supporting the notion that EGF acts as an autocrine/paracrine magnesiotropic hormone. A report by Groenestege et al. [94] indicates that by engaging its receptor in the basolateral domain of the distal convolute tubule, EGF activates TRPM6 at the apical domain of the cell and induces cellular Mg2+ accumulation. Point mutation in the pro-EGF sequence (P1070L) disrupts this cascade of events by retaining EGF to the apical membrane of the cell, ultimately resulting in the Mg2+ wasting typical of the syndrome termed isolated recessive renal hypomagnesaemia (IRH, OMIM 611718). The axis EGF/TRPM6/Mg2+ reabsorption is also altered in cancer patients undergoing treatment with antibodies anti-EGFR [95,96]. By blocking the EGF receptor, these antibodies antagonize the stimulation of TRPM6 activity and elicit renal Mg2+ wasting [95,96]. The mechanism by which EGF promotes TRPM6 activity and/or expression appears to involve ERK1/2 signaling. Ikari and collaborators have reported that addition of EGF to NRK-52E cells increases ERK1/2 phosphorylation and TRPM6 expression in a time-dependent manner [97] by modulating adaptin protein-1 (AP-1) [98]. Antagonists for integrin αvβ3, for MEK1/MEK2 activity, or the use of siRNA for TRPM6 all prevent the process [97]. Further studies are needed to elucidate how EGF, integrin, and ERK1/2 interact with each other to ultimately enhance TRPM6 expression. In this context, it is still unexplored whether the activation of this signaling axis is connected to the release of RACK1-mediated inhibition of TRPM6 activity by PKC as mentioned previously [93].
The physiological location of TRPM6 on the apical side of the intestinal and renal epithelium raises the question how Mg2+ is transported across the cytoplasm after entering the cell to be delivered to basolateral domain and be extruded into the blood stream. The general consensus is that Mg2+ enter the blood-stream via the operation of a yet-to be-cloned Na+/Mg2+ exchanger (see Section 3.2.1). More uncertainty exists as to whether Mg2+ moves freely through the cytoplasm or is instead transported bound to proteins. One hypothesis is that parvalbumin and calbindin-D28k, two proteins abundantly present within the cells of the distal convolute tubule of the nephron, mediate the trans-cellular transport of Mg2+ accumulated at the apical domain, or at least accelerate the delivery rate of the cation to the basolateral domain. However, as no detectable defects in Mg2+ excretion or homeostasis are observed in parvalbumin null mice [17], it remains questionable whether parvalbumin does play a role in transporting Mg2+ under physiological conditions, or other proteins can compensate for its absence in the null model.
3.1.2 Claudins
Paracellin-1 was the first Mg2+ transporting protein to be identified in mammals [99]. Its identification was based upon the genetic analysis of patients affected by Familial Hypomagnesaemia with Hypercalciuria and Nephrocalcinosis (FHHNC, OMIM 248250). This disease is characterized by massive renal Mg2+ and Ca2+ wasting that leads rapidly and irreversibly to renal failure [99]. At variance of what described for patients with TRPM6 mutations (Section 3.1.3.2), the symptoms and the progressive renal deterioration in FHHNC patients are not ameliorated by Mg2+ supplementation [99]. Lifton and collaborators identified the gene responsible for this disease and named it Paracellin-1 (PCLN-1) [99]. PCLN-1 encodes for paracellin-1 (PCLN-1), now renamed claudin-16. This protein is a member of the claudin family [100], which comprehends a group of tight junction proteins with 4 trans-membrane spans coordinated by 2 extracellular loops, and with both C- and N-termini on the cytoplasm side. More than 20 mutations affecting trafficking or permeability of claudin-16 have been currently identified [101].
Claudin-16 mediates paracellular Ca2+ and Mg2+ fluxes throughout the nephron. Yet, the modality by which these fluxes are generated is still controversial. Data obtained in LLC-PK1 (a renal cell line of porcine origin) indicate that claudin-16 mediates paracellular Na+-permeation which, in turn, generates a positive potential within the lumen of the nephron that acts as driving force for Mg2+ and Ca2+ reabsorption [102]. Data in MDCK cells, instead, point to an increase in Mg2+- and a decrease in Na+- permeability [103]. It is unclear whether these discrepancies reflect a different modus operandi in cell lines of differing origin, or depend on the experimental conditions utilized in the two studies. Either study, however, support the evidence that the expression of PCLN-1 is modulated by the concentration of magnesium present in the extracellular medium [104].
To properly function, claudin-16 has to be delivered to the tight junction where it interacts with the scaffolding protein ZO-1 [105]. The association and dissociation of claudin-16 and ZO-1 is regulated via PKA-mediated phosphorylation of Ser217 within claudin-16 sequence [105]. Dephosphorylation of this residue, which occurs upon activation of the Calcium Sensing Receptor (CaSR) [106] results in the dissociation of claudin-16 from ZO-1 and its accumulation within the lysosomal compartment [103]. The importance of Ser217 is highlighted by the observation that mutations of this residue accelerate claudin-16 turn-over and modulate its function. Mutation of Threo233 (T233R) also impairs the interaction between claudin-16 and ZO-1, and favors the accumulation of claudin-16 into lysosomes [103,105].
Recent evidence indicates the involvement of claudin-19, another claudin isoform, in mediating Mg2+ and Ca2+ reabsorption [107]. Claudin-19 forms a head-to-head complex with claudin-16 at the level of the tight junction, with specific cation-selectivity. While the functioning of claudin-16 as a channel does not appear to depend on its association with claudin-19, claudin-19 is indispensable to recruit claudin-16 to form a co-polymer at the level of the tight junction and to switch the channel selectivity from anion to cation [107]. The heteromeric association between claudin-16 and claudin-19 is dramatically affected by point mutations in claudin-16 (L145P, L151F, G191R, A209T, and F232C) and claudin-19 (L90P and G123R). Any of these mutations abolishes the physiological synergism between the two proteins and results in the development of FHHNC.
3.1.3 MagT1
Goytain and Quamme identified this protein in human epithelial cells, which up-regulate the protein encoding gene following exposure to low-Mg2+ concentrations in the culture medium [108]. This protein has an estimated molecular weight of 38 KDa and 5 trans-membrane domains in its immature form. Following the cleavage of the first trans-membrane segment located near the C-terminus, the mature protein contains only 4 trans-membrane spans. At variance of SLC41 (Section 3.3.1) and Mrs2 (discussed in the next Section), MagT1 does not present any significant degree of homology to prokaryotic Mg2+ transporters, but it exhibits some similarities with the oligosaccharide transferase complex OST3/OST6 that regulates protein glycosylation in the endoplasmic reticulum of yeast [109]. The murine orthologue of MagT1 is highly expressed in liver, heart, kidney and colon, with detectable levels in lung, brain and spleen [108]. For the most part, MagT1 levels in these tissues are consistent with the mRNA levels, the only exception being the liver in which a low protein level is detected [108]. At variance of the other transporters described in this section, MagT1 appears to be highly specificity for Mg2+ (Km = 0.23mM). The Mg2+-elicited currents are inhibited by Ni2+, Zn2+ and Mn2+ but not Ca2+. However, the inhibiting concentrations of any of the blocking cations are in excess of 0.2mM, far exceeding their physiological concentrations present in extracellular fluids. Nitrendipine at a concentration of ~10μM can inhibit MagT1-mediated Mg2+ current whereas the more common nifedipine does not, even at much higher concentrations [108]. Limited information is available about N33, a second member of the MagT family. Although able to transport Mg2+, this protein exhibits a much lower specificity for Mg2+ than MagT1. In addition, N33 can also mediate the transport of Fe2+, Mn2+ and Cu2+ [108].
The available information about MagT1 suggests that the protein possesses channel-like characteristics and high selectivity for Mg2+. Based upon the latter evidence, it would appear that this transporter is essential to regulate Mg2+ homeostasis in mammalian cells. This hypothesis is supported by the observation that knocking out of MagT1 and its human homolog TUSC3 in HEK-293 cells markedly reduces cellular Mg2+ content [110]. Either MagT1 or TUSC3 can complement the yeast Mg2+ transporter ALR1 [110]. Exposure of HEK-293 cells to low extracellular Mg2+ concentrations for 1–2 days increases the mRNA level of MagT1 but not that of TUSC3. In contrast, expression of either protein is not affected by incubating the cells in the presence of high extracellular Mg2+ concentration [110].
3.1.4 Mrs2
This protein was identified during a screening aimed at isolating nuclear genes suppressing RNA splicing defects in yeast mitochondrial introns [111]. The three main characteristics observed in Mrs2 deficient yeasts are: 1) a splicing phenotype, 2) a significant reduction in cytochromes content, and 3) a deficit in mitochondria respiration to the point that the yeasts become unable to grow on non-fermentable substrates.
Structurally, Mrs2 shows short regions of homology to the bacterial transporter CorA [112], and a similar membrane topology with 2 trans-membrane domains of this transporter. Mutant yeasts lacking Mrs2 present a decrease in total mitochondrial Mg2+ content and in matrix free Mg2+ level [113], and can be rescued by CorA fused to the mitochondrial N-terminus leader sequence of Mrs2, which guarantees proper insertion in the mitochondrial membrane. In contrast, over-expression of Mrs2 results in a rapid and marked increase in matrix free Mg2+ [113]. All together, this set of observation strongly suggests an essential role of Mrs2 in regulating mitochondrial Mg2+ homeostasis. At the functional level, Mrs2 operates as a channel, and it is modulated by mitochondrial Δψ and by inhibitors of F0-F1-ATPase or ANT, which substantially decrease the amplitude of Mg2+ influx. Highly conserved motifs in the middle region of the protein, corresponding to the coiled-coil portion of the channel, appear to be essential to form functional channels, or to gate the channel. One of the last publications by Schweyen’s group has confirmed in HEK293 cells some of the mitochondrial modifications previously observed in yeasts. HEK 293 cells deprived of Mrs2 lack expression of mitochondrial complex I and present reduced level of mitochondrial Mg2+ [114]. Furthermore, the cells show changes in morphology and an increased propensity to apoptosis, which completely abolishes cell viability within 2 weeks [114]. It still remains to be elucidated whether the decrease in mitochondrial Mg2+ depends on the absence of Mrs2, or is related to some extent to the absence of complex I, which affects mitochondrial Δψ and consequently Mg2+ retention within the organelle [115].
Mammalian cells express a single Mrs2 orthologue, which can be used to rescue Mg2+ deficient yeast strain [116]. Hence, it appears that the mammalian/human Mrs2 homologue mediates Mg2+ entry in mitochondria in a manner similar to the yeast homologue. Under conditions in which Mrs2p is absent or not functional, the operation of an alternative but much slower mitochondrial Mg2+ entry mechanism has been observed. Although this mechanism restores Mg2+ homeostasis only in part it rescues the phenotype of Mrs2 deficient yeast, ensuring their survival. No information is presently available about the identity, abundance, and regulation of this alternative transporter in mitochondria. Taken together, the data on Mrs2 suggest that this protein is essential but not indispensable to dynamically regulate Mg2+ level within the mitochondrion. In turn, the presence of a physiological level of Mg2+ within the mitochondrial matrix is essential to modulate mitochondrial dehydrogenases and oxygen consumption [117,118].
3.1.5 MMgTs
This gene family comprehends two proteins termed MMgT1 and MMgT2 (for membrane Mg2+ transporter 1 and 2). Goytain and Quamme identified these proteins by microarray analysis screening [119]. In the mouse, the chromosomal locations of these proteins are XA5 for MMgT1 and 11B2 for MMgT2. In the rat, the respective locations are Xq36 for MMgT1 and 10q23 for MMgT2. Human MMgT1 is instead located on Xq26.3 [120]. Immunohistochemistry evaluation indicates that MMgT1 and MMgT2 are located in the Golgi complex and post-Golgi vesicles, where they may contribute to the regulation of Mg2+ dependent enzymes involved in protein assembly and glycosylation [120]. This localization, however, does not exclude that these proteins may be transported via Golgi vesicles to the cell membrane or to other destinations downstream the Golgi network where they can play a role in modulating Mg2+ homeostasis. Widely distributed within tissues, these proteins are formed by 131 (MMgT1) and 123 (MMgT2) amino acids predicted to assemble into two trans-membrane domains. The relative small size of these proteins suggests that they can form homo-oligomeric and possibly heterooligomeric channels to favor Mg2+ permeation. MMgT-mediated Mg2+ uptake is a saturable process with a Km ≃1.5mM for MMgT1 and ≃0.6mM for MMgT2, values that do not vary significantly with voltage. Both MMgT1 and MMgT2 lack specificity for Mg2+ as they can transport other cations as well with some slight differences in cation permeation between the two isoforms. MMgT1 transports Sr2+, Fe2+, Co2+ and Cu2+ in addition to Mg2+ while MMgT2 favors Sr2+, Co2+, Cu2+, Ba2+ and Mn2+ transport [119]. So far, limited information is available about the specifics of MMgT1 expression and operation. We know that: 1) Mg2+-generated currents in MMgT1 are inhibited by Mn2+ (~0.2mM) but not by Gd3+ or Ni2+ [119], and 2) MMgT1 mRNA increases ~2.5 fold in the kidney cortex of mice on low-Mg2+ diet and ~3.5 fold in MDCT epithelial cells culture in low Mg2+ medium whereas MMgT2 mRNA increases ~1.5 fold in kidney cortex and ~3 fold in MDCT cells under similar experimental conditions [119]. These increases in expression are not specific for these transport mechanism as similar increase in expression have been reported for other Mg2+ entry mechanisms discussed previously.
The interested audience is referred to reviews by Touyz [63], Schmidt et al [120], Bindels [121], and Quamme [122] for a more detailed description of the intrinsic characteristics of TRPM7, TRPM6 and the other Mg2+ entry mechanisms summarily described in this Section.
3.2 Exchangers
While channels or channels-like mechanisms mediate Mg2+ entry, two exchange mechanisms mediate Mg2+ extrusion. Based upon the specific electrochemical requirements to favor Mg2+ extrusion, these mechanisms are referred to as Na+-dependent and Na+-independent Mg2+ exchanger, respectively (Table 1 and Figure 1). Because neither of these two mechanisms has been cloned, information about their operation, abundance and tissue specificity remains largely circumstantial or indirect based upon experimental conditions or pharmacological inhibition.
3.2.1 Na-dependent Exchanger (Na+/Mg2+ Exchanger)
Gunther and Vormann in 1984 provided the first evidence for the presence and operation of a Mg2+ transport mechanism in mammalian cells [123]. These authors were also the first to indicate that this transport mechanism elicits Mg2+ extrusion in a Na+-dependent, amiloride-inhibited manner [124]. Observed originally in chicken red blood cells, the operation of such a mechanism has been subsequently confirmed by other groups in mammalian red blood cells [125–127] including human erythrocytes [12, 128,129], and in various other mammalian cell types [see ref. 29 for a list]. Reports from Vormann and Gunther [33,130], Wolf and collaborators [25,131], and our laboratory [32,34,132–137] have provided compelling evidence that this Na+-dependent, amiloride-inhibited Mg2+ extrusion mechanisms is specifically activated by cAMP. Experimentally, it is irrelevant whether cellular cAMP increases via stimulation of β-adrenergic, glucagon, or PGE2 receptors, or via administration of forskolin or cellpermeant cyclic-AMP analogs [130–137]. In all these conditions, in fact, the Na+-dependent Mg2+ extrusion mechanism is activated via cAMP-mediated phosphorylation. Conversely, pre-treatment of the cells with inhibitors of adenylyl cyclase (e.g. Rp-cAMP) or PKA (e.g. PKI) completely blocks Mg2+ mobilization irrespective of the receptor or modality utilized to enhance cellular cAMP level [25].
The Mg2+ extrusion mediated by this exchanger requires the presence of a physiological concentration of Na+ in the extracellular milieu [132,134]. Thus, it is widely accepted that this Na+-dependent Mg2+ extrusion mechanisms is a Na+/Mg2+ exchanger. Because this Mg2+ extrusion mechanism has not been functionally cloned as yet, we lack detailed information about its membrane abundance, structure, proximity to other cellular transporters with whom it may possibly interact, and stoichiometry. Gunther and Vormann have suggested that the exchanger operates on electroneutral bases (2Na+ in:1 Mg2+ out) at least in chicken or turkey erythrocytes [123,124]. This hypothesis has been confuted by data obtained in mammalian erythrocytes including human red blood cells supporting an operation on electrogenic bases (1Na+ in:1 Mg2+ out) [126–128]. The reason for this discrepancy is not apparent, and it may depend on the experimental model (i.e. cell isolation vs. cultured cells), composition of the incubation medium, or modality of cell Mg2+ loading. In keeping with these possibilities, a recent report of ours suggests that the extrusion of Mg2+ via the Na+/Mg2+ exchanger is coupled to the outward movement of Cl− ions [138]. Under conditions in which Cl− is not present, the electrogenic exchange 1Na+ in:1Mg2+ out becomes electroneutral (2Na+ in:1Mg2+ out) [138]. Interestingly, only inhibitors of the Na+/Mg2+ exchanger block Cl− extrusion in addition to inhibiting Mg2+ mobilization whereas more or less specific inhibitors of Cl− transporters (e.g. flufenamic acid, DNDS, or DIDS) are ineffective at blocking Cl− movement. A role of cellular Cl− in stimulating the Na+/Mg2+ exchanger in erythrocytes has been reported by Gunther and collaborators [139]. Moreover, Rasgado-Flores and collaborators have reported Cl− transport following reverse activation of the Na+/Mg2+ exchanger in dialyzed squid axon [140]. Hence, the extrusion of Cl− in our model and in the squid axon can be interpreted as an attempt to equilibrate charge movement across the hepatocyte or the axon membrane. However, it remains unclear whether Cl− extrusion occurs through the Na+/Mg2+ exchanger directly or through Cl− channels, which are present in the hepatocyte membrane [141], activated by the exchanger.
Irrespective of the stoichiometry of exchange and the experimental model utilized the obtained results consistently indicate a Km for Na+ between 15 to 20 mM [142–144]. Pharmacological inhibition has done little to enhance our understanding of the modality of operation of the putative Na+/Mg2+ exchanger. Amiloride, imipramine and quinidine represent the three most commonly utilized inhibitors of the Na+-dependent Mg2+ extrusion [123,130,145]. However, their limited specificity does not clarify as to whether they inhibit the Na+/Mg2+ exchanger directly, or indirectly by operating on other transport mechanisms including Na+ and K+ channels which - in turn - alter the cell membrane potential and the driving force for Mg2+ transport across the plasma membrane.
Although the Na+/Mg2+ exchanger is not yet cloned, its presence and operation has been observed in various experimental models including the mammalian rumen. Also in the latter model, the kinetic parameters and characteristics of the exchanger are similar to those reported in other mammalian cells [146]. Using a hybridoma screening procedure, the group of Schweigel, Martens and colleagues has generated monoclonal antibodies against the Na+/Mg2+ exchanger present in porcine red blood cells. The addition of these antibodies to sheep rumen epithelial cells resulted in a marked inhibition of Mg2+ extrusion and the substantiation of a ~70 KDa mr protein band by Western blot analysis [146]. This is the first time that information about the molecular size of the elusive Na+/Mg2+ exchanger is obtained, and the utilization of these antibodies could represent an ideal tool to identify and recognize this transporter in mammalian tissue.
3.2.2 Na+-independent
In the absence of extracellular Na+ to support the operation of the Na+/Mg2+ exchanger, or in the presence of amiloride, imipramine or quinidine, which all block the exchanger, Mg2+ extrusion occurs via an alternative Na+-independent mechanism. The specificity of this mechanism, however, is far from defined. Different cations including Ca2+ or Mn2+, or anions such as HCO3 −, Cl−, or choline, [147,148] have been reported to be utilized by this mechanism to extrude Mg2+. Hence, it remains unclear whether we are in the presence of distinct transport mechanisms, or in the presence of a transporter that can operate as an antiporter for cations or a sinporter for cations and anions based upon the experimental conditions. On the other hand, Ebel and collaborators [148] have suggested that Na+-independent Mg2+ extrusion in red blood cells and hepatocytes occurs via the choline transporter, which can be inhibited rather specifically by cinchona alkaloids [148]. A second point of uncertainty is whether the Na+- independent pathway is activated by hormonal stimulation. Stimulation of liver cells by epinephrine, a mix adrenergic agonist, elicits an extrusion of Mg2+ that is equivalent to the sum of the amounts of Mg2+ mobilized by the separate stimulation of α1- and β-adrenergic receptors [134,135,149]. Moreover, the selective stimulation of α1-adrenergic receptors by phenylephrine requires the presence of physiological concentrations of both Na+ and Ca2+ in the extracellular medium to elicit Mg2+ extrusion from liver cells [134,135]. Phenylephrine stimulation would activate Ca2+-CaM signaling and capacitative Ca2+ entry to induce Mg2+ extrusion [135]. It is still undefined whether the Ca2+-CaM signaling pathway represents an alternative modality of activation of the Na+/Mg2+ exchanger, or it activates instead a different Mg2+ extrusion mechanism identifiable with the Na+-independent mechanism.
Lastly, it remains controversial whether ATP is required for the operation of the Na+-independent and Na+-dependent mechanisms. Reports by Gunther and collaborators [150,151] indicate a dependence of the Na+/Mg2+ exchanger on the presence of a physiological concentration of cellular ATP to the point that cellular Mg2+ efflux is reduced under conditions that markedly decrease cellular ATP level [150,151]. In the case of red blood cells, Mg2+ homeostasis and transport is affected changes in both ATP and 2,3 bisphosphoglycerate levels [11]. No regulatory effect of ATP on Mg2+ extrusion was observed in purified liver plasma membrane vesicles [144]. In the latter model, however, no Mg2+ extrusion was observed in alkaline phosphatase-treated basolateral liver plasma membrane vesicles in the absence of ATP [137]. The most likely explanation for this observation, however, would be that ATP was required to phosphorylate and activate the Na+/Mg2+ exchanger in the presence of PKA [137]. As for the Na+-independent Mg2+ extrusion mechanism no clear ATP dependence has been reported. This notion appears to be supported by the observation that this transporter continued to operate in alkaline-phosphatase treated apical liver plasma membrane even in the absence of ATP [137], suggesting that phosphorylation is not required for its functioning.
3.2.3 Mg2+/H+
This exchange mechanism was originally identified in A. thaliana and termed AtMHX, but its presence appears to be ubiquitous in plants [152]. This transporter presents 11 putative trans-membrane domains, it is exclusively localized in the plant vacuolar membrane, and it electrogenically exchanges Mg2+ or Zn2+ for protons. Following the ectopic over-expression of the transporter, tobacco plants become sensitize to grow in the presence of elevated concentrations of Mg2+ (or Zn2+) [152]. Presently, no corresponding gene and encoded protein have been identified in mammalian cells, although some experimental evidence suggests a direct or indirect exchange of Mg2+ for H+ under certain conditions [153]. An enhanced extrusion of cellular Mg2+ has been reported to occur in cells incubated in the presence of an acidic extracellular environment, which imposes an inwardly oriented H+ gradient, as long as extracellular Na+ is present [153,154]. Amiloride derivates, which inhibit the Na+/H+ exchanger with high affinity, are ineffective at blocking Mg2+ extrusion under these experimental conditions [155], thus excluding the involvement of the Na+/H+ exchanger in mediating Mg2+ extrusion either directly or indirectly through a coupling of this exchanger with the Na+/Mg2+ antiporter.
3.3 Carriers
This section enlists several novel Mg2+ transport mechanisms of murine or human origin identified as a result of low or deficient Mg2+ in the diet (diet restriction) or in the culture medium (medium restriction). The modus operandi of these transport mechanisms is plagued by limited information and even controversies. For practical reasons, these transport mechanisms are nondescriptively classified here as carriers.
3.3.1 SLC41
This family of Mg2+ transport mechanisms includes three members (A1, A2, and A3), all distantly related to prokaryotic MgtE channel [156]. Because no study has addressed function and structure of SLC41A3 isoform, all the available information provided here refers to SLC41A1 and A2 isoforms
SLC41A1 was the first member of this family to be identified [157]. The hydrophobic profile of this protein (~56 kDa Mr) predicts to presence of 10 trans-membrane domains, two of which presenting a discrete level of homology with MgtE [157]. Northern blot analysis indicates a broad distribution of the SLC41A1 gene, but its abundance varies markedly among tissues, the highest expression being in heart and testis and the lowest being in hematopoietic tissues and cells [157]. The expression of this gene is modest under basal conditions, but becomes markedly up-regulated in the renal cortex of mice fed low Mg2+ diet for several days [158]. Functional expression of mouse SLC41A1 in X. Laevis oocyte indicates that this protein can transport Mg2+ but also Fe2+, Cu2+, Zn2+ and Cd2+. In contrast, Ca2+ is not transported nor does it inhibit Mg2+ transport [158]. The initial observation of a Mg2+ generated current, which would be consistent with SLC41A1 operating as a channel [158], or possibly as an electrogenic antiporter along the line of the Na/Ca exchanger [159], contrasts with a recent report by Kolisek et al. [160]. These authors, in fact, strongly advocate for SLC41A1 operating as a carrier and actually promoting Mg2+ efflux rather than influx. The over-expression of SLC41A1 in HEK293 cells resulted in no detectable Mg2+ currents. Moreover, incubation of cells in Mg2+-free media resulted in a significant reduction of total Mg2+ content and [Mg2+]i, the amplitude of Mg2+ loss depending on the number of SLC41A1 molecules expressed in the membrane and the induction time. Lastly, the changes in [Mg2+]i were sensitive to the experimental temperature but insensitive to the Mg2+ channel blocker CoHexamine [160]. Kolisek and collaborators also suggested that SLC41A1 forms high molecular weight complexes within the cell membrane with molecular masses ranging between 720 and 1236 kDa (i.e. 720 kDa < C1, C2 < 1236 kDa) [160]. Addition of SDS resulted in the progressive degradation of the complexes in a step-wise manner until a protein band of ~56kDa is obtained, which corresponds to the molecular mass of the SLC41A1 monomer (i.e. 480 kDa < C3 < 720 kDa; 242 kDa <C4 <480 kDa, and M ~56 kDa) [160]. It is presently undefined whether SLC41A1 monomer aggregates to form large multimeric complexes or interacts with auxiliary proteins. The reason for the absence of Mg2+-generated currents in this study as compared to the original observation by Goytain and Quamme [158] is also not clear. One possibility could be that the murine [158] and human orthologs [160] operate differently although they are expected to operate in a similar manner based upon their high degree (>90%) of homology. Yet, the possibility that point mutations can dramatically alter SLC41A1 ion specificity and modality of function cannot be completely dismissed. Another point of difference is that while Goytain and Quamme did not report a dependency of SLC41A1 operation on Na+ or other cations or anions following expression in X. oocytes [158], Kolisek and collaborators reported a marked Cl− conductance following expression in HEK293 cells, which was abolished by DIDS [160]. Whether this reflects the operation of additional transport mechanisms or the presence of structural differences in the cell membrane of HEK293 cells [160] as compared to X. Laevis oocyte [158] are possibilities that need further investigation.
A second isoform labeled SLC41A2 has been identified in both humans and mice. SLC41A2 transports Mg2+ as well as other divalent cations albeit with a different selectivity and inhibition profile than SLC41A1 [161]. In addition to Mg2+, SLC41A2 can carry Ba2+, Ni2+, Co2+, Fe2+ and Mn2+ but not Ca2+, Cu2+ or Zn2+. At variance of SLC41A1, Mg2+ transport via SLC41A2 is inhibited by Ca2+ [161]. Both SLC41A1 and SLC41A2 generate Mg2+ currents in X. oocyte, and the ionic uptake is voltage dependent with an apparent affinity of 0.75 mM and 0.31 mM, respectively [158,161]. SLC41A2 is also widely expressed in mammalian tissues, but its expression is not affected by low Mg2+ diet [161]. At the structural level, SLC41A2 shares >70% homology with SLC41A1 and it is supposed to present 10 trans-membrane domains as well. A recent study by Scharenberg’s group, however, suggests a structural arrangement in 2 five trans-membrane spans linked together by a supplementary spanning motif [162]. Hydrophobicity analysis indicates that the C- and N- termini are located on different sites of the cell membrane [162], a configuration that will be consistent with a total of 11 trans-membrane segments.
3.3.2 ACDP2
The human ACDP gene family was identified by Wang and collaborators [163] as a possible candidate of the urofacial syndrome. Mapped to 10q23-10q24 chromosome, this gene family comprises 4 isoforms differentially located in human tissues. ACDP1 is essentially restricted to the brain. ACDP2 is more widely expressed, but still retains the highest expression in the brain while being absent in skeletal muscles. ACDP3 and ACDP4 are both ubiquitous, but have the highest expression in the heart [164]. The murine distribution of ACDP isoforms is very similar to that observed for the human orthologues [165]. Termed ancient conserved domain protein because all isoforms share one domain phylogenetically conserved from bacteria to man [163], these proteins are >50% homologous to the CorC transporter, which together with CorB and CorD plays a role in Mg2+ efflux in prokaryotes [166]. Over-expression of ACDP2 in X. oocytes indicates that this protein can transport a variety of divalent cations including Mg2+, Co2+, Mn2+, Sr2+, Ba2+, Cu2+, and Fe2+, whereas Zn2+ inhibit its activity [167]. Mg2+ transport via ACDP2 is voltage dependent, occurs with a Km of ~0.5mM, and does not require the presence of extracellular Na+ or Cl− [167]. Similarly to SLC41A1, ACDP2 gene becomes over-expressed following exposure to Mg2+ deficient diet [167].
3.3.3 NIPA
Located in the SPG6 locus of chromosome 15q11-q13, the NIPA1 gene is so called for ‘non-imprinted in Prader-Willi/Angelman syndrome, a disease characterized by a complex developmental disorder that affects numerous organs and systems [168]. Located among a set of approximately 30 genes linked to the disease [168], NIPA1 has also been implicated in autosomal dominant hereditary spastic paraplegia (HSP, OMIM 182600). The human and mouse genomes contain four members of the NIPA family, termed NIPA1 trough NIPA4, with an overall similarity of ~40%. Homology between human and mice proteins is also high at around 98%. Studies conducted by Goytain and Quamme indicate that NIPA1 [169] and NIPA2 [170] can both operate as Mg2+ transporters. Presenting a sequence of 323 (NIPA1) and 359 amino acids (NIPA2) arranged to form 9 and 8 trans-membrane spans, respectively, these two proteins transport Mg2+ in a saturable fashion, with different Km and specificity. In fact, NIPA1 has a Km ~0.66mM for Mg2+ [169], and can also transport Sr2+, Fe2+ or Co2+, albeit to a lesser extent [169] In contrast, NIPA2 is highly specific for Mg2+ with Km ~0.31mM [170]. NIPA3, instead, transports Sr2+, Ba2+, Fe2+ and Cu2+ whereas NIPA4 transport Sr2+ and Ba2+. Neither NIPA3 nor NIPA4 transports Mg2+. The insurgence of autosomal dominant HSP is based on specific point mutations in NIPA1 (i.e. G100R or T45R) [171]. Both glycine and threonine residues are conserved among ortholog NIPA1 channels in different species. There are no similar consensus sites in NIPA2, NIPA3 and NIPA4 paralogs, implying that the folding of these proteins might be different. In HSP patients, NIPA2 appears to be normal but it cannot functionally replace NIPA1 to ameliorate HSP symptoms, nor can NIPA3 or NIPA4 substitute for the defective NIPA1. This is surprising for NIPA2 as the encoding gene is part of the 30 genes cluster associated with the Prade-Willi syndrome together with NIPA1. Presently, there is no indication as to whether the Prade-Willi syndrome presents alteration in Mg2+ homeostasis.
3.3.4 Huntingtin
The use of oligonucleotide microarray analysis to screen for Mg2+-regulated transcripts in epithelial cells indicates that Huntingtin-interacting protein 14 (HIP14) and its related protein HIP14-like (HIP14L) are significantly up-regulated (~ 3fold) by low-extracellular Mg2+ [172]. Formed by ~532 amino acids arranged in 6 trans-membrane spans, HIP14 presents 69% homology to HIP14L and a strong sequence similarity to the ankyrin repeat protein Akr1p [173]. HIP14 also possesses a cytoplasmic DHHC cysteine-rich domain. Defined by Asp-His-His-Cys sequence motif this domain confers palmitoyl-acyltransferase activity to the protein, and gives it the ability to palmitoylate membrane components whereby modulating their structure. Mg2+ accumulation via HIP14 and HIP14L appears to be electrogenic, voltage-dependent, and saturable, with Km ~0.87 and ~0.74mM, respectively [172]. Inhibition of palmitoylation activity by 2-Br-palmitate, or deletion of the DHHC domain decreases HIP14 mediated Mg2+ accumulation by ~50%, suggesting that palmitoylation is not required for basal Mg2+ transport. The widespread tissue distribution and intracellular localization of HIP14 (nuclear and perinuclear regions, Golgi complex, mitochondria, microtubules, endosomes, clathrin-coated and non-coated vesicles, and plasma membrane [174]) implicates this protein in numerous cellular processes including transcriptional regulation, mitochondrial bioenergetics, structural scaffolding, vesicle trafficking, endocytosis, and dendrite formation [174]. Golgi and post-Golgi vesicles, however, appear to be the primary location of HIP14 [172,174]. Hence, it can be hypothesized that Mg2+ accumulation via this protein is associated with the role HIP14 plays in the physiological functioning of the cellular compartments in which the protein is located. The neuropathological manifestation of Huntington disease is characterized by progressive neurodegenerative disorders, cognitive deficits and choreic movements. All these manifestations are linked to the abnormal expansion of glutamine residues from less than 34 to more than 37 at the 18th amino acid position [173]. Presently, the mechanism responsible for the insurgence of these defects is unknown [173]. Similarly unknown is whether the poly-glutamine expansion alters Mg2+ transport, and whether perturbation of Mg2+ homeostasis plays any role in the insurgence of the neuronal defects typical of Huntington disease.
3.4 Mg2+ Transport in Purified Plasma Membrane Vesicles
In the absence of functional cloning information, several laboratories including ours have resorted to the use of plasma membrane vesicles to better characterize how different Mg2+ extrusion mechanisms operate in particular cell types. The plasma membrane model presents several advantages including: 1) a well defined ionic extra- and intra-vesicular milieu composition to determine the modality of operation of the various Mg2+ transporters under rigorous experimental conditions, and 2) the ability to investigate the operation of the different Mg2+ extrusion mechanisms in the absence of Mg2+ buffering by ATP, proteins or other cytosolic components, and partitioning within intracellular organelles. By purifying total liver plasma membrane or cardiac sarcolemmal vesicles as well as specific hepatic subpopulations enriched in basolateral or apical domains, our laboratory has been able to provide a better understanding of the selective location and specificity of operation of the Na+- dependent and Na+-independent Mg2+ extrusion mechanisms in both liver cells and cardiac myocytes.
In the hepatocyte, the Na+-dependent extrusion mechanism is specifically located in the basolateral domain [144], is selectively activated by Na+ [144,175], and is inhibited only by imipramine [175], and not by amiloride and amiloride derivates [175]. Moreover, the operation of the exchanger is completely abolished by pre-treatment of basolateral vesicles with alkaline phosphatase, but it can be restored by loading the vesicles with ATP and PKA-catalytic subunit [136,137], leaning further support to the notion that the Na+/Mg2+ exchanger is activated upon phosphorylation by cAMP (Figure 1). As this exchanger continues to operate in the presence of zero trans Mg2+ across the plasma membrane (i.e. 20 mM Mg2+ inside and outside the vesicles) [144], indication is there that Mg2+ extrusion strictly depends on the Na+ trans-membrane gradient, with a Km ≤ 20mM [144], in good agreement with kinetic data obtained in isolated hepatocytes [133] and other cell types [142]. Experiments based on TPP+ distribution have confirmed the electrogenicity of this exchange mechanism in plasma membrane vesicles, supporting a 1Na+ in:1Mg2+ out exchange ratio under the majority of experimental conditions tested [136,144,175]. Upon removal of intravesicular Cl−, the stoichiometry ratio of the exchanger switches from electrogenic to electroneutral (i.e. 2Na+ in:1Mg2+ out) [138]. Interestingly, in the presence of intra-vesicular Cl−, an extrusion of ~35nmol Cl−/mg protein is observed within 1min from the addition of external Na+, in concomitance with the extrusion of Mg2+ and the accumulation of external Na+ into the vesicles [138]. The Cl− extrusion is not inhibited by inhibitors of anion transport (e.g. DNDS, DIDS, or niflumic acid), of the Na/K/Cl cotransporter NKCC1 (e.g. bumetanide or furosemide) [138], thus excluding that Clmovement occurs via one of these mechanisms. The only agent able to block Cl− extrusion is imipramine [138], which specifically blocks the operation of the Na+/Mg2+ exchanger in the basolateral domain of the hepatocyte [175]. Hence, it would appear that Cl− is extruded via the Na+/Mg2+ exchanger or, alternatively, via Cl− channels for partial charge compensation [138]. The possibility that Cl− is extruded via the Na+/Mg2+ exchanger has been suggested by Rasgado-Flores et al. [140] in dialyzed squid axons, and it would be in good agreement with the observation by Gunther and collaborators that intracellular Cl− has a stimulatory role on the activity of the Na+/Mg2+ antiport in red blood cells [139].
The basolateral domain is not the only portion of the hepatocyte cell membrane in which the operation of a Mg2+ extrusion mechanism has been observed. Experiments carried out in liver plasma membrane vesicles enriched in apical domain indicate the presence of two apparently distinct and unidirectional Mg2+ transport mechanisms, which extrude intravesicular Mg2+ for extravesicular Na+ and Ca2+, respectively [175].
The apical Na+-dependent Mg2+ transporter presents several similarities to the basolateral transporter: 1) its Km for Na+ is comparable at ~15–20mM; 2) it selectively uses Na+ over other monovalent cations; 3) it electrogenically exchanges 1Na+ in:1Mg2+ out [175]. From the pharmacological stand-point, however, the apical exchanger can be distinguished form the basolateral exchanger based on its specific inhibition by amiloride [175], although it retains a significant level of inhibition by imipramine. In contrast, only imipramine can block the basolateral antiport [175]. The apical exchanger can also be distinguished from the basolateral antiport based on its inability to operate in reverse mode [175] and the non-requirement for cAMP-mediated phosphorylation to become active [136]. Based on preliminary observation, it would appear that this apical antiport does not transport Cl− as part of its operation (Romani, personal observation).
The apical domain of the hepatocyte also presents a Ca2+-dependent Mg2+ extrusion mechanism [136]. Specifically located in this domain, this exchanger is activated by micromolar Ca2+ concentrations (Km ≤50μM), and is insensitive to alkaline phosphatase pre-treatment [136,137]. The Mg2+ extrusion elicited by this antiport occurs on electroneutral basis (i.e. 1Ca2+ in:1Mg2+ out) [136]. The exchanger, however, is not Ca2+ specific, as Mg2+ extrusion can occur following the addition of micromolar concentrations of other divalent cations (Ca2+≫Co2+=Mn2+>Sr2+≫Ba2+>Cu2+≫Cd2+) to the extravesicular milieu [175]. Similarly to the apical Na+/Mg2+ antiport, the Ca2+-dependent mechanism is inhibited by amiloride or imipramine [175]. This observation raises the question as to whether we are in the presence of two distinct apical mechanisms, modulated by Na+ and cations, respectively. Several lines of evidence, however, do not fully support this possibility. First, the co-addition of Na+ and Ca2+ to a purified subpopulation of apical plasma membrane vesicles does not appear to enlarge Mg2+ extrusion to a significant extent (Romani, personal observation). Second, amiloride inhibits both Na+- and Ca2+- dependent Mg2+ extrusion processes to a comparable extent at a similar concentration [175]. Third, alkaline phosphatase treatment does not affect the Mg2+ extrusion elicited by either exchanger in apical liver plasma membrane vesicles [136]. Fourth, neither of these exchangers can operate in reverse at variance of the basolateral Na+/Mg2+ antiport. Taken together, these observations suggest the operation of a non-selective exchange mechanism able to utilize monovalent or divalent cations to promote Mg2+ extrusion. At the present time, the physiological implication for the operation of such an exchanger in the apical domain of the hepatocyte is not clear. Circumstantial evidence, however, might support a possible role of Mg2+ in limiting Ca2+ sedimentation in the bile with consequent formation of bile stones [176].
The operation of functionally similar Na+- and Ca2+-dependent Mg2+ extrusion mechanisms has also been observed in cardiac sarcolemma vesicles [137]. As in the case of liver plasma membrane vesicles, cardiac sarcolemma vesicles do not require intravesicular ATP to support the operation of Mg2+ transporters [137], and pretreatment of the vesicles with alkaline phosphatase specifically inhibits the reversible Na+-dependent Mg2+ extrusion mechanism but not the Ca2+-dependent Mg2+ extrusion pathway [137]. For technical reasons, it is presently undefined whether cardiac myocytes also possess two distinct Na+-dependent Mg2+-extrusion mechanisms in the sarcolemma, and whether the Ca2+/Mg2+ exchanger in sarcolemmal vesicles can also utilize Na+ to promote Mg2+ extrusion.
The operation of specific Mg2+ transport mechanisms has been observed in plasma membrane vesicles from brush border cells of rabbit ileum [177] and from rat duodenum and jejunum [178]. In these models, however, Mg2+ accumulation rather than extrusion has been observed. By using membrane vesicles from rabbit ileum and cell permeant and non-permeant Mag-Fura, Juttner and Ebel have observed the operation of a saturable Mg2+ uptake mechanism when the intracellular Na+ concentration is higher than the extracellular concentration [177]. The process becomes inoperative when the Na+ gradient is reversed (i.e., [Na+]i<[Na+]o), the vesicles are in zero trans condition for Na+, or external Na+ is removed. At variance with the Na+-Mg2+ antiporter operating in liver plasma membrane vesicles, the pathway in ileum vesicles is not reversible and appears to be electroneutral. Yet, it possess a Km for Na+ of 16mM, a value similar to the Km calculated in liver plasma membranes [144], in smooth muscle cells from guinea pig tenia caecum [142], and in chicken erythrocytes [179]. Another similarity with the transporter operating in basolateral liver plasma membranes is the lack of inhibition by amiloride analogs [177]. In good agreement with reports from Gunther and collaborators [139], the transporter characterized by Juttner and Ebel is modulated by intravesicular anions, especially Cl− and SCN−, and is markedly stimulated by antagonists of anion transport (e.g., H2-DIDS) [177].
The main difference between plasma membrane vesicles from duodenum and jejunum [178] is that a single Mg2+ uptake mechanism operates in the duodenum with a Km ~0.8mM, whereas two transporters operate in the jejunum with Km values of 0.15mM and 2.4mM, respectively. In both these experimental models, Mg2+ but not Ca2+ accumulation is reduced in the presence of alkaline phosphatase inhibitors [180], suggesting that Ca2+ and Mg2+ are transported via distinct pathways. This hypothesis is further supported by the observation that Mg2+ accumulation is inhibited by amiloride but not by Ca2+ channel antagonists. Consistent with the report by Juttner and Ebel [177], Mg2+ accumulation is stimulated by an intravesicular electronegative potential or an alkaline pHo [178]. The effect of external pH, however, is lost when [Mg2+]o>1mM [178]. Under the latter condition (i.e. [Mg2+]o>1mM), Mg2+ accumulation is enhanced by the presence of Na+ or K+ but it is inhibited by the presence of divalent cations (Co2+>Mn2+>Ca2+>Ni2+>Ba2+>Sr2+) in the extravesicular space [178].
4. Regulation of Mg2+ Transport and Homeostasis
The majority of mammalian cells retains their basal Mg2+ content virtually unchanged under resting conditions even when a major trans-membrane gradient is artificially imposed [1–3]. At the same time, compelling evidence supports the notion that different hormones induce the movement of large amounts of Mg2+ in either direction across eukaryotes’ cell membrane. As a result of these movements, changes in serum, total and - to a lesser extent - free Mg2+ content have been observed. Furthermore, these changes have resulted in detectable variations in Mg2+ level within organelles, especially mitochondria, with significant repercussions on cellular bioenergetics. A full understanding of the physiological relevance of these changes in cellular Mg2+ content is far from complete. Yet, a picture is slowly emerging, which relates changes in total Mg2+ content to energetic substrate utilization (e.g. glucose), cell cycle progression [65] or meaningful changes in Mg2+ content within discrete portions of the cell or cellular organelles. Hence, variations in total cellular Mg2+ content can effectively translate into changes in Mg2+ concentration within compartments that can modulate the activity of specific enzymes located therein.
4.1 Mg2+ Extrusion
Hormones like catecholamine or glucagon induce Mg2+ extrusion from various cell types or perfused tissues. The majority of these hormones have in common their ability to increase cellular cAMP level by activating G-protein coupled receptors (GPCR) at the cell membrane level, which ultimately converge on Gαs and adenylyl cyclase (Figure 1). While the Mg2+ extrusion elicited by these hormones depletes to a varying extent the Mg2+ pools present within the cytoplasm and the cellular compartments, the physical outward transport of Mg2+ across the cell membrane primarily occurs via the Na+/Mg2+ exchanger previously described (Figure 1). Yet, a (partial) contribution of the Na+-independent Mg2+extrusion mechanism cannot be completely excluded. Magnesium extrusion has also been observed following metabolic treatments that decrease cellular ATP content, the main Mg2+ buffering component. Interestingly, several of the hormones that induce Mg2+ extrusion from liver cells also elicit glucose output from the hepatocyte. Conversely, hormones that promote glycogen synthesis stimulate Mg2+ accumulation rather than extrusion (discussed in Section 4.2). Hence, it would appear that at least in liver cells Mg2+ extrusion is functionally associated with glucose transport and utilization.
4.1.1 Cyclic-AMP Dependent Mg2+ Extrusion
In 1974, Elliot and Rizack first reported a transport of Mg2+ across the plasma membrane of adipocytes stimulated by adrenocorticotrophic hormone [181]. In that case, Mg2+ was accumulated within the cells, but the authors did not elucidate the modality of transport or the mechanism involved. The first extensive characterization of hormone-induced Mg2+ transport was provided by Maguire and colleagues in S49 lymphoma cells and primary lymphocytes stimulated by β-adrenergic receptor agonist or PGE1 [182–185]. Maguire and Erdos [186] also provided the first observation that protein kinase C (PKC) activation enhances Mg2+ influx in S49 cells at variance of β-adrenergic receptor stimulation, which inhibits the process. Observation carried out in S49 cells lacking protein kinase A (PKA) or adenylyl cyclase (AC), however, indicated that the inhibitory effect of beta-adrenergic agonists was not mediated by cAMP [186,187]. At variance of what reported for primary lymphocytes [25], Mg2+ transport in S49 cells appears to be independent of extracellular Na+ concentration or membrane potential (Grubbs R.D. and Maguire M.E., unpublished observation). Further, Mg2+ turn-over in S49 requires more than 40 hours as compared to the much faster Ca2+ turn-over, which is completed in less than 3 hours [188].
These initial observations were followed by a long series of reports all supporting the notion that β-adrenergic agonists and other hormones control Mg2+ homeostasis in mammalian cells. In the majority of eukaryotic cells, hormones or agents that increase cellular cAMP level elicit a significant extrusion of Mg2+ into the extracellular space or the circulation [32–34]. This effect has been observed in cardiac ventricular myocytes [32,33,132,189], liver cells [34,35,133–135], red blood cells [31], lymphocytes [30] and Erhlich ascites cells [190] among other cells (see ref. 27 for a more comprehensive list), as well as in whole anesthetized animals [191,192]. In all the cell types tested so far, Mg2+ extrusion is a fast process that reaches the maximum within 8min from the application of the stimulus irrespective of the hormone (catecholamine, isoproterenol, glucagon, PGE1, or arachidonic acid) [30–35,189,190] or agent (i.e. forskolin or cell permeant cyclic AMP analogs) [30–34,133–135] utilized to increase cellular cAMP level (Figure 1). The key role of cAMP in modulating Mg2+ extrusion is further corroborated by the observation that pre-treatment of cells with hormones or agents that decrease cAMP production (e.g. carbachol [30–34, 133–135], insulin [193]) or prevent PKA activation (e.g. Rp-cAMP [25]) completely prevents cellular Mg2+ mobilization. In an open perfusion system, the amount of Mg2+ extruded from the organ (i.e. heart or liver) returns towards baseline level within 8 min from the application of the agonist irrespective of its dose or persistence in the perfusate [32,34]. This temporally limited extrusion suggests that Mg2+ is rapidly mobilized from a well defined cellular pool(s) that is(are) rapidly depleted. In support of this notion is the observation that sub-maximal doses of agonist sequentially infused within a few minutes from each other elicit Mg2+ extrusions of progressively decreasing amplitudes [30]. Under all these conditions, limited changes in cytosolic free [Mg2+]i have been observed [37,194], suggesting that Mg2+ is rapidly released from binding and buffering sites within the cytoplasm or cellular organelle(s) and extruded across the cell membrane. Irrespective of the hormone utilized, cAMP-mediated Mg2+ extrusion occurs via the putative Na+/Mg2+ exchanger described previously. In fact, either the removal of extra-cellular Na+ [143] or the cell pre-treatment with non-selective Na+ transport inhibitors like amiloride or imipramine [33,143], abolishes the Mg2+ extrusion almost completely. Under either inhibitory condition the reduced Mg2+ extrusion across the cell membrane originates a more sustained rise in cytosolic free [Mg2+]i [37,194], suggestive of the concept that blocking Na+-dependent transport mechanism prevents Mg2+ from being extruded across the cell membrane but not from being released from binding/buffering sites and/or cellular pool(s) into the cytoplasm. Two corollaries of this observation are that: 1) cAMP operates on at least two different levels (i.e. cellular organelle(s) and plasma membrane) to mobilize Mg2+ from the cell, and 2) only Mg2+ transport across the cell membrane is Na+-dependent whereas the mobilization from cellular organelle(s) is largely Na+-independent. Alternatively, it has to be postulated that cytosolic Na+ concentration, which ranges between 15 to 20mM in most cell types) is more than sufficient to favor Mg2+ transport across the membrane of cellular organelles.
4.1.2 Cyclic-AMP Independent Mg2+ Extrusion
In 1989, Jakob and collaborators reported that phenylephrine administration also promotes Mg2+ extrusion from liver cells via alpha1-adrenergic stimulation [36]. Subsequently, our laboratory [134,149] confirmed this observation and provided the first evidence that the co-stimulation of α1- and β-adrenergic receptor are not alternative but rather additive and complementary processes to induce Mg2+ extrusion from liver cells. This event is of particular relevance especially when the two classes of adrenergic receptors are stimulated by mix-adrenergic agonists such as epinephrine or norepinephrine [134,149]. Pre-infusion of insulin only abolishes β-adrenergic receptor mediated Mg2+ extrusion form liver cells, leaving unaffected the mobilization of Mg2+ mediated via α1-adrenergic receptors [149]. The inhibitory effect of insulin persists even in cells treated with cell-permeant cAMP analogs [149]. A similar inhibitory effect of insulin on β-adrenergic receptor mediated, cAMP-modulated, Mg2+ extrusion has been observed in cardiac myocytes [193]. These results have been attributed to an inhibitory effect of insulin on β-adrenergic receptor activation [195], and a stimulatory effect of the hormone on the cytosolic phosphodiesterase that degrades cAMP [196]. A report by Romero and collaborators [197], however, suggests also the possibility of a direct modulating effect of insulin on the Na+/Mg2+ exchanger, at least in erythrocytes.
Fagan and Romani [134,135] further investigated the modality of Mg2+ extrusion following α1- adrenergic receptor stimulation in liver cells. Their results indicate that phenylephrine-induced Mg2+ extrusion strictly depends on the activation of capacitative Ca2+ entry [135]. Inhibition of IP3-induced Ca2+ release from the endoplasmic reticulum, chelating of cytosolic Ca2+, or inhibition of Ca2+ entry at the plasma membrane level all result in the complete inhibition of Mg2+ extrusion from the hepatocyte [135]. The scant information available about possible binding of Mg2+ by cellular proteins prevented the authors from ascertaining whether Mg2+ extruded from the hepatocyte was mobilized from the ER, or displaced from cytosolic binding sites following the massive entry of Ca2+ across the cell membrane [135 and refs. therein]. Interestingly, extracellular Na+ and Ca2+ are both required for the phenylephrine-induced Mg2+ extrusion to occur [135]. In the absence of extracellular Ca2+, in fact, the amplitude of Mg2+ extrusion is decreased by ~15% to 20% whereas extracellular Na+ is responsible for the remaining 80%–85% of the extrusion. It is presently unclear whether Mg2+ extrusion occurs via the Ca2+-activated, Na+-dependent mechanism observed in the apical domain of the hepatocyte, or whether Na+ is required to maintain membrane potential and facilitate Ca2+ entry across the hepatocyte cell membrane. It has to be noted, however, that in the absence of receptor activation, thapsigargin administration can mimic phenylephrine stimulation and elicit Mg2+ extrusion from the hepatocyte, even in the absence of extracellular Ca2+ [135], although to a lesser extent. Hence, it would appear that an optimal level of cytosolic Ca2+ has to be attained in order for Mg2+ extrusion to occur via displacement from cellular binding sites or via a Ca2+- calmodulin-activated mechanism [135]. Interestingly, the group of Schweyen has provided evidence that in yeast Mg2+ deprivation accelerates Ca2+ accumulation. In turn, this translates into a more rapid activation of Ca2+-mediated signaling [198].
4.1.3 Mg2+ Homeostasis and Glucose
The presence of redundant Mg2+ extrusion mechanisms or modalities of activation of a common Mg2+ extrusion pathway points to a basic question: What is the physiological significance of Mg2+ mobilization in mammalian cells?
The general answer is that Mg2+ extrusion can have a different significance in different cells due to the physiological differentiation and function of the various cell types. In the case of cardiac myocytes, for example, an increase in extracellular Mg2+ level has been associated with a modulating effect on the open probability of the L-type Ca2+-channels [199] and a temporary decrease in SA node action potential [189]. In the case of liver cells, instead, Mg2+ transport appears to be associated with a regulatory role on glucose transport and utilization (Figure 1). Hormones like catecholamine [134,149] or glucagon [134], and adrenergic agonists like isoproterenol or phenylephrine [134,149], which elicit Mg2+ extrusion from liver cells, all activate glycogenolysis and promote release of hepatic glucose into the blood-stream within a similar time frame [134]. Interestingly, the presence of amiloride or imipramine inhibits both Mg2+ extrusion and hepatic glucose output [134]. The converse is also true. Inhibition of glucose transporter activity by phlorethin results in a qualitatively similar inhibition of Mg2+ extrusion from liver cells [134]. The presence of a close functional ‘link’ between glucose and Mg2+ homeostasis is corroborated by the observation that overnight starvation completely depletes hepatic glycogen and glucose, and concomitantly decreases to a significant extent (minus 15%) total hepatic Mg2+ content as a consequence of pro-glycemic hormones (i.e. catecholamine and glucagon) activation [200]. Noteworthy, this decrease in hepatic Mg2+ content is equivalent in amplitude to that elicited via in vitro stimulation of perfused livers by the same hormones [200], or that observed to occur in livers of type-I diabetic animals [201], which are markedly depleted in cellular glycogen. This functional link between glucose and Mg2+ homeostasis can also be observed under conditions in which glucose accumulation and glycogen synthesis are stimulated by insulin administration to cardiac ventricular myocytes [193] or pancreatic beta cells [202]. In both experimental models, the amount of Mg2+ accumulated within the cells is directly proportional to the amplitude of glucose accumulation. Conversely, decreasing extracellular Mg2+ concentration directly reduces the amount of glucose accumulated within the cells [134,193]. A role of Mg2+ in regulating glucose homeostasis is underlined by the observation that several glycolytic enzymes, including hexokinase, phosphofructokinase, phosphoglycerate mutase, phosphoglycerate kinase, enolase and pyruvate kinase, show activation at low, and inhibition at high Mg2+ concentrations [203,204].
Diabetic conditions provide a clear albeit indirect proof of the glucose/Mg2+ relationship. Work by Altura’s group [205] and more recently by Resnick and Barbagallo [206,207] indicate that cellular Mg2+ content is markedly decreased under type-I and type-II diabetes. Originally observed in red blood cells [206,207], the decrease has been reported to occur also in several other tissues including skeletal muscles [201], liver [201], and heart [208]. Interestingly, Mg2+ extrusion via β-adrenergic signaling remains operative and is actually up-regulated in hepatocytes from type-I diabetic rats [201] while it is markedly inhibited in cardiac myocytes from the same animals [208]. Whether this reflects a differential operation and modulation of β2-adrenoceptors in liver cells versus β1-adrenoceptors in cardiac cells has not been clarified. Both cell models, however, show a marked inhibition of the Mg2+ entry mechanism(s), which persists also in liver plasma membrane vesicles [201,209]. Addition of glucose or glycogen to plasma membrane vesicles from diabetic animals renormalizes the amplitude of Mg2+ extrusion, but is ineffective at restoring Mg2+ accumulation via the operation in reverse of the Na+/Mg2+ exchanger [209]. The defect in total cellular Mg2+ content appears to be strongly associated with the decrease in protein synthesis and ATP production detected in the cells [208]. Supplementation of exogenous insulin restores protein synthesis and ATP production as well as Mg2+ homeostasis and extrusion provided that insulin is administered for at least two weeks [208]. As indicated previously, it appears that the role of insulin in modulating Mg2+ homeostasis is not restricted to controlling glucose homeostasis and accumulation or the release of pro-glycemic hormones like glucagon, but extends to a direct modulation of the Na+/Mg2+ exchanger [197]. This effect would directly increase cellular Mg2+ content and would also impact on the insulin receptor itself. Data obtained in animals maintained on low Mg2+ diet indicate that a decrease in cellular Mg2+ content affects the ability of the insulin receptor to properly phosphorylate the downstream insulin receptor substrate (IRS) and propagate the signaling within muscle cells [210]. This result might be of relevance to explain – at least in part - the decrease in glucose accumulation observed in skeletal muscles under diabetic conditions [210].
4.1.4 Mg2+ Homeostasis and ATP
Although hormones represent the most dynamic mechanism by which a cell can rapidly extruded 10% to 15% of its total cellular Mg2+ content within a few minutes from the application of the hormones, Mg2+ can also be extruded from the cell following exposure to various agents or conditions that markedly decrease cellular ATP content and production. Cyanide [154,211], mitochondrial uncouplers [38,115], fructose [212], ethanol [213], or hypoxia [214] are just some of the agents or conditions that affect cellular ATP level and Mg2+ homeostasis. All these agents, in fact, act by decreasing ATP content by preventing the mitochondrial electron chain from generating ATP (cyanide or uncouplers), by acting as an ATP trap (fructose), or by altering the redox state of pyridine nucleotide within the cytoplasm and the mitochondrion (ethanol). Because ATP represents the major Mg2+ buffering component within the cell (Figure 1) [8,9], a decrease in its content or its degradation into ADP or AMP results in an increase dissociation of Mg2+ form the binding and an increase in cytosolic free [Mg2+]i. Ultimately, such an increase in cytosolic Mg2+ level originates a detectable Mg2+ extrusion from the cell [154,211–214]. Such an extrusion can be observed in erythrocytes, which possess limited cellular buffering capacity for Mg2+ and no compartmentation [215], as well as in cells that possess additional Mg2+ buffering due to the presence of proteins or cellular organelles in addition to ATP and phosphonucleotides [154,211–214]. In several cases, such as fructose addition [212], the changes in cytosolic [Mg2+]i can elicit glycogenolysis via activation of glycogen phosphorylase and glucose utilization to restore cellular ATP levels [212]. The majority of these experimental conditions promote a modest increase in cytosolic free [Mg2+]i, which is considerably lower than the increase expected to occur based upon the corresponding decrease in ATP level. This observation strongly supports the notion that the majority of Mg2+ released from ATP and other binding sites is extruded from the cell. Furthermore, because ATP level decreases following changes in pyridine nucleotide ratio or mitochondria poisoning, it would appear that not phosphorylation but the rise in cytosolic Mg2+, even if modest, is sufficient to activate Mg2+ extrusion and limit the rise in cytosolic free Mg2+ concentration to approximately 100–200μM at the most [211]. Hence, it can be presumed that such an increase is sufficient to activate enzymes and metabolic reactions controlled by Mg2+.
On the other hand, cellular ATP regulates Mg2+ extrusion in ways other than acting as a buffering component. Evidence for ATP additional role has been provided by experiments in giant squid axon [216], mammalian hepatocytes [217], or erythrocytes [11]. In squid axon, the Na+-dependent Mg2+ extrusion requires a physiological level of ATP to operate, and as the level of ATP decreases so does the amplitude of Mg2+ extrusion [217]. In erythrocytes and hepatocytes, instead, ATP appears to regulate the Na+-independent Mg2+ extrusion process [11,217]. The modality by which ATP regulates the Mg2+ extrusion process is unclear, but it appears to be unrelated to the operation of an ATPase mechanism. This notion is supported by the observation that a decrease in cellular ATP level as it occurs for example under diabetic or alcoholic conditions paradoxically results in an increased extrusion of Mg2+ via the Na+- dependent mechanism in a manner directly proportional to the decrease in ATP level [201,213]. Hence, it appears that the role of ATP is predominantly that of a ligand for Mg2+ both in the cytoplasm and the mitochondrial matrix [8,9], and that a decrease in ATP results in an increase in free Mg2+ and its consequent extrusion from the cell (Figure 1).
4.2 Mg2+ Accumulation
The identification of several Mg2+ entry mechanisms strongly support the hypothesis that cellular Mg2+ is dynamically maintained through the operation of entry and exit mechanisms that are differentially regulated by hormones and metabolic conditions. A striking difference is there, however, between the Mg2+ exit and the Mg2+ entry mechanisms. In the case of Mg2+ extrusion mechanisms we have a good understanding of the signaling activating their operation but we lack any information about the structure of the mechanisms themselves. In the case of Mg2+ entry mechanisms, instead, we do have structural information about several of these mechanisms but for the most part we lack detailed information about their individual activation by hormones or second messenger, and their possible cooperation under specific conditions.
4.2.1 Role of Protein Kinase C
Experimental evidence indicates that mammalian cells can accumulate large amounts of Mg2+ as a result of hormonal stimulation (Figure 1). Administration of hormones like carbachol, vasopressin, angiotensin-II, or insulin to various cell types results in the inhibition of cAMP-mediated Mg2+ extrusion and/or the reversal of Mg2+ extrusion into Mg2+ accumulation [32,193]. The list of cells that respond to hormonal stimulation by accumulating Mg2+ is quite long [see ref. 29 for a list], and includes cardiac myocytes [32,193], smooth muscle cells [218], hepatocytes [34,219], platelets [220], lymphocytes [221], fibroblasts [222], and pancreatic beta cells [202], just to name a few. In addition to inhibiting cAMP production, several of the hormones indicated above activate protein kinase C (PKC) as part of their cellular signaling. Evidence supporting a role of PKC in mediating Mg2+ accumulation has been provided by several laboratories. Maguire and collaborators have reported that administration of phorbol-myristate acetate (PMA), which directly activates PKC, elicits a marked accumulation of Mg2+ in S49 lymphoma cells [184]. A similar effect of PMA has been reported by Somogyi’s group in thymocytes [223], and by our laboratory in cardiac myocytes [219] and hepatocytes [219]. Furthermore, our group has reported that down-regulation of PKC by exposure to a large dose of PMA for 3 hours completely abolishes the ability of cardiac and liver cells to accumulate Mg2+ while leaving unaffected the responsiveness of these cells to adrenergic agonists [219]. A similar inhibition of Mg2+ accumulation has been observed following treatment of cells with the PKC inhibitors calphostin [218] or staurosporine [224]. Alteration in PKC distribution and activity associated with a defective accumulation of Mg2+ have been observed in arterial smooth muscle cells [225] and hepatocytes [226] isolated from animals exposed to alcohol, or in liver cells of diabetic animals [227].
Protein kinase C activation is only part of the integral response of hormones like angiotensin-II or vasopressin. The interaction of these hormones with their receptor, in fact, activates phospholipase C which, in turn hydrolyses PIP2 to generate diacyl-glycerol (DAG) and IP3. These two molecules would then activate PKC and IP3 receptor in the ER, respectively. Activation of IP3 receptor results in a marked but transient increase in cytosolic Ca2+ followed by a more sustained Ca2+ entry across the plasma membrane through store-operated channels (SOC). Thus, Ca2+ signaling is an integral component of the cellular response elicited by these hormones. Yet, the contribution of Ca2+ increase and signaling in mediating Mg2+ accumulation is poorly defined. Liver cells loaded with Bapta-AM, which chelates cytosolic Ca2+, are unable to extrude and accumulate Mg2+ following stimulation by phenylephrine and PMA, respectively [133]. Administration of thapsigargin, which inhibits the SRCA pumps and increases cytosolic Ca2+ by favoring its release from the endoplasmic reticulum, also prevents Mg2+ accumulation [133] and actually induces a Mg2+ extrusion from the liver cell if applied for more than 3–5 min [133, 135]. Because of the different time-scale and amplitude of the changes in cellular Ca2+ and Mg2+ content [133], it is difficult to properly correlate these variations. Cytosolic free Ca2+ transiently increases several orders of magnitude above its resting level. In contrast, cytosolic free Mg2+, which is already in the millimolar or sub-millimolar range, increases by 10% to 15% [37] at the most, although in absolute terms this amount far exceeds the overall change in cytosolic Ca2+ mass.
An unresolved point of inconsistency in the role of Ca2+ and PKC signaling in regulating Mg2+ accumulation is provided by the reports that the administration of phenylephrine, which activates PKC signaling in addition to inositol 1,4,5 trisphosphate and Ca2+ signaling, does not elicit Mg2+ accumulation but induces an Mg2+ extrusion from liver cells [135]. These results raise the question as to what modulates the different cellular response to the administration of phenylephrine or vasopressin. One possibility could be that different PKC isoforms are activated under one condition but not the other. For example, hepatocytes possess 3 classical and at least 2 novel PKC isoforms [227]. Thus, it can be envisaged that one isoform (or class of isoforms) is involved in mediating Mg2+ accumulation while another isoform (or class of isoforms) is involved in modulating Mg2+ extrusion. Consistent with this hypothesis, recent data from our laboratory suggests that PKCε is essential for Mg2+ accumulation to occur [226]. Under conditions in which the expression of this isoform is inhibited by antisense, or its translocation to the cell membrane is prevented - for example - by ethanol administration, no Mg2+ accumulation is observed in liver cells [226]. Interestingly, this PKC isoform has the highest affinity for Mg2+ among all PKC isoenzymes, with a Km ~1mM [228], close to the physiological free [Mg2+]i measured in the cytoplasm of the hepatocyte [37,229] and other mammalian cells as well [218]. Although the mechanism ultimately responsible for the accumulation of Mg2+ within the hepatocyte has not been identified, it is worth considering the recent observation by Bindels and collaborators that in the absence of PKC activation or following RACK1 over-expression, RACK1 can bind to TRPM6, and possibly TRPM7, at the level of the kinase domain and inhibit the channel activity [86].
4.2.2 Role of MAPKs
Several lines of evidence indicate that additional signaling pathways (e.g. MAPKS) are involved in determining differing cellular responses under seemingly similar stimulatory conditions. Reports from Altura and collaborators in arterial muscle cells [230], Touyz’s laboratory in vascular smooth muscle cells [231], and our group in liver cells [232] indicate that pharmacological inhibition of ERK1/2 and p38 MAPKs abolishes PKC mediated Mg2+ accumulation [232]. In addition, inhibition of MAPKs signaling hampers Mg2+ accumulation and affects cyclin activity in vascular smooth muscle cells [231], preventing the cells from progressing in the cell cycle [231]. This effect may occur via changes in nuclear functions directly regulated by Mg2+, as proposed by Rubin [233], and/or changes in nuclear signaling by ERK2, which depends on Mg2+ level to properly dimerize, translocate and activate specific nuclear targets [234]. The role of ERK1/2 in regulating Mg2+ homeostasis is further emphasized by the evidence that increased ERK1/2 phosphorylation and TRPM6 expression have been observed following EGF administration to renal epithelial cells [97,98]. The role of MAPKs in Mg2+ homeostasis, however, is far from clear as ERK1/2 appears to be involved in mediating also Mg2+ extrusion [230,235].
4.2.3 Role of EGF
As mentioned earlier, direct and indirect evidence implicates EGF in regulating Mg2+ accumulation, at least in kidney cells. The administration of EGF controls TRPM6 channel expression and operation in the apical domain of renal epithelial cells to promote Mg2+ accumulation [97,98,236]. Point mutations in the EGF sequence limit TRPM6 functioning and Mg2+ accumulation within the cells [237]. The modulation of TRPM6 expression appears to occur via ERK1/2 signaling coupled to activator protein-1 (AP-1) [98]. Indirect evidence of Mg2+ homeostasis regulation by EGF is provided by the observation that antibodies against EGF used in several form of colon cancer [95,96] induce Mg2+ wasting and hypomagnesaemia.
5. Serum Mg2+ Level and Mg2+ Sensing Mechanism
Humans and many mammals present a circulating Mg2+ level of ~1.2–1.4 mEq/L [19, 239]. Clinical and experimental evidence indicates that serum Mg2+ level decreases in humans and animals in several chronic diseases [201]. Yet, there is a remarkable lack of information as to whether serum Mg2+ undergoes circadian fluctuations following hormonal or non-hormonal stimuli (e.g., fasting or exercise). The infusion of catecholamine [239–241] or [191,192,242] results in a marked dose- and time-dependent increase in circulating Mg2+ content. This increase is maximal within 20min from the agent administration [191], remaining unchanged for up to 2 hours following the removal of the agonist [191]. Considering this time frame of changes, the pre-infusion level of serum Mg2+, the glomerular filtration rate (1.62 mL/min), and the fractional excretion (17%) [243], it is evident that the increase in serum Mg2+ level is independent of the hemodynamic changes elicited by the β-adrenergic agonist [191] and renal excretion [243]. Consistent with the whole body distribution of β2 versus β1 adrenergic receptors [244,245], the increase in serum Mg2+ occurs can be mimicked by specific β2-adreno-ceptor agonist and inhibited by specific β2-blocker [191]. The amplitude of the increase in circulating Mg2+ level suggests that the adrenergic agonist mobilizes Mg2+ from various tissues [191], including bone [192]. The latter hypothesis is supported by the observation that the infusion of carbonic anhydrase inhibitor prevents the increase in serum Mg2+ level elicited by isoproterenol administration in anesthetized rats [192]. It is interesting to note that the hormones that increase plasma Mg2+ by mobilizing the cation from different organs or tissues are also responsible for increasing Mg2+ reabsorption in the Henle’s loop, thus preventing a net Mg2+ loss.
Presently, no specific Mg2+ sensing mechanism in the circulation has been identified. However, the Ca2+ sensing receptor [246] can detect changes in circulating Mg2+ level in a range of concentrations higher than those of Ca2+ [247] and consistent with the increase in serum Mg2+ levels reported in the literature [191,192]. The observation that in cells of the distal convoluted tubule (MDCT) of the mouse the Ca2+-sensing receptor can be activated by extracellular Ca2+ and Mg2+ with comparable sensitivity [248] suggests interesting hypotheses in terms of whole body physiology. The activation of this sensor mechanism would inhibit glucagon- or vasopressin-mediated Mg2+ accumulation into the cells [249] and favor its urinary elimination, possibly explaining the clinical and experimental evidence that hypomagnesaemia and hypocalcaemia inhibit hormone-stimulated cAMP-mediated reabsorption of Mg2+ and Ca2+ along the different segments of the nephron [250]. In addition, the Ca2+ sensing receptor would represent a distal regulatory mechanism to restore magnesaemia to a physiological level following the increase observed in anesthetized animals infused with adrenergic agonists [191,192]. It is still an open question as to whether this sensing mechanism or associated modulating components are altered under diabetic conditions in which a significant loss of tissue Mg2+ content and increased magnesuria are observed.
At variance of hypercalcaemia, which is associated with muscle weakness and arrhythmia, an increase in serum Mg2+ level appears to be well tolerated under in vivo conditions. Rats infused with boluses of Mg2+ that increase serum Mg2+ level by 50% do not exhibit significant systemic hemodynamic changes but show a marked increase in coronary artery flow [251]. Baboons infused with pharmacological doses of Mg2+ sufficient to prevent epinephrine-induced cardiac arrhythmias show a significant attenuation of epinephrine-induced increase in mean arterial pressure and systemic vascular resistance [252]. It would appear, therefore, that an increase in extracellular Mg2+ concentration regulates catecholamine release from peripheral and adrenal sources [253] and consequently cardiac contractility [188]. Taken together, these observations suggest that an increase in serum Mg2+ level following adrenergic stimulation can: 1) act as a feed-back mechanism to modulate catecholamine release and activity, and 2) contribute to improved blood flow and O2 delivery to the heart and possibly other tissues at a time when an increase in energy production is expected.
The presence of a Mg2+ sensor at the cell level is also debated. The presence and operation of such a sensor mechanism is supported by several lines of evidence. First, prolonged exposure to 0mM [Mg2+]o decreases cytosolic free Mg2+ concentration by approximately 50% in cardiac ventricular myocytes [254], MDKC [255], or MDCT cells [256]. This reduced cytosolic Mg2+ level is maintained as long as the cells are incubated in the presence of 0mM [Mg2+]o, but returns to normal level as soon as [Mg2+]o is increased in a time-frame that is directly proportional to the extracellular Mg2+ concentration utilized [254–256]. The presence of L-type Ca2+-channel inhibitors (e.g. verapamil or nifedipine) or La3+ in the extra-cellular milieu prevents the restoration of Mg2+ level [254]. The concomitant absence of significant changes in cytosolic [Ca2+]i excludes that Ca2+ may act as a regulatory mechanism, and suggests a direct effect of these inhibitory agents on the Mg2+ entry mechanism [254]. As TRPM7 operation is affected by gadolinium [54], these results anticipate the presence and operation of the Mg2+ specific channels TRPM7 [42] and TRPM6 [43].
A second line of evidence for the presence of a Mg2+ sensor in eukaryotic cells is provided by the occurrence of Mg2+ extrusion in all the conditions in which cellular ATP decreases as a result of chemical hypoxia [211], or exposure to fructose [212], ethanol [213], or cyanide [154]. Under all these conditions, Mg2+ extrusion only occurs when extracellular Na+ is available to be exchanged for cellular Mg2+. In the absence of external Na+, almost no Mg2+ extrusion occurs [154,213,257], and a significant increase in cytosolic Mg2+ can be detected [154, 257]. Hence, a scenario can be envisage whereby release of Mg2+ from cellular organelles [282] or from binding moieties such as ATP [154,213] results in an increase in cytosolic Mg2+ content that is detected by the sensor which, in turn, activates the Mg2+ extrusion mechanism. The nature of this sensor is still undefined. Because almost all the metabolic conditions mentioned above are characterized by changed in the ratio between reduced and oxidized pyridine nucleotide levels (e.g. ethanol [213]), it is an appealing albeit unproved hypothesis that the concentrations of these nucleotides (or their ratio) acts as a Mg2+ sensor in eukaryotic cells.
Similarly to Mg2+ extrusion, cellular Mg2+ accumulation also requires proper ion distribution, especially phosphate [256] and potassium [258], across the cell membrane. The role of potassium is of particular relevance as it suggests that Mg2+ is accumulated for charge compensation as the result of changes in membrane potential [259–261]. Especially in polarized epithelia (e.g. nephron and intestine) Mg2+ entry mechanisms such as TRPM7 and TRPM6 are located on the apical side, counterbalancing the operation of the Na+/Mg2+ exchanger and the Na+/K+-ATPase on the basolateral domain of the cell (see ref. 121 for a review). Whether the effect on K+ occurs through changes in membrane potential, or indirectly via a reduced operation of the Na+/K+-ATPase coupled to the operation in reverse of the Na+/Mg2+ exchanger [262] is topic for future investigation. In the particular case of K+, it has also to be noted that pathological conditions characterized by a marked decrease in tissue Mg2+ content (e.g., diabetes, [201]) are also characterized by an inability of the tissue to properly transport potassium [263,264]. This effect is the direct results of insulin absence or ineffectiveness coupled to a reduced activity rate of the Na+/K+-ATPase. It remains to be determined as to whether changes in pyridine nucleotide levels (or ratio), this time in an opposite direction, promote Mg2+ accumulation.
6. Physiological Role of Intracellular Mg2+
One of the conclusions generated by the data presented in the previous sections is that Mg2+ acts an indispensable regulatory cation for enzymes, phosphometabolites, and channels [1,265] (Figure 1). Several glycolytic enzymes, including hexokinase, phosphofructokinase, phosphoglycerate mutase, phosphoglycerate kinase, enolase and pyruvate kinase, show activation at low, and inhibition at high Mg2+ concentrations [203,204]. Adenylyl cyclase represents the best example of an enzyme directly regulated by Mg2+. As suggested by Maguire’s data (reviewed in ref. 266), Mg2+ exerts this effect by acting at two different sites: one site is on the guanyl nucleotide coupling protein, where it regulates agonist affinity as well as the interaction with the catalytic subunit. The second site is on the catalytic subunit and regulates the activity of this subunit.
The regulation of adenylyl cyclase and other cellular enzymes (such as those involved in glucose homeostasis [203,204] occur at Mg2+ concentrations between 0.5 to 1mM, which are well within the fluctuations in free [Mg2+]i measured in the cytoplasm of various cells including hepatocyte [229]. With the exception of the glycolytic enzymes, however, studies attempting to evidence a regulatory role of Mg2+ for cytosolic enzymes have been disappointing, mostly because of the underlying assumption that Mg2+ would operate as Ca2+ in modulating enzyme activity. While Ca2+ presents a major concentration gradient between cytoplasm and extracellular space and between cytoplasm and endoplasmic (and sarcoplasmic) reticulum lumen, free Mg2+ concentrations in the cytoplasm and the extracellular fluid are very similar, both being in the millimolar or sub-millimolar range. Consequently, an increase or a decrease in cytosolic Mg2+ level of an amplitude equivalent to those observed for Ca2+ will remain largely undetected by fluorescent or 31P-NMR techniques. Heretofore, a role of Mg2+ as transient regulator of cytosolic enzymes appears to be unlikely. It has to be noted that even under conditions in which hormonal and non-hormonal stimuli elicit major fluxes of Mg2+ across the cell plasma membrane in either direction, massive translocations of Mg2+ that increase or decrease total cellular Mg2+ content by 1–2mM (equivalent to 5%–10% of the total cell Mg2+ content) result in limited or no changes in cytosolic [Mg2+]i [193,211]. This disconnect can be explained by assuming that the source or destination of the transported Mg2+ is a cellular organelle, or a major binding site, or that Mg2+ is rapidly buffered by phosphonucleotides, phospholipids, or G proteins. Therefore, regulation of cellular functions by Mg2+ should not be necessarily expected to occur in the cytosol, like for Ca2+, but within organelles and plasma where Mg2+ concentration can rapidly increase or decrease by more than 20% [191,192].
The following pages will highlight what is known about the regulatory effect of extracellular or intracellular Mg2+ on cation channels activity at the plasma membrane level, as well as on mitochondria respiration and integrity following changes in Mg2+ concentration within the organelle.
6.1 Ca2+- and K+-Channels
One field in which a clear physiological role of Mg2+ has been identified is the regulation of ion channels, namely Ca2+ channels and K+ channels.
White and Hartzell were the first to report a regulatory effect of intracellular free Mg2+ on calcium channels [267]. These authors observed that increasing intracellular free [Mg2+]i from 0.3 to 3.0mM in cardiac ventricular myocytes by internal perfusion it decreased to a small extent basal L-type Ca2+-channels current (ICa) and by more than 50% the cAMP-mediated enhancement in ICa amplitude [267]. This effect was due to a direct action of Mg2+ on the phosphorylated channel or on dephosphorylation rate of the channel rather than to changes in cAMP concentration or cAMP-dependent phosphorylation [267]. Similar results were reported in guinea pig cardiac myocytes by Agus and Morad who observed Mg2+-induced block on Ca2+ current by direct effect on the inactivation state of the channel [268]. The block persisted in the presence of cAMP, and was not reversed by elevation of extracellular Ca2+ concentration or addition of catecholamine [268]. Similar effects of Mg2+ on Ca2+-channels have been observed in vascular smooth muscle cells and endothelial cells from human placenta [269], in which MgCl2 (but also MgSO4) acts at an extracellular site of the voltage-gated Ca2+ channels, and on T-type Ca2+-channels [270]. Recent evidence by Catterall and his group proposes a modulating effect of Mg2+ on the EF-hand motif located in the C-terminus of Cav1.2 channels [271].
Additional Ca2+ channels modulated by extracellular Mg2+ are the store-operated Ca2+ channels (SOC) and the store-operated calcium release-activated Ca2+ (CRAC) channels.
In the case of SOCs, Mg2+ prevents or reverses the vasoconstriction elicited by phenylephrine administration but not that induced by K+ depolarization [272]. This observation would suggest that Mg2+ contributes to regulate both the myogenic tone and the α1-adrenoceptor-induced, Ca2+-mediated vasoconstriction occurring through SOCs. This effect on the vasculature could be lost to a significant extent under hypertensive conditions, in which a decrease in plasma Mg2+ and a vasoconstriction hypertone have been observed.
As for CRACs, the effect of Mg2+ is more at the intracellular level [273]. CRAC channels are highly selective for Ca2+ under physiological conditions whereas removal of extracellular divalent cations makes them freely permeable to monovalent cations, in particular Na+. Experimental evidence indicates that intracellular Mg2+ can modulate the activity and selectivity of these channels therefore affecting monovalent cation permeability. A report by Prakriya and Lewis [273], however, argues that the channels modulated by intracellular Mg2+ are not CRAC channels, but a different class of channels that open when Mg2+ is washed out of the cytosol. These channels have been termed Mg2+-inhibited cation (MIC) channels, and could be distinguished by CRAC channels based upon modality of inhibition, regulation, ion permeation and selectivity [272]. These results, however, do not exclude the possibility of an inhibitory effect of intracellular Mg2+ on CRAC channels.
Potassium channels are also targets for Mg2+. Matsuda [274] has reported that cytosolic Mg2+ block the outward currents of inwardly rectifying K+ channels without affecting the inward currents. However, the Mg2+ block is achieved at a half-saturating concentration of 1.7μM, a concentration far from the physiological Mg2+ level in the cytoplasm. Hence, it is difficult to envision the occurrence of a similar regulatory effect under normal conditions without invoking Mg2+ micro compartmentalization. More realistic would be the occurrence of a regulatory role of intracellular Mg2+ on Kv channels in vascular smooth muscle cells [275]. In this case, in fact, an increase in intracellular Mg2+ - in a range of concentrations consistent with its physiological variations – slows down the kinetic of activation of the Kv channel, causing also inward rectification at positive membrane potentials and a shift in voltagedependent inactivation [275]. Intracellular Mg2+ also modulates large-conductance (BK type) Ca2+- dependent K+ channels either by blocking the pore of BK channels in a voltage-dependent manner, or by activating the channels independently of changes in Ca2+ and voltage through preferential binding to the channel open conformation at a site different from Ca2+ sites. Interestingly, Mg2+ may also bind to Ca2+ sites and competitively inhibit Ca2+-dependent activation [276].
The inhibitory effect of Mg2+ is not restricted to channels in the cell membrane. Experimental evidence by Bednarczyk et al. [277] indicates that Mg2+ within the mitochondrial matrix can modulate gating and conductance of mitochondrial KATP channels, which play a key role in promoting mitochondrial recovery and cell survival under ischemia/reperfusion conditions.
6.2 Mitochondrial Dehydrogenases
Mitochondria represent one of the major cellular Mg2+ pools. The concentration of Mg2+ within the organelle ranges between 14 to 16 mM [278], and circumstantial evidence from this [279] and other laboratories [257,280,281] suggests that Mg2+ can be mobilized from mitochondria under various conditions including hormonal stimulation through a not fully elucidated mechanism. Regulation of mitochondrial Mg2+ homeostasis has been analyzed in detail in several recent reviews [16,20,278], and we direct the interest reader to them for further information. In this section, we will focus on the role of intra- and extra-mitochondrial Mg2+ in modulating the activity of specific proteins within the organelle.
It is commonly accepted that changes in matrix Ca2+ can affect the activity rate of mitochondrial dehydrogenases and consequently the respiration rate [282,283]. Experimental evidence supports a similar role for Mg2+ as the activity of several mitochondrial dehydrogenases has been observed to increase within minutes from the application of hormonal or metabolic stimuli in the absence of a detectable increase in mitochondrial Ca2+ [284,285]. In particular, the results indicate that a decrease in mitochondrial Mg2+ increases several fold the activity of succinate and glutamate dehydrogenases while leaving unaffected the activity of α-ketoglutarate dehydrogenase and pyruvate dehydrogenase [117,118]. This evidence would support the concept that changes in matrix Mg2+ content (in combination with, or in alternative to changes in mitochondrial Ca2+) can control mitochondria respiration, at least under well defined conditions (Figure 1). In this respect, mitochondrial Mg2+ content appears to change quite significantly during transition from state 3 to state 4 [286], affecting the amplitude of mitochondria respiration. In addition, data from our laboratory [279], Zhang and Melvin [281], and Kubota et al. [257] all suggest that catecholamine stimulation can mobilize mitochondrial Mg2+ via a direct effect of cAMP on mitochondria. Hence, catecholamine administration will enhance mitochondrial respiration via cAMP-mediated modulation of mitochondrial Mg2+, which - in turn - will directly stimulate succinate and glutamate dehydrogenases while sensitizing other dehydrogenases to changes in mitochondrial Ca2+ concentrations.
Additional mitochondrial function modulated by changes in Mg2+ within the organelle are anion channels present in the mitochondrial membrane [287] as well as the opening of the permeability transition pore [288]. The mitochondrial inner membrane anion channel (IMAC) transports various anions, and is involved in regulating the organelle volume in conjunction with the K+/H+ antiporter. Although its fine regulation is not fully elucidated as yet, experimental evidence suggests that matrix Mg2+ and protons maintain the channel in its closed state [287]. Kinetic studies by Beavis and collaborators support a main role of Mg2+ in maintaining the channel in a conformation that would allow fine modulation by small changes in pH and proton distribution under physiological conditions [287]. The end results will be the maintenance of an optimal proton gradient and Δψ across the mitochondrial membrane, essential to retain proper organelle function and intra-mitochondrial Mg2+ content [115].
Perturbance of mitochondrial Δψ, Ca2+ content or ATP level all result in the opening of the permeability transition pore (PTP) in the inner mitochondrial membrane [288] and the rapid reequilibration of intra-mitochondrial ions and solutes down their concentration gradient. While it is well established that an increase in mitochondrial Ca2+ content facilitates PTP opening, an increase in mitochondrial Mg2+ antagonizes it. This effect can be appreciated well in yeasts, which do not possess a canonical PTP [289]. Creatine kinase also regulates PTP opening by tightly associating to the mitochondrial membrane and remaining in an active state [290]. Both the binding and activity state of the protein are Mg2+-dependent, and removal of Mg2+from the extra-mitochondrial environment results in a decline in creatine kinas activity and PTP opening [290].
Hence, it appears that Mg2+ regulates volume, ion composition, and ATP production within the mitochondrion, modulating the metabolic interaction between the organelle and the hosting cell.
6.3 Reticular G6Pase
The Endoplasmic Reticulum (ER) represents another major Mg2+ pool within the cell, with a total concentration estimated to be between 14 to 18mM [1]. Yet, no information is available about the modality by which Mg2+ enter and exit the organelle and how is buffered within the ER lumen. Limited information is also available about any major role of luminal Mg2+ on reticular functions other than protein synthesis [233].
Work by Volpe and collaborators [291,292], Gusev and Niggli [293], and Laver and Honen [294] suggests that cytosolic and perhaps luminal Mg2+ concentration have a major effect in limiting Ca2+ uptake into the ER/SR and its release from the organelle via IP3 [292] and ryanodine receptor (RyR) [294]. While a direct effect of Mg2+ on RyR opening has been observed [293, 294], it is unclear whether a similar effect takes place on the IP3 receptor. Recently, our laboratory has reported that cytosolic Mg2+ can have a regulatory role on the activity of reticular glucose 6-phosphatase (G6Pase) in liver cells [295]. This effect is biphasic, with an optimal stimulatory effect at ~0.5mM [Mg2+]i and an inhibitory effect at higher Mg2+ concentrations [295]. The Mg2+ effect appears to be at the level of the glucose 6-phosphate (G6Pi) transport component of the G6Pase enzymatic complex in that it is abolished by EDTA (as Mg2+ chelating agent) or taurocholic acid, which permeabilizes the ER membrane allowing for the direct delivery of G6Pi to the catalytic site of the G6Pase within the ER lumen bypassing the transport mechanism [295]. The modulating effect of Mg2+ on G6Pase hydrolysis rate is also observed in purified microsomes [296] isolated from livers of animals exposed for 2 weeks to a Mg2+ deficient diet [296]. Also in microsomes, the G6Pi hydrolysis rate is dynamically decreased by addition of Mg2+ at a concentration similar to that reported to be present in the hepatocyte cytoplasm, or increased by EDTA addition [296]. It is presently undetermined whether Mg2+ exerts a similar modulating effect on other reticular enzymatic activities.
6.4 Cell pH and Volume
Cells exposed to cyanide [154], fructose [212], hypoxia, [211,214], ethanol [213], or choline chloride [133] undergo a marked cellular acidification, decrease in cellular ATP content, and a large Mg2+ extrusion. This extrusion is the consequence of a decrease in buffering capacity (ATP loss) and binding affinity within the cytoplasm. Recently, Yamaguchi and Ishikawa [297] reported that a cytosolic [Mg2+]i of ~1mM (a physiological Mg2+ concentration measured in the cytosol of various cells [229,265]), inhibits by ~50% the current generated by the electrogenic Na+-HCO3− cotransporter NBCe1-B. Increasing the free Mg2+ concentration to 3 completely abolishes NBCe1-B current. This regulatory effect is exerted by Mg2+ and not Mg*ATP, and occurs at the N-terminus of the transporter [297]. It is still unresolved whether Mg2+ binds the N-terminus of the transporter directly or exerts its effects via an intermediate, Mg2+-modulated regulatory protein [297]. On the other hand, increasing cellular Mg2+ content has a stimulatory role on the expression of aquaporin 3 in CaCo-3 cells [298]. This isoform of aquaporin is highly expressed in the gastro-intestinal tract, in which it absorbs water, glycerol and urea. The effect of Mg2+ on aquaporin mRNA expression appears to involve cAMP/PKA/CREB signaling, as well as MEK1/2 and MSK1 [298], suggesting the occurrence of both short- and long-term regulation on the protein activity and expression. As aquaporin 3 is highly expressed in brain, erythrocytes, kidney, and skin, in addition to the gastro-intestinal tract, the occurrence of a modulating effect of Mg2+ on aquaporin 3 expression in these tissues may be highly relevant for various physiological and pathological conditions including brain swelling following traumatic injury. It remains to be determined whether Mg2+ exerts a similar regulatory role on other aquaporin isoforms.
Taken together, these two sets of information emphasize a role of Mg2+ in regulating directly pH, volume, and cation concentration, especially Na+ within the cell, and indirectly fatty acid metabolism via aqauporin 3-mediated glycerol accumulation.
6.5 Cell Cycle
Cell cycle [231,299,300], cell proliferation [301], and cell differentiation [302–304] have all been associated with the maintenance of an optimal Mg2+ level. Under conditions in which cellular Mg2+ accessibility is restricted or reduced, cell proliferation and cell cycle progression are markedly impaired (Figure 1) as well as cell differentiation [302–304]. The mechanisms by which a decrease in cellular Mg2+ content affects these cellular processes revolve around defective MAPKs [231] and p27 [300] signaling, increased oxidative stress level [302], and decreased Mg*ATP levels [233,304]. Because cellular Mg*ATP level is at a level optimal for protein synthesis [233], any alteration in this metabolic parameter will have major repercussion on the proper functioning of the cell. In addition, extracellular Mg2+ levels regulate integrins signaling, de facto modulating the interaction among cells and between cells and extracellular matrix [305]. All together, these observations support the notion that an optimal Mg2+ level is essential to guarantee cell cycle progression and retention of proper cell morphology and function, and prevent the undesired progression towards cell death or neoplastic destiny [306].
7. Conclusions
In the last few years, our understanding of the mechanisms regulating cellular and whole body Mg2+ homeostasis has advanced significantly. Although in terms of overall understanding the field still lags behind the knowledge available for other ions such as Ca2+, H+, K+ or Na+, the identification of Mg2+ channels and transport mechanisms in the membrane of cells and cellular organelles, and a better comprehension of the various signaling pathways and conditions regulating Mg2+ transport are providing new tools to address essential questions about the relevance of Mg2+ for various cell functions under physiological and pathological conditions.
Magnesium is essential to regulate numerous cellular functions and enzymes.
Magnesium is highly compartmentalized within cytoplasm and cellular organelles.
Metabolic or hormonal stimuli elicit major Mg2+ fluxes across the cell membrane.
A major loss of cellular and serum Mg2+ occurs in alcoholism and other diseases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Romani A, Scarpa A. Arch Biochem Biophys. 1992;298:1–12. doi: 10.1016/0003-9861(92)90086-c. [DOI] [PubMed] [Google Scholar]
- 2.Wolf FI, Torsello A, Fasanella S, Cittadini A. Mol Asp Med. 2003;24:11–26. doi: 10.1016/s0098-2997(02)00088-2. [DOI] [PubMed] [Google Scholar]
- 3.Wolf FI, Cittadini A. Mol Asp Med. 2003;24:3–9. doi: 10.1016/s0098-2997(02)00087-0. [DOI] [PubMed] [Google Scholar]
- 4.Jung DW, Apel L, Brierley GP. Biochemistry. 1990;29:4121–4128. doi: 10.1021/bi00469a015. [DOI] [PubMed] [Google Scholar]
- 5.Rutter GA, Osbaldeston NJ, McCormack JG, Denton RM. Biochem J. 1990;271:627–634. doi: 10.1042/bj2710627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Somlyo AV, McClellan G, Gonzalez-Serratos H, Somlyo AP. J Biol Chem. 1985;260:6801–6807. [PubMed] [Google Scholar]
- 7.Hofer AM, Machen TE. Proc Natl Acad Sci USA. 1993;90:2598–2602. doi: 10.1073/pnas.90.7.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scarpa A, Brinley FJ. Fed Proc. 1981;40:2646–252. [PubMed] [Google Scholar]
- 9.Lüthi D, Günzel D, McGuigan JA. Exp Physiol. 1999;84:231–252. [PubMed] [Google Scholar]
- 10.Flatman PW, Lew VL. J Physiol. 1981;315:421–446. doi: 10.1113/jphysiol.1981.sp013756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Günther T, Vormann J, McGuigan JA. Biochem Mol Biol Int. 1995;37:871–875. [PubMed] [Google Scholar]
- 12.Raftos JE, Lew VL, Flatman PW. Eur J Biochem. 1999;263:635–645. doi: 10.1046/j.1432-1327.1999.00506.x. [DOI] [PubMed] [Google Scholar]
- 13.Oki S, Ikura M, Zhang M. Biochemistry. 1997;36:4309–4316. doi: 10.1021/bi962759m. [DOI] [PubMed] [Google Scholar]
- 14.Wang S, George SE, Davis JP, Johnson JD. Biochemistry. 1998;37:14539–14544. doi: 10.1021/bi9814641. [DOI] [PubMed] [Google Scholar]
- 15.Allouche D, Parello J, Sanejouand YH. J Mol Biol. 1999;285:857–873. doi: 10.1006/jmbi.1998.2329. [DOI] [PubMed] [Google Scholar]
- 16.Ogoma Y, Kobayashi H, Fujii T, Kondo Y, Hachimori A, Shimizu T, Hatano M. Int J Biol Macromol. 1992;14:279–286. doi: 10.1016/s0141-8130(05)80041-8. [DOI] [PubMed] [Google Scholar]
- 17.Bogucka K, Wojtczak L. Biochem Biophys Res Commun. 1971;44:1330–1337. doi: 10.1016/s0006-291x(71)80231-0. [DOI] [PubMed] [Google Scholar]
- 18.Belge H, Gailly P, Schwaller B, Loffing J, Debaix H, Riveira-Munoz E, Beauwens R, Devogelaer JP, Hoenderop JG, Bindels RJ, Devuyst O. Proc Natl Acad Sci USA. 2007;104:14849–14854. doi: 10.1073/pnas.0702810104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lentner C, editor. Geigy Scientific Tables. Ciba-Geigy; Basel: Switzerland: 1984. [Google Scholar]
- 20.Flatman PW. J Membr Biol. 1984;80:1–14. doi: 10.1007/BF01868686. [DOI] [PubMed] [Google Scholar]
- 21.Romani A. Arch Biochem Biophys. 2007;458:90–102. doi: 10.1016/j.abb.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Polimeni PI, Page E. Recent advances in study in cardiac cells and metabolism. In: Dhalle NS, editor. IV. University Park Press; Baltimore: 1974. pp. 217–232. [Google Scholar]
- 23.Rogers TA, Haven FL, Mahan PE. J Natl Cancer Inst. 1960;25:887–888. doi: 10.1093/jnci/25.4.887. [DOI] [PubMed] [Google Scholar]
- 24.Rogers TA. J Cell Comp Physiol. 1961;57:119–121. doi: 10.1002/jcp.1030570209. [DOI] [PubMed] [Google Scholar]
- 25.Wolf FI, Di Francesco A, Covacci V, Cittadini A. Arch Biochem Biophys. 1997;344:397–403. doi: 10.1006/abbi.1997.0199. [DOI] [PubMed] [Google Scholar]
- 26.Maguire ME, Erdos JJ. J Biol Chem. 1978;253:6633–6636. [PubMed] [Google Scholar]
- 27.Grubbs RD, Maguire ME. Magnesium. 1987;6:113–127. [PubMed] [Google Scholar]
- 28.Romani AM, Maguire ME. Biometals. 2002;15:271–283. doi: 10.1023/a:1016082900838. [DOI] [PubMed] [Google Scholar]
- 29.Romani A, Scarpa A. Front Biosci. 2000;5:D720–D734. doi: 10.2741/romani. [DOI] [PubMed] [Google Scholar]
- 30.Günther T, Vormann J. Magnes Trace Elem. 1990;9:279–282. [PubMed] [Google Scholar]
- 31.Matsuura T, Kanayama Y, Inoue T, Takeda T, Morishima I. Biochim Biophys Acta. 1993;1220:31–36. doi: 10.1016/0167-4889(93)90093-5. [DOI] [PubMed] [Google Scholar]
- 32.Romani A, Scarpa A. Nature. 1990;346:841–844. doi: 10.1038/346841a0. [DOI] [PubMed] [Google Scholar]
- 33.Vormann J, Gunther T. Magnesium. 1987;6:220–224. [PubMed] [Google Scholar]
- 34.Romani A, Scarpa A. FEBS Lett. 1990;269:37–40. doi: 10.1016/0014-5793(90)81113-3. [DOI] [PubMed] [Google Scholar]
- 35.Gunther T, Vormann J, Hollriegl V. Magnes Bull. 1991;13:122–124. [Google Scholar]
- 36.Jakob A, Becker J, Schottli G, Fritzsch G. FEBS Lett. 1989;246:127–130. doi: 10.1016/0014-5793(89)80267-4. [DOI] [PubMed] [Google Scholar]
- 37.Fatholahi M, Lanoue K, Romani A, Scarpa A. Arch Biochem Biophys. 2000;374:395–401. doi: 10.1006/abbi.1999.1619. [DOI] [PubMed] [Google Scholar]
- 38.Kubota T, Shindo Y, Tokuno K, Komatsu H, Ogawa H, Kudo S, Kitamura Y, Suzuki K, Oka K. Biochim Biophys Acta. 2005;1744:19–28. doi: 10.1016/j.bbamcr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Kehres DG, Lawyer CH, Maguire ME. Microb Comp Genomics. 1998;3:151–159. doi: 10.1089/omi.1.1998.3.151. [DOI] [PubMed] [Google Scholar]
- 40.Moncrief MB, Maguire ME. J Biol Inorg Chem. 1999;4:523–527. doi: 10.1007/s007750050374. [DOI] [PubMed] [Google Scholar]
- 41.Preston RR. Science. 1990;250:285–288. doi: 10.1126/science.2218533. [DOI] [PubMed] [Google Scholar]
- 42.Nadler MJ, Hermosura MC, Inabe K, Perraud A-L, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 43.Schlingmann KP, Weber S, Peters M, Niemann NL, Vitzthum H, Klingel K, Kratz E, Haddad M, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M. Nat Genet. 2002;31:166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi H, Matsushita M, Nairn A, Kuriyan J. Mol Cell. 2001;7:1047–1057. doi: 10.1016/s1097-2765(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 45.Ryazanova LV, Pavur KS, Petrov AN, Dorovkov MV, Ryazanov AG. Mol Biol (Mosk) 2001;35:321–332. [PubMed] [Google Scholar]
- 46.Ryazanov AG. FEBS Lett. 2002;514:26–29. doi: 10.1016/s0014-5793(02)02299-8. [DOI] [PubMed] [Google Scholar]
- 47.Runnels LW, Yue L, Clapham DE. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 48.Bessac BF, Fleig A. J Physiol. 2007;582:1073–1086. doi: 10.1113/jphysiol.2007.130534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. J Gen Physiol. 2003;121:49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voels T, Nilliues B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 51.Chubanov V, Waldegger S, Mederos Y, Schnitzler M, Vitzthum H, Sassen MC, Seyberth HW, Konrad M, Gudermann T. Proc Natl Acad Sci USA. 2004;101:2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitz C, Dorovkov MV, Zhao X, Davenport BJ, Ryazanov AG, Perraud A-L. J Biol Chem. 2005;280:37763–37771. doi: 10.1074/jbc.M509175200. [DOI] [PubMed] [Google Scholar]
- 53.Chubanov V, Gudermann T, Schlingmann KP. Pflugers Arch. 2005;451:228–234. doi: 10.1007/s00424-005-1470-y. [DOI] [PubMed] [Google Scholar]
- 54.Li M, Jiang J, Yue L. J Gen Physiol. 2006;127:525–537. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li M, Du J, Jiang J, Ratzan W, Su LT, Runnels LW, Yue L. J Biol Chem. 2007;282:25817–25830. doi: 10.1074/jbc.M608972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gwanyanya A, Amuzescu B, Zakharov SI, Macianskiene R, Sipido KR, Bolotina VM, Vereecke J, Mubagwa K. J Physiol. 2004;559:761–776. doi: 10.1113/jphysiol.2004.067637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang J, Li M, Yue L. J Gen Physiol. 2005;126:137–150. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Runnels LW, Yue L, Clapham DE. Nat Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 59.Takezawa R, Schmitz C, Demeuse P, Scharenberg AM, Penner R, Fleig A. Proc Natl Acad Sci USA. 2004;101:6009–6014. doi: 10.1073/pnas.0307565101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langeslag M, Clark K, Moolenaar WH, van Leeuwen FN, Jalink K. J Biol Chem. 2007;282:232–239. doi: 10.1074/jbc.M605300200. [DOI] [PubMed] [Google Scholar]
- 61.Macianskiene R, Gwanyanya A, Vereecke J, Mubagwa K. Cell Physiol Biochem. 2008;22:109–118. doi: 10.1159/000149788. [DOI] [PubMed] [Google Scholar]
- 62.Gwanyanya A, Sipido KR, Vereecke J, Mubagwa K. Am J Physiol. 2006;291:C627–C635. doi: 10.1152/ajpcell.00074.2006. [DOI] [PubMed] [Google Scholar]
- 63.Touyz RM, He Y, Montezano ACI, Yao G, Chubanov V, Gudermann T, Callera GE. Am J Physiol. 2006;290:R73–R78. doi: 10.1152/ajpregu.00515.2005. [DOI] [PubMed] [Google Scholar]
- 64.Sahni J, Scharenberg AM. Cell Metab. 2008;8:84–93. doi: 10.1016/j.cmet.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sahni J, Tamura R, Sweet IR, Scharenberg AM. Cell Cycle. 2010;9:3565–3574. doi: 10.4161/cc.9.17.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Middelbeek J, Clark K, Venselaar HV, Huynen MA, van Leeuwen FN. Cell Mol Life Sci. 2010;67:875–890. doi: 10.1007/s00018-009-0215-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmitz C, Perraud A-L, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- 68.Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, Moolenaar WH, Jalink K, van Leeuwen FN. EMBO J. 2006;25:290–301. doi: 10.1038/sj.emboj.7600931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, Mazur A, Fleig A, Ryazanov AG. Nature Commun. 2010;1:109. doi: 10.1038/ncomms1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clark K, Middelbeek J, Morrice NA, Figdor CG, Lasonder E, van Leeuwen FN. PLoS One. 2008;3:e1876, 1–10. doi: 10.1371/journal.pone.0001876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dorovkov MV, Ryazanov AG. J Biol Chem. 2004;279:50643–50646. doi: 10.1074/jbc.C400441200. [DOI] [PubMed] [Google Scholar]
- 72.Clark K, Middelbeek J, Lasonder E, Dulyaninova NG, Morrice NA, Ryazanov AG, Bresnick AR, Figdor CG, van Leeuwen FN. J Mol Biol. 2008;378:790–803. doi: 10.1016/j.jmb.2008.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su L-T, Agapito MA, Li M, Simpson W, Huttenlocher A, Habas R, Yue L, Runnels LW. J Biol Chem. 2006;281:11260–11270. doi: 10.1074/jbc.M512885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paravicini TM, Yogi A, Mazur A, Touyz RM. Hypertension. 2009;53:423–429. doi: 10.1161/HYPERTENSIONAHA.108.124651. [DOI] [PubMed] [Google Scholar]
- 75.Parente L, Solito E. Inflamm Res. 2004;53:125–132. doi: 10.1007/s00011-003-1235-z. [DOI] [PubMed] [Google Scholar]
- 76.Montezano AC, Zimmerman D, Yusuf H, Burger D, Chignalia AZ, Wadhera V, van Leeuwen FN, Touyz RM. Hypertension. 2010;56:453–462. doi: 10.1161/HYPERTENSIONAHA.110.152058. [DOI] [PubMed] [Google Scholar]
- 77.Perraud A-L, Zhao X, Ryazanov AG, Schmitz C. Cell Signal. 2011;23:587–593. doi: 10.1016/j.cellsig.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 79.Wei WL, Sun HS, Olah ME, Sun X, Czerwinska E, Czerwinski W, Mori Y, Orser BA, Xiong ZG, Jackson MF, Tymianski M, MacDonald JF. Proc Natl Acad Sci USA. 2007;104:16323–16328. doi: 10.1073/pnas.0701149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang H, Tian SL, Zeng Y, Li LL, Shi J. Brain Res Bull. 2008;76:124–130. doi: 10.1016/j.brainresbull.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 81.Chen H-S, Xie J, Zhang Z, Su L-T, Yue L, Runnels LW. PLoS One. 2010;5:e11161. doi: 10.1371/journal.pone.0011161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brauchi S, Krapivinsky G, Krapivinsky L, Clapham DE. Proc Natl Acad Sci USA. 2008;105:8304–8308. doi: 10.1073/pnas.0800881105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abed E, Moreau R. Cell Prolif. 2007;40:849–865. doi: 10.1111/j.1365-2184.2007.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abed E, Martineau C, Moreau R. Calcif Tissue Int. 2011;88:246–253. doi: 10.1007/s00223-010-9455-z. [DOI] [PubMed] [Google Scholar]
- 85.Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC. Nat Genet. 2002;31:171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 87.Walder RY, Yang B, Stokes JB, Kirby PA, Cao X, Shi P, Searby CC, Husted RF, Sheffield VC. Human Mol Gen. 2009;18:4367–4375. doi: 10.1093/hmg/ddp392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woudenberg-Vrenken TE, Sukinta A, van der Kemp AW, Bindels RJ, Hoenderop JG. Nephron Physiol. 2011;117:p11–p19. doi: 10.1159/000320580. [DOI] [PubMed] [Google Scholar]
- 89.Thébault S, Cao G, Venselaar H, Xi Q, Bindels RJ, Hoenderop JG. J Biol Chem. 2008;283:19999–20007. doi: 10.1074/jbc.M800167200. [DOI] [PubMed] [Google Scholar]
- 90.Cao G, van der Wijst J, van der Kemp A, van Zeeland F, Bindels RJ, Hoenderop JG. J Biol Chem. 2009;284:14788–14795. doi: 10.1074/jbc.M808752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Groenestege WM, Hoenderop JG, van den Heuvel L, Knoers N, Bindels RJ. J Am Soc Nephrol. 2006;17:1035–1043. doi: 10.1681/ASN.2005070700. [DOI] [PubMed] [Google Scholar]
- 92.Rondón LJ, Groenestege WM, Rayssiguier Y, Mazur A. Am J Physiol. 2008;294:R2001–R2007. doi: 10.1152/ajpregu.00153.2007. [DOI] [PubMed] [Google Scholar]
- 93.Cao G, Thébault S, van der Wijst J, van der Kemp A, Lasonder E, Bindels RJ, Hoenderop JG. Curr Biol. 2008;18:168–176. doi: 10.1016/j.cub.2007.12.058. [DOI] [PubMed] [Google Scholar]
- 94.Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, van den Heuvel LP, van Cutsem E, Hoenderop JG, Knoers NV, Bindels RJ. J Clin Invest. 2007;117:2260–2267. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, van Cutsem E. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 96.Dimke H, van der Wjist J, Alexander TR, Mejier IM, Mulder GM, van Goor H, Tejpar S, Hoenderop JG, Bindels RJ. J Am Soc Nephrol. 2010;21:1309–1316. doi: 10.1681/ASN.2009111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ikari A, Okude C, Sawada H, Yamazaki Y, Sugatani J, Miwa M. Biochem Biophys Res Commun. 2008;369:1129–1133. doi: 10.1016/j.bbrc.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 98.Ikari A, Sanada A, Okude C, Sawada H, Yamazaki Y, Sugatani J, Miwa M. J Cell Physiol. 2010;222:481–487. doi: 10.1002/jcp.21988. [DOI] [PubMed] [Google Scholar]
- 99.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodrigues-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Science. 1999;285:103–1106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 100.Lal-Nag M, Morin PJ. Genome Biol. 2009;10:235. doi: 10.1186/gb-2009-10-8-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kausalya PJ, Amasheh S, Gunzel D, Wurps H, Muller D, Fromm M, Hunziker W. J Clin Invest. 2006;116:878–891. doi: 10.1172/JCI26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hou J, Paul DL, Goodenough DA. J Cell Sci. 2005;118:5109–118. doi: 10.1242/jcs.02631. [DOI] [PubMed] [Google Scholar]
- 103.Ikari A, Matsumoto S, Harada H, Takagi K, Hayashi H, Suzuki Y, Degawa M, Miwa M. J Cell Sci. 2006;119:1781–1789. doi: 10.1242/jcs.02901. [DOI] [PubMed] [Google Scholar]
- 104.Efrati E, Arsentiev-Rozenfeld J, Zelikovic I. Am J Physiol. 2005;288:F272–F283. doi: 10.1152/ajprenal.00021.2004. [DOI] [PubMed] [Google Scholar]
- 105.Muller D, Kausalya PJ, Claverie-Martin F, Meij IC, Eggert P, Garcia-Nieto V, Hunziker W. Am J Hum Genet. 2003;73:1293–1301. doi: 10.1086/380418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Khan MA, Conigrave AD. Br J Pharmacol. 2010;159:1039–1050. doi: 10.1111/j.1476-5381.2009.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hou J, Renigunta A, Gomes AS, Hou M, Paul DL, Waldegger S, Goodenough DA. Proc Natl Acad Sci USA. 2009;106:15350–15355. doi: 10.1073/pnas.0907724106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goytain A, Quamme GA. BMC Genomics. 2005;6:48. doi: 10.1186/1471-2164-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shibatani T, David LL, McCormack AL, Frueh K, Skach WR. Biochemistry. 2005;44:5982–5992. doi: 10.1021/bi047328f. [DOI] [PubMed] [Google Scholar]
- 110.Zhou H, Clapham DE. Proc Natl Acad Sci USA. 2009;106:15750–15755. doi: 10.1073/pnas.0908332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wiesenberger G, Waldherr M, Schweyen RJ. J Biol Chem. 1992;267:6963–6969. [PubMed] [Google Scholar]
- 112.Bui DM, Gregan J, Jarosch E, Ragnini A, Schweyen RJ. J Biol Chem. 1999;274:20438–20443. doi: 10.1074/jbc.274.29.20438. [DOI] [PubMed] [Google Scholar]
- 113.Kolisek M, Zsurka G, Samaj J, Weghuber J, Schweyen RJ, Schweigel M. EMBO J. 2003;22:1235–1244. doi: 10.1093/emboj/cdg122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Piskacek M, Zotova L, Zsurka G, Schweyen RJ. J Cell Mol Med. 2009;13:693–700. doi: 10.1111/j.1582-4934.2008.00328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Akerman KE. J Bioenerg Biomembr. 1981;13:133–139. doi: 10.1007/BF00763835. [DOI] [PubMed] [Google Scholar]
- 116.Zsurka G, Gregan J, Schweyen RJ. Genomics. 2001;72:158–168. doi: 10.1006/geno.2000.6407. [DOI] [PubMed] [Google Scholar]
- 117.Panov A, Scarpa A. Biochemistry. 1996;35:427–432. doi: 10.1021/bi952101t. [DOI] [PubMed] [Google Scholar]
- 118.Panov A, Scarpa A. Biochemistry. 1996;35:12849–12856. doi: 10.1021/bi960139f. [DOI] [PubMed] [Google Scholar]
- 119.Goytain A, Quamme GA. Am J Physiol. 2008;294:C495–502. doi: 10.1152/ajpcell.00238.2007. [DOI] [PubMed] [Google Scholar]
- 120.Schmitz C, Deason F, Perraud A-L. Magnes Res. 2007;20:6–18. [PubMed] [Google Scholar]
- 121.Alexander RT, Hoenderop JG, Bindels RJ. J Am Soc Nephrol. 2008;19:1451–1458. doi: 10.1681/ASN.2008010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Quamme GA. Am J Physiol. 2010;298:407–429. [Google Scholar]
- 123.Gunther T, Vormann J, Forster R. Biochem Biophys Res Commun. 1984;119:124–131. doi: 10.1016/0006-291x(84)91627-9. [DOI] [PubMed] [Google Scholar]
- 124.Gunther T, Vormann J. Biochem BiophysRes Commun. 1985;130:540–545. doi: 10.1016/0006-291x(85)90450-4. [DOI] [PubMed] [Google Scholar]
- 125.Feray J-C, Garay R. Biochim Biophys Acta. 1986;856:76–84. doi: 10.1016/0005-2736(86)90012-x. [DOI] [PubMed] [Google Scholar]
- 126.Flatman PW, Smith LM. J Physiol. 1990;431:11–25. doi: 10.1113/jphysiol.1990.sp018318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu W, Willis JS. J Membr Biol. 1994;141:277–287. doi: 10.1007/BF00235137. [DOI] [PubMed] [Google Scholar]
- 128.Ludi H, Schatzmann HJ. J Physiol. 1987;390:367–382. doi: 10.1113/jphysiol.1987.sp016706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vormann J, Magdorf K, Günther T, Wahn U. Eur J Clin Chem Clin Biochem. 1994;32:833–836. doi: 10.1515/cclm.1994.32.11.833. [DOI] [PubMed] [Google Scholar]
- 130.Gunther T, Vormann J. FEBS Lett. 1992;297:132–134. doi: 10.1016/0014-5793(92)80343-f. [DOI] [PubMed] [Google Scholar]
- 131.Wolf FI, Di Francesco A, Covacci V, Corda D, Cittadini A. Arch Biochem Biophys. 1996;331:194–200. doi: 10.1006/abbi.1996.0298. [DOI] [PubMed] [Google Scholar]
- 132.Romani A, Marfella C, Scarpa A. Circ Res. 1993;72:1139–1148. doi: 10.1161/01.res.72.6.1139. [DOI] [PubMed] [Google Scholar]
- 133.Romani A, Marfella C, Scarpa A. J Biol Chem. 1993;268:15489–15495. [PubMed] [Google Scholar]
- 134.Fagan TE, Romani a. Am J Physiol. 2000;279:G943–G950. doi: 10.1152/ajpgi.2000.279.5.G943. [DOI] [PubMed] [Google Scholar]
- 135.Fagan TE, Romani A. Am J Physiol. 2001;280:G1145–G1156. doi: 10.1152/ajpgi.2001.280.6.G1145. [DOI] [PubMed] [Google Scholar]
- 136.Cefaratti C, Ruse C. Mol Cell Biochem. 2007;297:209–214. doi: 10.1007/s11010-006-9325-1. [DOI] [PubMed] [Google Scholar]
- 137.Cefaratti C, Romani A. Mol Cell Biochem. 2007;303:63–72. doi: 10.1007/s11010-007-9456-z. [DOI] [PubMed] [Google Scholar]
- 138.Cefaratti C, Romani A. Mol Cell Biochem. 2011;351:133–142. doi: 10.1007/s11010-011-0720-x. [DOI] [PubMed] [Google Scholar]
- 139.Ebel H, Gunther T. FEBS Lett. 2003;543:103–107. doi: 10.1016/s0014-5793(03)00417-4. [DOI] [PubMed] [Google Scholar]
- 140.Rasgado-Flores H, Gonzalez-Serratos H, DeSantiago J. Am J Physiol. 1994;266:C1112–C1117. doi: 10.1152/ajpcell.1994.266.4.C1112. [DOI] [PubMed] [Google Scholar]
- 141.Aromataris EC, Roberts ML, Barritt GJ, Rychkov GY. Glucagon activates Ca2+ and Cl− channels in rat hepatocytes. J Physiol. 2006;573.3:611–625. doi: 10.1113/jphysiol.2006.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tashiro M, Konishi M. Biophys J. 1997;73:3371–3384. doi: 10.1016/S0006-3495(97)78361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gunther T. Magnes Bull. 1996;18:2–6. [Google Scholar]
- 144.Cefaratti C, Romani A, Scarpa A. Am J Physiol. 1998;275:C995–C1008. doi: 10.1152/ajpcell.1998.275.4.C995. [DOI] [PubMed] [Google Scholar]
- 145.Feray J-C, Garay R. Naunyn-Schmied Arch Pharmacol. 1988;338:332–337. doi: 10.1007/BF00173409. [DOI] [PubMed] [Google Scholar]
- 146.Schweigel M, Vormann J, Martens H. Am J Physiol. 2000;278:G400–G408. doi: 10.1152/ajpgi.2000.278.3.G400. [DOI] [PubMed] [Google Scholar]
- 147.Günther T. Miner Electrolyte Metab. 1993;19:259–265. [PubMed] [Google Scholar]
- 148.Ebel H, Hollstein M, Gunther T. Biochim Biophys Acta. 2002;1559:135–144. doi: 10.1016/s0005-2736(01)00445-x. [DOI] [PubMed] [Google Scholar]
- 149.Keenan D, Romani A, Scarpa A. FEBS Lett. 1996;395:241–244. doi: 10.1016/0014-5793(96)01051-4. [DOI] [PubMed] [Google Scholar]
- 150.Günther T, Vormann J, Höllriegl V. Biochim Biophys Acta. 1990;1023:455–461. doi: 10.1016/0005-2736(90)90139-f. [DOI] [PubMed] [Google Scholar]
- 151.Ebel H, Hollstein M, Günther T. Biochim Biophys Acta. 2004;1667:132–140. doi: 10.1016/j.bbamem.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 152.Shaul O, Hilgemann DW, de-Almeida-Engler J, Van Montagu M, Inz D, Galili G. EMBO J. 1999;18:3973–3980. doi: 10.1093/emboj/18.14.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Günther T, Vormann J. FEBS Lett. 1990;265:55–58. doi: 10.1016/0014-5793(90)80882-j. [DOI] [PubMed] [Google Scholar]
- 154.Dalal P, Romani A. Metabolism. 2010;59:166301671. doi: 10.1016/j.metabol.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Günzel D, Schlue WR. J Physiol. 1996;491:595–608. doi: 10.1113/jphysiol.1996.sp021242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Smith RL, Thompson LJ, Maguire ME. J Bacteriol. 1995;177:1233–1238. doi: 10.1128/jb.177.5.1233-1238.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wabakken T, Rian E, Kveine M, Aasheim H-C. Biochem Biophys Res Commun. 2003;306:718–724. doi: 10.1016/s0006-291x(03)01030-1. [DOI] [PubMed] [Google Scholar]
- 158.Goytain A, Quamme GA. Physiol Genomics. 2005;21:337–342. doi: 10.1152/physiolgenomics.00261.2004. [DOI] [PubMed] [Google Scholar]
- 159.Quednau BD, Nicoll DA, Philipson KD. Pflugers Arch. 2004;447:543–548. doi: 10.1007/s00424-003-1065-4. [DOI] [PubMed] [Google Scholar]
- 160.Kolisek M, Launay P, Beck A, Sponder G, Serafini N, Brenkus M, Froschauer EM, Martens H, Fleig A, Schweigel M. J Biol Chem. 2008;283:16235–16247. doi: 10.1074/jbc.M707276200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Goytain A, Quamme GA. Biochem Biophys Res Commun. 2005;330:701–705. doi: 10.1016/j.bbrc.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 162.Sahni J, Nelson B, Scharenberg AM. Biochem J. 2007;401:505–513. doi: 10.1042/BJ20060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang CY, Davoodi-Semiromi A, Shi JD, Yang P, Huang YQ, Agundez JA, Moran JM, Ochoa B, Hawkins-Lee B, She JX. Am J Med Genet A. 2003;119:9–14. doi: 10.1002/ajmg.a.20042. [DOI] [PubMed] [Google Scholar]
- 164.Wang CY, Shi JD, Yang P, Kumar PG, Li QZ, Run QG, Su YC, Scott HS, Kao KJ, She JX. Gene. 2003;306:37–44. doi: 10.1016/s0378-1119(02)01210-6. [DOI] [PubMed] [Google Scholar]
- 165.Wang CY, Yang P, Chi JD, Purohit S, Guo D, An H, Gu JG, Ling J, Dong Z, She JX. BMC Genomics. 2004;5:7. doi: 10.1186/1471-2164-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kehres DG, Maguire ME. Biometals. 2002;15:261–270. doi: 10.1023/a:1016078832697. [DOI] [PubMed] [Google Scholar]
- 167.Goytain A, Quamme GA. Physiol Genomics. 2005;22:382–389. doi: 10.1152/physiolgenomics.00058.2005. [DOI] [PubMed] [Google Scholar]
- 168.Butler MG. Am J Med Genet. 1990;35:319–332. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Goytain A, Hines RM, El-Husseini A, Quamme GA. J Biol Chem. 2007;282:8060–8068. doi: 10.1074/jbc.M610314200. [DOI] [PubMed] [Google Scholar]
- 170.Goytain A, Hines RM, Quamme GA. Am J Physiol. 2008;295:C944–C953. doi: 10.1152/ajpcell.00091.2008. [DOI] [PubMed] [Google Scholar]
- 171.Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK. Am J Hum Genet. 2003;73:967–971. doi: 10.1086/378817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Goytain A, Hines RM, Quamme GA. J Biol Chem. 2008;283:33365–33374. doi: 10.1074/jbc.M801469200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li S-H, Li X-J. Trends Genet. 2004;20:146–152. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 174.Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, Hayden MR. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cefaratti C, Romani A, Scarpa A. J Biol Chem. 2000;275:3772–3780. doi: 10.1074/jbc.275.6.3772. [DOI] [PubMed] [Google Scholar]
- 176.Moore EW. Hepatology. 1990;12:206S–214S. [PubMed] [Google Scholar]
- 177.Juttner R, Ebel H. Biochim Biophys Acta. 1998;1370:51–63. doi: 10.1016/s0005-2736(97)00242-3. [DOI] [PubMed] [Google Scholar]
- 178.Baillien M, Cogneau M. Magnesium. 1995;8:331–339. [PubMed] [Google Scholar]
- 179.Schatzmann HJ. Biochim Biophys Acta. 1993;1148:15–18. doi: 10.1016/0005-2736(93)90155-s. [DOI] [PubMed] [Google Scholar]
- 180.Baillien M, Wang H, Cogneau M. Magnesium. 1995;8:315–329. [PubMed] [Google Scholar]
- 181.Elliot DA, Rizack MA. J Biol Chem. 1974;249:3985–3990. [PubMed] [Google Scholar]
- 182.Bird SJ, Maguire ME. J Biol Chem. 1978;253:8826–8834. [PubMed] [Google Scholar]
- 183.Erdos JJ, Maguire ME. Mol Pharmacol. 1980;18:379–383. [PubMed] [Google Scholar]
- 184.Erdos JJ, Maguire ME. J Physiol. 1983;337:351–371. doi: 10.1113/jphysiol.1983.sp014628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Grubbs RD, Wetherill CA, Kutschke K, Maguire ME. Am J Physiol. 1984;248:C51–C57. doi: 10.1152/ajpcell.1985.248.1.C51. [DOI] [PubMed] [Google Scholar]
- 186.Maguire ME, Erdos JJ. J Biol Chem. 1978;253:6633–6636. [PubMed] [Google Scholar]
- 187.Maguire ME, Erdos JJ. J Biol Chem. 1980;255:1030–1035. [PubMed] [Google Scholar]
- 188.Grubbs RD, Collins SD, Maguire ME. J Biol Chem. 1985;259:12184–12192. [PubMed] [Google Scholar]
- 189.Howarth FC, Waring J, Hustler BI, Singh J. Magnes Res. 1994;7:187–197. [PubMed] [Google Scholar]
- 190.Wolf FI, Di Francesco A, Cittadini A. Arch Biochem Biophys. 1994;308:335–341. doi: 10.1006/abbi.1994.1048. [DOI] [PubMed] [Google Scholar]
- 191.Keenan D, Romani A, Scarpa A. Circ Res. 1995;77:973–983. doi: 10.1161/01.res.77.5.973. [DOI] [PubMed] [Google Scholar]
- 192.Gunther T, Vormann J. Magnes Bull. 1992;14:122–125. [Google Scholar]
- 193.Romani AM, Matthews VD, Scarpa A. Circ Res. 2000;86:326–333. doi: 10.1161/01.res.86.3.326. [DOI] [PubMed] [Google Scholar]
- 194.Amano T, Matsubara T, Watanabe J, Nakayama S, Hotta N. Brit J Pharmacol. 2000;130:731–738. doi: 10.1038/sj.bjp.0703361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Karoor V, Baltensperger K, Paul H, Czech MC, Malbon CC. J Biol Chem. 1995;270:25305–25308. doi: 10.1074/jbc.270.43.25305. [DOI] [PubMed] [Google Scholar]
- 196.Smoake JA, Moy G-MM, Fang B, Solomon SS. Arch Biochem Biophys. 1995;323:223–232. doi: 10.1006/abbi.1995.9971. [DOI] [PubMed] [Google Scholar]
- 197.Ferreira A, Rivera A, Romero JR. J Cell Physiol. 2004;199:434–440. doi: 10.1002/jcp.10463. [DOI] [PubMed] [Google Scholar]
- 198.Wiesenberger G, Steinleitner K, Malli R, Graier WF, Vormann J, Schweyen RJ, Stadler JA. Eukaryot Cell. 2007;6:592–599. doi: 10.1128/EC.00382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang M, Berlin JR. Am J Physiol. 2006;291:C83–C92. doi: 10.1152/ajpcell.00579.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Torres LM, Youngner J, Romani A. Am J Physiol. 2005;288:G195–G206. doi: 10.1152/ajpgi.00488.2003. [DOI] [PubMed] [Google Scholar]
- 201.Fagan TE, Cefaratti C, Romani A. Am J Physiol. 2004;286:E184–E193. doi: 10.1152/ajpendo.00200.2003. [DOI] [PubMed] [Google Scholar]
- 202.Henquin JC, Tamagawa T, Nenquin M, Cogneau M. Nature. 1983;301:73–74. doi: 10.1038/301073a0. [DOI] [PubMed] [Google Scholar]
- 203.Garfinkel D, Garfinkel L. Magnesium. 1988;7:249–261. [PubMed] [Google Scholar]
- 204.Otto M, Heinrich R, Kuhn B, Jacobasch G. Eur J Biochem. 1974;49:169–178. doi: 10.1111/j.1432-1033.1974.tb03822.x. [DOI] [PubMed] [Google Scholar]
- 205.Resnick LM, Altura BT, Gupta RK, Laragh JH, Alderman MH, Altura BM. Diabetologia. 1993;36:767–770. doi: 10.1007/BF00401149. [DOI] [PubMed] [Google Scholar]
- 206.Resnick LM. Am J Hypertens. 1993;6:123S–134S. doi: 10.1093/ajh/6.4s.123s. [DOI] [PubMed] [Google Scholar]
- 207.Barbagallo M, Dominguez LJ. Arch Biochem Biophys. 2007;458:40–47. doi: 10.1016/j.abb.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 208.Reed G, Cefaratti C, Berti-Mattera LN, Romani A. J Cell Biochem. 2008;104:1034–1053. doi: 10.1002/jcb.21690. [DOI] [PubMed] [Google Scholar]
- 209.Cefaratti C, McKinnis A, Romani A. Mol Cell Biochem. 2004;262:145–154. doi: 10.1023/b:mcbi.0000038230.86485.52. [DOI] [PubMed] [Google Scholar]
- 210.Suarez A, Pulido N, Casla A, Casanova B, Arrieta FJ, Rovira A. Diabetologia. 1995;38:1262–1270. doi: 10.1007/BF00401757. [DOI] [PubMed] [Google Scholar]
- 211.Harman AW, Nieminen AL, Lemasters JJ, Herman B. Biochem Biophys Res Commun. 1990;170:477–483. doi: 10.1016/0006-291x(90)92116-h. [DOI] [PubMed] [Google Scholar]
- 212.Gaussin V, Gailly P, Gillis J-M, Hue L. Biochem J. 1997;326:823–827. doi: 10.1042/bj3260823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tessman PA, Romani A. Am J Physiol. 1998;275:G1106–G1116. doi: 10.1152/ajpgi.1998.275.5.G1106. [DOI] [PubMed] [Google Scholar]
- 214.Gasbarrini A, Borle AB, Farghali H, Bender C, Francavilla A, van Thiel D. J Biol Chem. 1992;267:6654–6663. [PubMed] [Google Scholar]
- 215.Hwa J, Romani A, Marfella C, Scarpa A. Biophys J. 1993;63:A307. [Google Scholar]
- 216.Di Polo R, Beauge’ L. Biochim Biophys Acta. 1988;946:424–428. doi: 10.1016/0005-2736(88)90418-x. [DOI] [PubMed] [Google Scholar]
- 217.Günther T, Höllriegl V. Biochim Biophys Acta. 1993;1149:49–54. doi: 10.1016/0005-2736(93)90023-s. [DOI] [PubMed] [Google Scholar]
- 218.Touyz RM, Schiffrin EL. J Biol Chem. 1996;271:24353–24358. doi: 10.1074/jbc.271.40.24353. [DOI] [PubMed] [Google Scholar]
- 219.Romani A, Marfella C, Scarpa A. FEBS Lett. 1992;296:135–140. doi: 10.1016/0014-5793(92)80364-m. [DOI] [PubMed] [Google Scholar]
- 220.Hwang DL, Yen CF, Nadler JL. J Clin Endocrinol Metab. 1993;76:549–553. doi: 10.1210/jcem.76.3.8445010. [DOI] [PubMed] [Google Scholar]
- 221.Grubbs RD, Maguire ME. J Biol Chem. 1986;261:12550–12554. [PubMed] [Google Scholar]
- 222.Ishijima S, Tatibana M. J Biochem. 1994;115:730–737. doi: 10.1093/oxfordjournals.jbchem.a124403. [DOI] [PubMed] [Google Scholar]
- 223.Csermely P, Fodor P, Somogyi J. Carcinogenesis. 1987;8:1663–1666. doi: 10.1093/carcin/8.11.1663. [DOI] [PubMed] [Google Scholar]
- 224.Günther T, Vormann J. Biochim Biophys Acta. 1995;1234:105–110. doi: 10.1016/0005-2736(94)00267-s. [DOI] [PubMed] [Google Scholar]
- 225.Yang ZW, Wang J, Zheng T, Altura BT, Altura BM. Stroke. 2001;32:249–257. doi: 10.1161/01.str.32.1.249. [DOI] [PubMed] [Google Scholar]
- 226.Torres LM, Konopnika B, Berti-Mattera LN, Liedtke C, Romani A. Alcohol Clin Exp Res. 2010;34:1659–1669. doi: 10.1111/j.1530-0277.2010.01252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tang EY, Parker PJ, Beattie J, Houslay MD. FEBS Lett. 1993;326:117–123. doi: 10.1016/0014-5793(93)81774-t. [DOI] [PubMed] [Google Scholar]
- 228.Konno Y, Ohno S, Akita Y, Kawasaki H, Suzuki K. J Biochem. 1989;106:673–678. doi: 10.1093/oxfordjournals.jbchem.a122915. [DOI] [PubMed] [Google Scholar]
- 229.Corkey BE, Duszynski J, Rich TL, Matchinsky B, Williamson JR. J Biol Chem. 1986;261:2567–2574. [PubMed] [Google Scholar]
- 230.Yang Z, Wang J, Altura BT, Altura BM. Pflugers Arch. 2000;439:240–247. doi: 10.1007/s004249900179. [DOI] [PubMed] [Google Scholar]
- 231.Touyz RM, Yao G. J Cell Physiol. 2003;197:326–335. doi: 10.1002/jcp.10393. [DOI] [PubMed] [Google Scholar]
- 232.Torres LM, Cefaratti C, Perry B, Romani A. Mol Cell Biochem. 2006;288:191–199. doi: 10.1007/s11010-006-9139-1. [DOI] [PubMed] [Google Scholar]
- 233.Rubin H. Adv Cancer Res. 2005;93:1–58. doi: 10.1016/S0065-230X(05)93001-7. [DOI] [PubMed] [Google Scholar]
- 234.Waas WF, Dalby KN. Biochemistry. 2003;42:2960–2970. doi: 10.1021/bi027171w. [DOI] [PubMed] [Google Scholar]
- 235.Kim SJ, Kang HS, Kang MS, Yu X, Park SY, Kim IS, Kim NS, Kim SZ, Kwak YG, Kim JS. Biochem Biophys Res Commun. 2005;333:1132–1138. doi: 10.1016/j.bbrc.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 236.Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. J Am Soc Nephrol. 2009;20:78–85. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.van der Wjist J, Hoenderop JG, Bindels RJ. Magnes Res. 2009;22:127–132. [PubMed] [Google Scholar]
- 238.Mudge GH. In: The Pharmacological Basis of Therapeutics. Goodman Gilman A, Godman LS, Rall TW, Murad F, editors. MacMillian; New York: 1989. [Google Scholar]
- 239.Joborn H, Akerstrom G, Ljunghall S. Clin Endocrinol. 1985;23:219–226. doi: 10.1111/j.1365-2265.1985.tb00217.x. [DOI] [PubMed] [Google Scholar]
- 240.Bailly C, Imbert-Teboul M, Roinel N, Amiel C. Am J Physiol. 1990;258:F1224–F1231. doi: 10.1152/ajprenal.1990.258.5.F1224. [DOI] [PubMed] [Google Scholar]
- 241.Rayssiguier Y. Horm Metab Res. 1977;9:309–314. doi: 10.1055/s-0028-1093519. [DOI] [PubMed] [Google Scholar]
- 242.Guideri G. Arch Intern Pharmacodyn Therap. 1992;14:122–125. [PubMed] [Google Scholar]
- 243.Shafik IM, Quamme GA. Am J Phyiol. 1989;257:F974–F977. doi: 10.1152/ajprenal.1989.257.6.F974. [DOI] [PubMed] [Google Scholar]
- 244.Molinoff PB. Drugs. 1984;28:1–14. doi: 10.2165/00003495-198400282-00002. [DOI] [PubMed] [Google Scholar]
- 245.Barnes PJ. Am J Respir Crit Care Med. 1995;152:838–860. doi: 10.1164/ajrccm.152.3.7663795. [DOI] [PubMed] [Google Scholar]
- 246.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Herbert SC. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 247.Nemeth EF, Scarpa A. J Biol Chem. 1987;262:5188–5196. [PubMed] [Google Scholar]
- 248.Bapty BW, Dai LJ, Ritchie G, Jirik F, Canaff L, Hendy GN, Quamme GA. Kidney Int. 1998;53:583–592. doi: 10.1046/j.1523-1755.1998.00790.x. [DOI] [PubMed] [Google Scholar]
- 249.Bapty BW, Dai LJ, Ritchie G, Canaff L, Hendy GN, Quamme GA. Am J Physiol. 1998;275:F353–F360. doi: 10.1152/ajprenal.1998.275.3.F353. [DOI] [PubMed] [Google Scholar]
- 250.Quamme GA, Dirks JH. Am J Physiol. 1980;238:F187–F198. doi: 10.1152/ajprenal.1980.238.3.F187. [DOI] [PubMed] [Google Scholar]
- 251.Dipette DJ, Simpson K, Guntupalli J. Magnesium. 1987;6:136–149. [PubMed] [Google Scholar]
- 252.Stanbury JB. J Pharmacol Exp Ther. 1984;93:52–62. [PubMed] [Google Scholar]
- 253.James MFM, Cork RC, Harlen GM, White JF. Magnesium. 1988;7:37–43. [PubMed] [Google Scholar]
- 254.Quamme GA, Rabkin SW. Biochem Biophys Res Commun. 1990;167:1406–1412. doi: 10.1016/0006-291x(90)90679-h. [DOI] [PubMed] [Google Scholar]
- 255.Quamme GA, Dai L-S. Am J Physiol. 1990;259:C521–C525. doi: 10.1152/ajpcell.1990.259.3.C521. [DOI] [PubMed] [Google Scholar]
- 256.Dai L-J, Friedman PA, Quamme GA. Kidney Inter. 1997;51:1710–1718. doi: 10.1038/ki.1997.236. [DOI] [PubMed] [Google Scholar]
- 257.Kubota T, Shindo Y, Tokuno K, Komatsu H, Ogawa H, Kudo S, Kitamura Y, Suzuki K, Oka K. Biochim Biophys Acta. 2005;1744:19–28. doi: 10.1016/j.bbamcr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 258.Dai L-J, Friedman PA, Quamme GA. Kidney Inter. 1991;51:1008–1017. doi: 10.1038/ki.1997.141. [DOI] [PubMed] [Google Scholar]
- 259.Tashiro M, Tursun P, Miyazaki T, Watanabe M, Konishi M. Jpn J Physiol. 2002;52:541–551. doi: 10.2170/jjphysiol.52.541. [DOI] [PubMed] [Google Scholar]
- 260.Schweigel M, Lang I, Martens H. Am J Physiol. 1999;277:G976–G982. doi: 10.1152/ajpgi.1999.277.5.G976. [DOI] [PubMed] [Google Scholar]
- 261.Schweigel M, Martens H. Am J Physiol. 2003;285:G45–G53. doi: 10.1152/ajpgi.00396.2002. [DOI] [PubMed] [Google Scholar]
- 262.Günther T, Vormann J. Biochim Biophys Acta. 1995;1234:105–110. doi: 10.1016/0005-2736(94)00267-s. [DOI] [PubMed] [Google Scholar]
- 263.Mondon CE, Dolkas CB, Olefsky JM, Reaven GM. Diabetes. 1974;24:225–229. doi: 10.2337/diab.24.2.225. [DOI] [PubMed] [Google Scholar]
- 264.Taylor R, Agius L. Biochem J. 1988;250:625–640. doi: 10.1042/bj2500625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Grubbs RD, Maguire ME. Magnesium. 1987;6:113–127. [PubMed] [Google Scholar]
- 266.Maguire ME. Trends Pharmacol Sci. 1984;5:73–77. [Google Scholar]
- 267.White RE, Hartzell HC. Science. 1988;239:778–780. doi: 10.1126/science.2448878. [DOI] [PubMed] [Google Scholar]
- 268.Agus ZS, Morad M. Annu Rev Physiol. 1991;53:299–307. doi: 10.1146/annurev.ph.53.030191.001503. [DOI] [PubMed] [Google Scholar]
- 269.Bara M, Guiet-Bara A. Magnes Res. 2001;14:11–18. [PubMed] [Google Scholar]
- 270.Serrano J, Dashti SR, Perez-Reyes E, Jones SW. Biophys J. 2000;79:3052–3062. doi: 10.1016/S0006-3495(00)76540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Brunet S, Scheuer T, Klevit R, Catterall WA. J Gen Physiol. 2006;126:311–323. doi: 10.1085/jgp.200509333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Zhang J, Wier G, Blaustein MP. Am J Physiol. 2002;283:H2692–H2705. doi: 10.1152/ajpheart.00260.2002. [DOI] [PubMed] [Google Scholar]
- 273.Prakriya M, Lewis RS. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Matsuda H. Annu Rev Physiol. 1991;53:289–298. doi: 10.1146/annurev.ph.53.030191.001445. [DOI] [PubMed] [Google Scholar]
- 275.Tammaro P, Smith AL, Crowley BL, Smirnov SV. Cardiovasc Res. 2005;65:387–396. doi: 10.1016/j.cardiores.2004.10.035. [DOI] [PubMed] [Google Scholar]
- 276.Shi J, Krishnamoorthy G, Wang Y, Hu L, Chaturvedi N, Harilal D, Qin J, Cui J. Nature. 2002;418:876–880. doi: 10.1038/nature00941. [DOI] [PubMed] [Google Scholar]
- 277.Bednarczyk P, Dolowy K, Szewczyk A. FEBS Lett. 2005;579:1625–1630. doi: 10.1016/j.febslet.2005.01.077. [DOI] [PubMed] [Google Scholar]
- 278.Gunther T. Magnesium. 1986;5:53–59. [PubMed] [Google Scholar]
- 279.Romani A, Dowell E, Scarpa A. J Biol Chem. 1991;266:24376–24384. [PubMed] [Google Scholar]
- 280.Zhang GH, Melvin JE. J Biol Chem. 1992;267:20721–20727. [PubMed] [Google Scholar]
- 281.Zhang GH, Melvin JE. J Biol Chem. 1996;271:29067–29072. doi: 10.1074/jbc.271.46.29067. [DOI] [PubMed] [Google Scholar]
- 282.McCormack JG, Halestrap A, Denton RM. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 283.Hansford RG. J Bioenerg Biomembr. 1994;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- 284.Moravec CS, Bond M. Am J Physiol. 1991;260:H989–H997. doi: 10.1152/ajpheart.1991.260.3.H989. [DOI] [PubMed] [Google Scholar]
- 285.Moravec CS, Bond M. J Biol Chem. 1992;267:5310–5316. [PubMed] [Google Scholar]
- 286.Brierley GP, Davis M, Jung DW. Arch Biochem Biophys. 1987;253:322–332. doi: 10.1016/0003-9861(87)90185-8. [DOI] [PubMed] [Google Scholar]
- 287.Beavis AD, Powers M. J Biol Chem. 2004;279:4045–4050. doi: 10.1074/jbc.M310475200. [DOI] [PubMed] [Google Scholar]
- 288.Bernardi P. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 289.Bradshaw PC, Pfeiffer DR. BMC Biochem. 2006;7:4. doi: 10.1186/1471-2091-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Dolder M, Walzel B, Speer O, Schlattner U, Wallimann T. J Biol Chem. 2003;278:17760–17766. doi: 10.1074/jbc.M208705200. [DOI] [PubMed] [Google Scholar]
- 291.Volpe P, Alderson-Lang BH, Nickols GA. Am J Physiol. 1990;258:C1077–C1085. doi: 10.1152/ajpcell.1990.258.6.C1077. [DOI] [PubMed] [Google Scholar]
- 292.Volpe P, Vezú L. Magnes Res. 1993;6:267–274. [PubMed] [Google Scholar]
- 293.Gusev K, Niggli E. J Gen Physiol. 2008;132:721–730. doi: 10.1085/jgp.200810119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Laver DR, Honen BN. J Gen Physiol. 2008;132:429–446. doi: 10.1085/jgp.200810001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Doleh L, Romani A. Arch Biochem Biophys. 2007;467:283–290. doi: 10.1016/j.abb.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Barfell A, Crumbly A, Romani A. Arch Biochem Biophys. 2011;509:157–163. doi: 10.1016/j.abb.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Yamaguchi S, Ishikawa T. Biochem Biophys Res Commun. 2008;376:100–104. doi: 10.1016/j.bbrc.2008.08.104. [DOI] [PubMed] [Google Scholar]
- 298.Okahira M, Kubota M, Iguchi K, Usui S, Hirano K. Eur J Pharmacol. 2008;588:26–32. doi: 10.1016/j.ejphar.2008.03.063. [DOI] [PubMed] [Google Scholar]
- 299.Maguire ME. Ann NY Acad Sci. 1988;551:215–217. doi: 10.1111/j.1749-6632.1988.tb22338.x. [DOI] [PubMed] [Google Scholar]
- 300.Sgambato A, Wolf FI, Faraglia B, Cittadini A. J Cell Physiol. 1999;180:245–254. doi: 10.1002/(SICI)1097-4652(199908)180:2<245::AID-JCP12>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 301.Wolf FI, Trapani V, Simonacci M, Boninsegna A, Mazur A, Maier JA. Nutr Cancer. 2009;61:131–136. doi: 10.1080/01635580802376360. [DOI] [PubMed] [Google Scholar]
- 302.Covacci V, Bruzzese N, Sgambato A, Di Francesco A, Russo MA, Wolf FI, Cittadini A. J Cell Biochem. 1998;70:313–322. doi: 10.1002/(sici)1097-4644(19980901)70:3<313::aid-jcb4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 303.Wolf FI, Covacci V, Bruzzese N, Di Francesco A, Sachets A, Cord D, Cittadini A. J Cell Biochem. 1998;71:441–448. doi: 10.1002/(sici)1097-4644(19981201)71:3<441::aid-jcb12>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 304.Di Francesco A, Desnoyer RW, Covacci V, Wolf FI, Romani A, Cittadini A, Bond M. Arch Biochem Biophys. 1998;360:149–157. doi: 10.1006/abbi.1998.0937. [DOI] [PubMed] [Google Scholar]
- 305.Trache A, Trzeciakowski JP, Meininger GA. J Mol Recognit. 2010;23:316–321. doi: 10.1002/jmr.985. [DOI] [PubMed] [Google Scholar]
- 306.Wolf FI, Cittadini AR, Maier JA. Cancer Treat Rev. 2009;35:378–382. doi: 10.1016/j.ctrv.2009.01.003. [DOI] [PubMed] [Google Scholar]