

Abstract
Objectives
We studied the development and fitness cost of 2-deoxystreptamine aminoglycoside resistance of Mycobacterium abscessus.
Methods
Spontaneous 2-deoxystreptamine aminoglycoside-resistant mutants were selected and the frequency of their appearance was determined. The 3′ part of the rrs gene was sequenced to characterize mutations. Additionally, we determined the MICs of aminoglycoside drugs for the different mutants obtained. The dominance/recessivity traits of the different mutations were examined and we explored the potential cost conferred by the mutations selected in vitro on the fitness of these isolates compared with the wild-type strain.
Results
The in vitro mutation rate for 2-deoxystreptamine aminoglycoside resistance was ∼10−7 mutations/cell division. In addition to the known rrs A→G substitution at position 1408 (Escherichia coli numbering), which confers kanamycin resistance (KanR), three new substitutions in rrs were identified in M. abscessus KanR mutants, i.e. T→A at 1406, C→T at 1409 and G→T at 1491. Heterodiploids carrying genomic mutations T→A at 1406 and A→G at 1408 with the wild-type rrs gene carried by the pNBV1 vector showed a resistant phenotype. In contrast, heterodiploids carrying genomic mutations C→T at 1409 and G→T at 1491 with the wild-type rrs gene carried by the pNBV1 vector had a susceptible phenotype. No burden on fitness was observed for the different mutations.
Conclusion
Mutations in the rrs gene that confer high-level 2-deoxystreptamine aminoglycoside resistance on M. abscessus differ in their dominance/recessivity traits and have no biological cost under our experimental conditions.
Keywords: fitness, antibiotic resistance, evolution
Introduction
Mycobacterium abscessus is a rapidly growing mycobacterium (RGM) that has been associated with a variety of different diseases, including a chronic lung disease that can be found in patients with bronchiectasis and young adults with cystic fibrosis.1,2 In addition, M. abscessus along with Mycobacterium chelonae account for most of the disseminated cutaneous infections caused by RGM.1,3
These organisms have been involved in several healthcare-associated disease outbreaks, including surgical wound infections and post-tympanostomy tube placement infections.4,5 M. abscessus represents one of the most antibiotic-resistant RGM species,1,6 with only a small number of useful antibiotics available.7 This species thrives in hostile environments, a feature associated with its capacity to cause outbreaks of healthcare-associated disease. The organism is also resistant to major anti-tuberculosis drugs, including isoniazid, rifampicin, ethambutol and pyrazinamide, and to most antimycobacterial drugs, including tetracycline, fluoroquinolones and sulphonamides.8 However, this species is naturally susceptible to amikacin and clarithromycin/azithromycin, which are used in combination for clinical treatment.8 The naturally antibiotic-resistant phenotype of M. abscessus may result from weak permeability of the cell wall.9 In addition, the genome contains many potential intrinsic drug resistance determinants such as an Ambler class A β-lactamase, a rifampicin ADP-ribosyl transferase, a ribosome methyltransferase capable of conferring resistance to macrolides,10 an aminoglycoside 2′-N-acetyltransferase and around 12 homologues of aminoglycoside phosphotransferases.9
Only a few studies have reported the genetic basis of acquired resistance to drugs used for the treatment of M. abscessus and most of them involve mutations in the single rRNA operon. Clarithromycin resistance in M. abscessus is conferred by a single point mutation in 23S rRNA, involving adenine at position 2058 or adenine at position 2059 (Escherichia coli numbering will be used in this article).11 Resistance to 2-deoxystreptamine aminoglycosides has been shown to be conferred by a single point mutation in the rrs gene (MAB_5051, encoding 16S rRNA) at position 1408,12 which is also responsible for a high level of resistance to amikacin, kanamycin and gentamicin (MICs >1024 mg/L). M. abscessus is known to contain only one copy of the ribosomal rRNA operon,12 which makes it an ideal organism to study the nature of aminoglycoside drug resistance at the molecular level. Aminoglycosides are highly potent, broad-spectrum antibiotics that are used for the treatment of life-threatening infections. Most aminoglycosides are naturally occurring substances produced by actinomycetes of the genera Streptomyces or Micromonospora. The bactericidal activity of aminoglycosides is primarily a consequence of inhibition of protein synthesis. Aminoglycosides bind to the bacterial 30S ribosomal subunit, causing a lack of fidelity in the translation process, and this results in cell death.13 This class of antibiotic is characterized by a broad antimicrobial spectrum, rapid bactericidal action and the ability to act synergistically with other drugs.14 Recent studies on the molecular structure of aminoglycosides complexed with bacterial site A of rRNA has shown that alteration to critical positions within the rRNA structure may be responsible for the high-level drug resistance to this class of antibiotic.15 In the present study, we investigated rrs gene sequences of spontaneous kanamycin-resistant mutants of M. abscessus for the presence of different mutations and assessed their recessivity/dominance phenotype as well as their potential fitness cost.
Materials and methods
Bacterial strains and media
Strains used in this study are listed in Table S1 (available as Supplementary data at JAC Online). E. coli DH5α cells were used for cloning as previously described.16 M. abscessus CIP104536T17 strains were grown on Luria–Bertani (LB) or Mueller–Hinton agar (MHA) or brain heart infusion (BHI) broth (Difco). Aminoglycoside antibiotics used were purchased from Sigma-Aldrich, and Zeocin was purchased from Invitrogen. When required, Zeocin was added to the medium at the following final concentration: 25 mg/L for E. coli and 50 mg/L for M. abscessus. Bacterial strains were stored at −80°C as described.18
In vitro selection of spontaneous resistant mutants and mutational frequency measurement
Spontaneous drug-resistant mutants were selected in vitro as previously described.12 In brief, aliquots of 1 mL of stationary phase culture of M. abscessus CIP104536T were plated on LB agar containing 100 mg/L of 2-deoxystreptamine aminoglycosides (kanamycin, amikacin, gentamicin and tobramycin). In parallel, serial dilutions were plated on antibiotic-free LB medium. The plates were incubated for 5–7 days at 37°C. The colonies were counted and mutational frequency was defined as the number of resistant isolates divided by the total colony count. Thirty-five kanamycin-resistant M. abscessus colonies were selected for further studies.
Susceptibility testing
Aminoglycoside susceptibility of spontaneous kanamycin-resistant mutants of M. abscessus with parental M. abscessus CIP104536T (wild-type) as a control, were tested on MHA (Difco) as previously described.19 The spontaneous mutants were grown at 37°C in LB broth to 0.6 optical density (OD) at 600 nm. Final concentrations of 104–105 cfu/mL were spotted on MHA plates containing different concentrations of drugs and growth was observed after 5–7 days of incubation at 37°C.
rrs gene sequencing
Genomic DNA was extracted20 from spontaneous kanamycin-resistant mutants and the wild-type strain. This genomic DNA was used as a template for PCR amplification of the last 341 bp of the rrs gene, including positions from 1200 to 1501, where mutations related with aminoglycoside resistance have been described in other mycobacterial species,21 using rrs1-F primer (5′-ATGACGTCAAGTCATCATGCC-3′), corresponding to rrs positions 1160–1180, and rrs1-R (5′-AGGTGATCCAGCCGCACCTTC-3′), corresponding to rrs positions 1484–1504. The PCR product was purified and sequenced using rrs1-F primer.
Generation of M. abscessus heterozygote strains carrying wild-type and mutated rrs gene
First, the mycobacterial replicative low-copy vector pNBV122 was used as a backbone in which the hygromycin resistance cassette was replaced by a Zeocin resistance cassette to construct a plasmid termed pNBV1-ZeoR.
Genomic DNA of M. abscessus was used as template for PCR amplification of the whole-length wild-type rrs gene with 300 bp upstream and downstream to the gene, using primer pair rrs2-F (5′-AGCTTCTAGAGTCGCTCGGAAGAGCGAAAGTCG-3′) and rrs2-R (5′-CGCTCTAGAAGTGCCAAGGCATTCACCATG-3′), both containing an XbaI restriction site (underlined). The PCR product was purified, digested by XbaI enzyme and cloned into XbaI-digested vector pNBV1-ZeoR, resulting in plasmid pWT (see Table S2, available as Supplementary data at JAC Online). Electrocompetent M. abscessus kanamycin-resistant mutants MUT1406 (T1406A), MUT1408 (A1408G), MUT1409(C1409T) and MUT1491 (G1491T) were prepared by growing a bacterial culture to mid-log phase (OD600 0.6–0.8), harvesting the bacterial cells and washing them three times in 10% glycerol and resuspending them in the same buffer. Plasmid pWT was introduced by electroporation of 200 μL of electrocompetent cells with 100 ng of DNA using a Bio-Rad Gene Pulser II set at 200 Ω, 2.5 kV, 25 μF. Cells were recovered in 1 mL of LB broth at 37°C for 8 h and spread onto LB agar plates with Zeocin. The transformants, named H1 series, (Table 1) were further tested for resistance against aminoglycoside antibiotics.
Table 1.
Summary of dominance/recessivity phenotype of M. abscessus strains
| Phenotype |
||||
|---|---|---|---|---|
| M. abscessus strains | Genotype | Kanamycin | Zeocin | Kanamycin MIC (mg/L)a |
| WT | WT | S | S | <2 |
| M.T1406A | M | R | S | >1000 |
| M.A1408G | M | R | S | >1000 |
| M.C1409T | M | R | S | >1000 |
| M.G1491T | M | R | S | >1000 |
| H1.T1406A/WT | H | R | R | >1000 |
| H1.A1408G/WT | H | R | R | >1000 |
| H1.C1409T/WT | H | S | R | <2 |
| H1.G1491T/WT | H | S | R | <2 |
| H2.WT/T1406A | H | S | R | <2 |
| H2.WT/A1408G | H | S | R | <2 |
| H2.WT/C1409T | H | S | R | <2 |
| H2.WT/G1491T | H | S | R | <2 |
R, resistant; S, susceptible; WT, wild-type; M, mutant; H, heterodiploids; H1, heterodiploids with mutation in the chromosomal rrs and wild-type rrs on plasmid; H2, heterodiploids with chromosomal rrs and rrs mutations carried on plasmid.
aMICs determined from three experiments.
Genomic DNA of the M. abscessus kanamycin-resistant mutants MUT1406, MUT1408, MUT1409 and MUT1491 was used as a template for PCR amplification of the mutated rrs genes and the amplicons were cloned into pNBV1-ZeoR as described above, resulting in plasmids pA1408G, pT1406A, pC1409T and pG1491T, respectively (Table S2). These plasmids were electroporated into M. abscessus (wild-type) and the transformants, named H2 series (Table 1), were tested for their resistance phenotype as described above.
Determination of bacterial fitness
The cost of a resistance mutation was determined by direct competition against the drug-susceptible parental strain, as described previously.23 Briefly, equal densities of drug-susceptible and drug-resistant strains were mixed and incubated in antibiotic-free BHI broth and 0.05 mL of the culture was transferred into 5 mL of fresh BHI broth for growth. Aliquots were plated every 72 h onto drug-free BHI agar to count the total number of colonies. In parallel, the number of drug-resistant bacterial cells was determined by plating aliquots on BHI agar containing kanamycin (100 mg/L). The number of parental drug-susceptible cells was calculated as the total number of bacterial cells minus the number of drug-resistant cells. The experiments were performed in triplicate with three independent cultures. The difference in fitness between two competing strains at time t was computed by using the following function:
where r and s represent the number of drug-resistant and drug-susceptible cells, respectively, at a given time and t, rt − 1 and st − 1 denote the number of drug-resistant and drug-susceptible cells at the preceding timepoint (t − 1). The quotient of the ratios of the cell numbers at the two consecutive timepoints t−1 and t was standardized with the exponent 1/7 because cell numbers were determined every seven generations. St is called the selection coefficient at time t. St is equal to 0 if there is no difference in fitness between the competing strains. If drug resistance reduces bacterial fitness, St is negative. St is positive if resistance increases bacterial fitness. The relative bacterial fitness is calculated as: fitt = 1 + St. The cost per generation is calculated as: cpg = 1 − eSt.
Results
Selection of aminoglycoside-resistant mutants and nucleotide sequence analysis of 3′ part of 16S rRNA gene
M. abscessus (wild-type) was plated on different 2-deoxystreptamine aminoglycosides (kanamycin, amikacin, gentamicin and tobramycin) at a concentration of 100 mg/L. M. abscessus mutants were obtained at frequencies around 10−7. Thirty-five in vitro mutants resistant to kanamycin underwent DNA sequencing to locate the possible genetic alterations in rrs. Four different mutations were found among the kanamycin-resistant isolates. Twelve out of 35 (35%) had a guanine for adenine substitution at position 1408 (A→G). Seventeen isolates (48%) harboured the substitution thymine for guanine at position 1491 (G→T), and five resistant isolates (14%) had a thymine for cytosine substitution at position 1409 (C→T). Only one mutant (3%) had a substitution from thymine to adenine at position 1406 (T→A) (Figure 1). All the spontaneous resistant mutants analysed carried mutations in the part of rrs gene sequenced.
Figure 1.
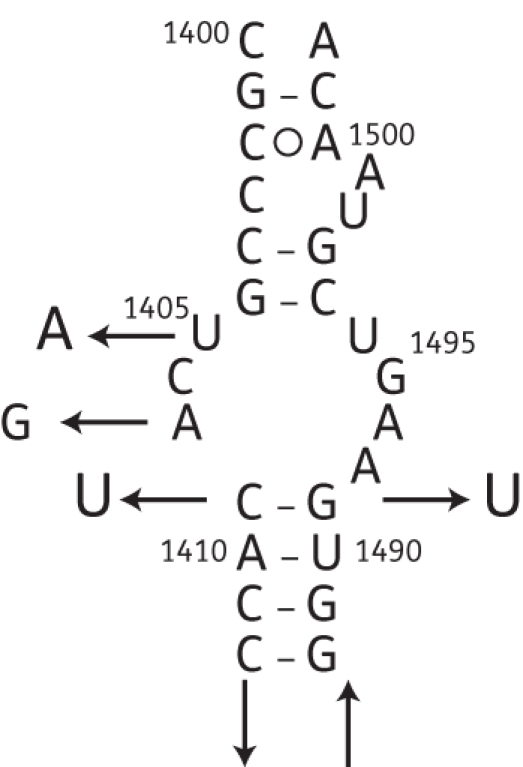
Secondary structure model of E. coli 16S rRNA containing the four mutations conferring 2-deoxystreptamine aminoglycoside resistance in M. abscessus.
Antibiotic susceptibility testing
All four M. abscessus kanamycin-resistant mutants, namely MUT1406, MUT1408, MUT1409 and MUT1491, were highly resistant to kanamycin (MICs >1000 mg/L, Table 1) and also to other 2-deoxystreptamine aminoglycosides tested (amikacin and gentamicin) (MICs >1000 mg/L) (data not shown). The parental strain M. abscessus CIP104536T had low MICs (<2 mg/L) for all the 2-deoxystreptamine aminoglycoside antibiotics tested.
Dominance/recessivity of M. abscessus kanamycin-resistant mutants
We investigated recessivity or dominance of 2-deoxystreptamine aminoglycoside-resistant mutations by constructing a series of heterodiploid M. abscessus strains (termed H1). M. abscessus kanamycin-resistant strains independently carrying the four mutations (MUT1406, MUT1408, MUT1409 and MUT1491) were transformed with pWT (Table S2). The transformants H1.T1406A/WT, H1.A1408G/WT, H1.C1409T/WT and H1.G1491T/WT were selected on Zeocin (50 mg/L) and then tested for their resistance/susceptibility to the 2-deoxystreptamine aminoglycosides by determining the MICs of kanamycin. The results showed that the heterodiploids H1.T1406A/WT and H1.A1408G/WT were kanamycin resistant (MICs >1000 mg/L), whereas the heterodiploids H1.C1409T/WT and H1.G1491T/WT had a kanamycin-susceptible phenotype (MICs <2 mg/L) (Table 1).
To gain further insights, a second series of heterodiploids (termed H2) was constructed. For these experiments, the four rrs mutated genes, each carrying a distinct mutation, were cloned into pNBV1-ZeoR to construct plasmids pT1406A, pA1408G, pC1409T and pG1491T. Individually, each plasmid was electroporated into the M. abscessus (wild-type) strain. The transformants H2 were selected on Zeocin (50 mg/L) and tested for their MICs of kanamycin as described above. The results are shown in Table 1. All four transformants H2 (H2.WT/T1406A, H2.WT/A1408G, H2.WT/C1409T and H2.WT/G1491T) had a kanamycin-susceptible phenotype (MICs <2 mg/L). In order to confirm that the H1 and H2 heterodiploids were heterozygous for the rrs gene and to rule out the possibility that the resistance or susceptible phenotype observed resulted from a possible reversion of the mutated nucleotide to wild-type, the rrs alleles in the four H1 and H2 transformants were sequenced. For each H1 and H2 transformant, sequence analysis confirmed the presence of both the wild-type and the mutant nucleotide (i.e. T and A at position 1406, A and G at position 1408, C and T at position 1409, and G and T at position 1491) (data not shown).
Fitness cost of spontaneous mutations
Bacterial fitness was investigated by measuring bacterial generation in standard competition experiments to determine the mutation-mediated cpg (defined as the percentage difference in growth rate during competition). Our results showed that the cpg was very similar for the four mutations (T1406A, A1408G, C1409T and G1491T) and ranged between 0.008 and 0.081 (Table 2). Thus, in the experimental conditions used in this study, the mutations confer no cost on the fitness of M. abscessus.
Table 2.
Determination of bacterial fitness by competitive growth between M. abscessus wild-type and kanamycin-resistant mutants (estimated from three experiments)
| Mutation | Median St | Median cpg | Median fitt |
|---|---|---|---|
| rrs T1406A | −3.1 × 10−1–2.5 × 10−1 | 0.012 | 0.989 |
| rrs T1408G | −4.6 × 10−1–3.6×10−1 | 0.008 | 0.985 |
| rrs C1409T | −2.7 × 10−1–5.4 × 10−1 | 0.056 | 0.912 |
| rrs G1491T | −7.6 × 10−1–7.9 × 10−1 | 0.081 | 0.899 |
St, selection coefficient at time t, estimated from t0 (day 1) to t7 (21 days); cpg, cost per generation; fitt, relative bacterial fitness.
Discussion
An initial study of aminoglycoside resistance in RGM24 demonstrated that high-level resistant mutants to amikacin (MICs >1024 mg/L) could be obtained in M. chelonae and M. abscessus (identified at that time as M. chelonei) at a frequency of 10−5–10−7. Prammananan et al.12 reported that amikacin-resistant clinical isolates of M. abscessus from patients with post-tympanostomy tube placement otitis media or patients with cystic fibrosis who had received aminoglycoside therapy had a single mutation in the rrs gene (adenine substituted for guanine at position 1408, A1408G). This substitution is responsible for a high level of resistance (MICs >1024 mg/L) to amikacin and other 2-deoxystreptamine aminoglycosides (kanamycin, gentamicin, tobramycin and neomycin) in M. abscessus. In the current study we investigated the rrs gene sequences of M. abscessus for mutations affecting 2-deoxystreptamine aminoglycoside resistance. Spontaneous resistant mutants were obtained in vitro at a frequency of 10−7 after three independent experiments, which is consistent with a single-step mutational event and with mutational frequencies reported previously.24 After sequencing the last 300 bp region of the rrs gene from kanamycin-resistant in vitro mutants, we found four different single mutations in this part of the gene. In addition to the known A→G substitution at position 1408, we found three new mutations not previously described for M. abscessus, that were identified at position 1406 (T→A substitution), position 1409 (C→T substitution) and position 1491 (G→T substitution). The isolates were highly resistant (MICs >1000 mg/L) to kanamycin and other 2-deoxystreptamine aminoglycosides (gentamicin, tobramycin and amikacin). Evidence for the importance of these key positions in the interaction with aminoglycoside antibiotics has been reported.15,25,26 Mutations affecting these key positions have been associated with an antibiotic resistance phenotype in other bacteria. In M. smegmatis, mutations at positions 1408, 1409 and 1491 within the rrn gene (coding for 16S rRNA) confer kanamycin and other aminoglycoside resistance phenotypes.27 In E. coli, mutations at positions 1408, 1409 and 1491 caused resistance to kanamycin, paromomycin and other aminoglycoside antibiotics.28,29 Maus et al.19 showed that three rrs mutations (A1408G, C1409T and G1491T) in M. tuberculosis confer kanamycin resistance and were associated with a cross-resistance pattern to cyclic peptides (capreomycin and viomycin). M. abscessus and M. tuberculosis possess a single rRNA operon and our results showed that the same positions in this gene resulted in a similar drug-resistant phenotype in both organisms.
Aminoglycosides inhibit translation and promote the generation of truncated proteins, resulting in the death of the organism. The question of recessivity versus dominance of aminoglycoside resistance mutations can be an ambiguous matter.30 Apirion and Schlessinger30 showed that kanamycin resistance in E. coli is recessive. By using a conjugation system, Prammananan et al.12 demonstrated that resistance due to rrs resistant alleles is recessive and that recombinase RecA-mediated gene conversion was responsible for the aminoglycoside-resistant phenotype in a heterozygote (rRNAwt/rRNAmut) strain of M. smegmatis. To determine the implication of the rrs gene mutations in kanamycin-resistant phenotypes of M. abscessus, we constructed heterodiploid strains of M. abscessus containing the chromosomal kanamycin-resistant mutated rrs alleles and the plasmid-borne wild-type allele. The heterodiploid strains H1.T1406A/WT and H1.A1408G/WT had a kanamycin-resistant phenotype (MICs >1000 mg/L), suggesting that mutations T1406A and A1408G are dominant. In contrast, the heterodiploid strains H1.C1409T/WT and H1.G1491T/WT had a kanamycin-sensitive phenotype (MICs <2 mg/L), possibly due to the effect of the drug on ribosomes containing the susceptible wild-type rrs allele that resulted in a sufficient number of translation errors to induce a lethal phenotype. Since the susceptibility of heterodiploids H1.C1409T/WT and H1.G1491T/WT was restored by the introduction of the wild-type rrs gene, our results suggest that mutations C1409T and G1491T are recessive. In another experiment, we transformed M. abscessus (wild-type) with the plasmids pT1406A, pA1408G, pC1409T and pG1491T, containing each one of the four mutated rrs gene characterized in this work. All heterodiploids of the H2 series (WT/A1406T, WT/A1408G, WT/C1409T and WT/G1491T) were susceptible to kanamycin (Table 1). This result confirmed the recessivity of mutations C1409T and G1491T.
The susceptibility to kanamycin of H2 heterodiploids WT/A1406T and WT/A1408G was unexpected given the apparent dominance trait observed for these two mutations in the H1 series of heterodiploids. It is conceivable that the original mutants MUT1406 and MUT1408 could have additional mutations in genes other than rrs which contribute to their kanamycin-resistant phenotype. The absence of these mutations within the plasmids pT1406A and pA1408G would result in the susceptible phenotype observed in the H2 series of heterodiploids. A recent report has described mutations affecting the promoter of the eis gene, a previously uncharacterized aminoglycoside acetyltransferase responsible for kanamycin resistance in M. tuberculosis clinical strains harbouring the wild-type version of the rrs gene.31 Although the M. abscessus genome lacks an orthologue of the eis gene,32 a similar phenomenon could contribute to kanamycin resistance in M. abscessus. The shift of a heterogeneous population of ribosomes could also be a major factor in determining the antibiotic resistance/sensitivity phenotype. It was reported in E. coli that cells expressing a ribosome with mutation A1408G were resistant to aminoglycosides, with a mixed population of 60% mutant and 40% wild-type ribosomes.33 If expression levels of the genomic rrs locus with T1406A and A1408G mutations were higher than those of the wild-type rrs gene, which is under the control of the hsp60 promoter harboured by the vector pNBV1-ZeoR, the shift towards mutant ribosomes may be responsible for the resistant phenotype of the heterodiploids H1 (T1406A/WT and A1408G/WT).
In order to establish whether the different mutations generate a fitness disadvantage or advantage, we investigated the cost of resistance of rrs mutations. In vitro competition assays using the drug-susceptible wild-type strain of M. abscessus and our spontaneous drug-resistant mutants indicated that chromosomal resistance mutations appeared to be cost neutral (Table 2). The cost per generation is very similar for the four mutations studied (T1406A, A1408G, C1409T and G1491T) and is close to 0%. Our results suggest that these mutations are stabilized in the population in order to confer a high-level drug resistance phenotype to M. abscessus. However, it is possible that a fitness cost could be revealed under different environmental conditions. Compensatory mutations are unlikely to be involved in maintaining drug resistance if the resistance mutations carry only very small or no fitness cost. Given that in our system the M. abscessus mutations are cost-neutral, there is presumably no selective pressure for any such compensatory mutation to occur. The absence of clinical isolates carrying mutations T1406A, C1409T and G1491T may be the consequence of some cost in vivo, since the critical trait that influences the spread of resistance is the fitness cost of drug resistance.34 Further studies using experimental models reproducing environmental conditions are required to determine why such mutations have so far not been found in M. abscessus isolates associated with clinical infections. Other researchers have reported low fitness costs associated with mutations affecting genes coding for rRNA in other bacterial species. In a recent report, Shcherbakov et al.27 generated single mutants at position A1408, C1409 and C1491 in a Mycobacterium smegmatis single rrnB operon variant and observed that the mutation A1408G is associated with high-level resistance to aminoglycosides with a low fitness cost in the mutant strain. Similarly, the mutations C1409T and G1491T conferred intermediate resistance to aminoglycosides; however, the mutant strains had a high fitness cost. Our results and a previous study27 might lead to speculation that mutations in rrs genes conferring high resistance to aminoglycoside antibiotics are linked to low or no cost in terms of mycobacterial fitness.
In summary, we report new mutations of the rrs gene in in vitro-selected kanamycin-resistant mutants of M. abscessus, which confer resistance to kanamycin and to other 2-deoxystreptamine aminoglycosides. This information could be useful for diagnosis of aminoglycoside resistance mutations in clinical isolates of M. abscessus.
Supplementary data
Tables S1 and S2 are available as Supplementary data at JAC Online (http://jac.oxfordjournals.org/).
Funding
This work was supported by National Institute of Health and Medical Research- Inserm, Pasteur Institute and University Paris 7 Denis-Diderot.
Transparency declarations
None to declare.
Supplementary Material
Acknowledgements
We thank Ivan Matic, Peter Sanders and José-Antonio Ainsa for very helpful advice.
References
- 1.Brown-Elliott BA, Wallace RJ., Jr Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev. 2002;15:716–46. doi: 10.1128/CMR.15.4.716-746.2002. doi:10.1128/CMR.15.4.716-746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Groote MA, Huitt G. Infections due to rapidly growing mycobacteria. Clin Infect Dis. 2006;42:1756–63. doi: 10.1086/504381. doi:10.1086/504381. [DOI] [PubMed] [Google Scholar]
- 3.Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367–416. doi: 10.1164/rccm.200604-571ST. doi:10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 4.Lowry PW, Jarvis WR, Oberle AD, et al. Mycobacterium chelonae causing otitis media in an ear-nose-and-throat practice. N Engl J Med. 1988;319:978–82. doi: 10.1056/NEJM198810133191504. doi:10.1056/NEJM198810133191504. [DOI] [PubMed] [Google Scholar]
- 5.Ozluer SM, De'Ambrosis BJ. Mycobacterium abscessus wound infection. Australas J Dermatol. 2001;42:26–9. doi: 10.1046/j.1440-0960.2001.00468.x. doi:10.1046/j.1440-0960.2001.00468.x. [DOI] [PubMed] [Google Scholar]
- 6.Medjahed H, Gaillard JL, Reyrat JM. Mycobacterium abscessus: a new player in the mycobacterial field. Trends Microbiol. 2010;18:117–23. doi: 10.1016/j.tim.2009.12.007. doi:10.1016/j.tim.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Griffith DE. The talking Mycobacterium abscessus blues. Clin Infect Dis. 2011;52:572–4. doi: 10.1093/cid/ciq252. doi:10.1093/cid/ciq252. [DOI] [PubMed] [Google Scholar]
- 8.Petrini B. Mycobacterium abscessus: an emerging rapid-growing potential pathogen. APMIS. 2006;114:319–28. doi: 10.1111/j.1600-0463.2006.apm_390.x. doi:10.1111/j.1600-0463.2006.apm_390.x. [DOI] [PubMed] [Google Scholar]
- 9.Ripoll F, Pasek S, Schenowitz C, et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One. 2009;4:e5660. doi: 10.1371/journal.pone.0005660. doi:10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nash KA, Brown-Elliott BA, Wallace RJ., Jr A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. 2009;53:1367–76. doi: 10.1128/AAC.01275-08. doi:10.1128/AAC.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace RJ, Jr, Meier A, Brown BA, et al. Genetic basis for clarithromycin resistance among isolates of Mycobacterium chelonae and Mycobacterium abscessus. Antimicrob Agents Chemother. 1996;40:1676–81. doi: 10.1128/aac.40.7.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prammananan T, Sander P, Brown BA, et al. A single 16S ribosomal RNA substitution is responsible for resistance to amikacin and other 2-deoxystreptamine aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J Infect Dis. 1998;177:1573–81. doi: 10.1086/515328. doi:10.1086/515328. [DOI] [PubMed] [Google Scholar]
- 13.Kotra LP, Haddad J, Mobashery S. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother. 2000;44:3249–56. doi: 10.1128/aac.44.12.3249-3256.2000. doi:10.1128/AAC.44.12.3249-3256.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen LF, Kaye D. Current use for old antibacterial agents: polymyxins, rifamycins, and aminoglycosides. Infect Dis Clin North Am. 2009;23:1053–75. doi: 10.1016/j.idc.2009.06.004. doi:10.1016/j.idc.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Hobbie SN, Pfister P, Brull C, et al. Analysis of the contribution of individual substituents in 4,6-aminoglycoside-ribosome interaction. Antimicrob Agents Chemother. 2005;49:5112–8. doi: 10.1128/AAC.49.12.5112-5118.2005. doi:10.1128/AAC.49.12.5112-5118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 17.Catherinot E, Clarissou J, Etienne G, et al. Hypervirulence of a rough variant of the Mycobacterium abscessus type strain. Infect Immun. 2007;75:1055–8. doi: 10.1128/IAI.00835-06. doi:10.1128/IAI.00835-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortes MA, Nessar R, Singh AK. Laboratory maintenance of Mycobacterium abscessus. Curr Protoc Microbiol. 2010 doi: 10.1002/9780471729259.mc10d01s18. Chapter 10: Unit 10D 1. [DOI] [PubMed] [Google Scholar]
- 19.Maus CE, Plikaytis BB, Shinnick TM. Molecular analysis of cross-resistance to capreomycin, kanamycin, amikacin, and viomycin in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2005;49:3192–7. doi: 10.1128/AAC.49.8.3192-3197.2005. doi:10.1128/AAC.49.8.3192-3197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pelicic V, Reyrat JM, Gicquel B. Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on mycobacteria. J Bacteriol. 1996;178:1197–9. doi: 10.1128/jb.178.4.1197-1199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki Y, Katsukawa C, Tamaru A, et al. Detection of kanamycin-resistant Mycobacterium tuberculosis by identifying mutations in the 16S rRNA gene. J Clin Microbiol. 1998;36:1220–5. doi: 10.1128/jcm.36.5.1220-1225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howard NS, Gomez JE, Ko C, et al. Color selection with a hygromycin-resistance-based Escherichia coli-mycobacterial shuttle vector. Gene. 1995;166:181–2. doi: 10.1016/0378-1119(95)00597-x. doi:10.1016/0378-1119(95)00597-X. [DOI] [PubMed] [Google Scholar]
- 23.Sander P, Springer B, Prammananan T, et al. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother. 2002;46:1204–11. doi: 10.1128/AAC.46.5.1204-1211.2002. doi:10.1128/AAC.46.5.1204-1211.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wallace RJ, Jr, Hull SI, Bobey DG, et al. Mutational resistance as the mechanism of acquired drug resistance to aminoglycosides and antibacterial agents in Mycobacterium fortuitum and Mycobacterium chelonei. Evidence is based on plasmid analysis, mutational frequencies, and aminoglycoside-modifying enzyme assays. Am Rev Respir Dis. 1985;132:409–16. doi: 10.1164/arrd.1985.132.2.409. [DOI] [PubMed] [Google Scholar]
- 25.Hobbie SN, Bruell C, Kalapala S, et al. A genetic model to investigate drug-target interactions at the ribosomal decoding site. Biochimie. 2006;88:1033–43. doi: 10.1016/j.biochi.2006.04.008. doi:10.1016/j.biochi.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 26.Pfister P, Hobbie S, Vicens Q, et al. The molecular basis for A-site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and X-ray crystal structures. Chembiochem. 2003;4:1078–88. doi: 10.1002/cbic.200300657. doi:10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]
- 27.Shcherbakov D, Akbergenov R, Matt T, et al. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in-vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol. 2010;77:830–40. doi: 10.1111/j.1365-2958.2010.07218.x. doi:10.1111/j.1365-2958.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- 28.De Stasio EA, Moazed D, Noller HF, et al. Mutations in 16S ribosomal RNA disrupt antibiotic–RNA interactions. EMBO J. 1989;8:1213–6. doi: 10.1002/j.1460-2075.1989.tb03494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baudin F, Ehresmann C, Romby P, et al. Higher-order structure of domain III in Escherichia coli 16S ribosomal RNA, 30S subunit and 70S ribosome. Biochimie. 1987;69:1081–96. doi: 10.1016/0300-9084(87)90008-3. doi:10.1016/0300-9084(87)90008-3. [DOI] [PubMed] [Google Scholar]
- 30.Apirion D, Schlessinger D. Coresistance to neomycin and kanamycin by mutations in an Escherichia coli locus that affects ribosomes. J Bacteriol. 1968;96:768–76. doi: 10.1128/jb.96.3.768-776.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaunbrecher MA, Sikes RD, Jr, Metchock B, et al. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2009;106:20004–9. doi: 10.1073/pnas.0907925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei J, Dahl JL, Moulder JW, et al. Identification of a Mycobacterium tuberculosis gene that enhances mycobacterial survival in macrophages. J Bacteriol. 2000;182:377–84. doi: 10.1128/jb.182.2.377-384.2000. doi:10.1128/JB.182.2.377-384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Recht MI, Puglisi JD. Aminoglycoside resistance with homogeneous and heterogeneous populations of antibiotic-resistant ribosomes. Antimicrob Agents Chemother. 2001;45:2414–9. doi: 10.1128/AAC.45.9.2414-2419.2001. doi:10.1128/AAC.45.9.2414-2419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol. 2006;9:461–5. doi: 10.1016/j.mib.2006.07.002. doi:10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.