

Abstract
β-thalassemias are heterogeneous hereditary anemias characterized by a reduced output of β-globin chains. The disease is most frequent in the temperate regions of the world, where it represents an important health problem. In the last decades, several programs, aimed at controlling the birth rate of thalassemia newborns by screening and prenatal diagnosis of populations with high risk of β-thalassemia, have been successful accomplished. Bone marrow transplantation has offered a definitive cure for the fraction of patients with available donors. In the same time, steady improvements were made in the traditional clinical management of β-thalassemia patients. The introduction of the oral iron chelators deferiprone that preferentially chelates hearth iron and the development of novel NMR diagnostic methods has led to reduced morbility, increased survival and improved quality of life. More recently, major advances have being made in the discovery of critical modifier genes, such as Myb and especially BCL11A (B cell lymphoma 11A), a master regulator of HbF (fetal hemoglobin) and hemoglobin switching. Polimorphysms of BCL11A, Myb and γ-globin genes account for most of the variability in the clinical phenotypes in β-thalassemia and sickle cell anemia patients. Finally, the year 2010 has brought in the first successful experiment of gene therapy in a β-thalassemia patient, opening up the perspective of a generalized cure for all β- thalassemia patients.
Key words: β-thalassemia, gene therapy, iron chelation, GWAS.
Introduction
β-thalassemias, a group of autosomal recessive disorders resulting from reduced or absent production of β-globin chains from the β-globin locus, are very heterogeneous at the molecular level (for review see Weatherall & Clegg, 2001 and Cao & Galanello).1,2 More than 200 disease-causing mutations have been described to date. The large majority of mutations are simple nucleotide substitutions or deletions or insertions of oligonucleotides leading to frameshift. Rarely the β-thalassemias are the result of gross gene deletions. Homozygosity for β-thalassemia usually leads to the severe transfusion-dependent phenotype of thalassemia major. Treatment with a regular transfusion program and chelation therapy, aimed at reducing the transfusion iron-overload allows for normal growth and development and extends the life expectancy into the third to fifth decade. Bone marrow or cord blood transplantation (BMT) from an HLA (human leukocyte antigen) -identical sib represent an alternative to traditional transfusion and chelation therapy. More rarely the homozygous state for β-thalassemia leads to the phenotype of thalassemia intermedia. Individuals with thalassemia intermedia present later, and have milder anemia and only rarely require transfusion. However also patients with thalassemia intermedia are at risk to develop iron overload secondary to increased intestinal iron absorption because of ineffective erythropoiesis.
In the last few years there has been a dramatic advancement in the field of chelation therapy, genetic factors ameliorating the clinical phenotype, hemoglobin switching, gene therapy with lentiviral vectors and development of embryonic-like stem cells obtained by transfection and reprogramming differentiated somatic cells with key regulatory factors. All these developments, which are the topics of this review, may lead in the long term to advances in the traditional and in the gene and stem-cell-based therapy.
Genetic modifier factors in β-thalassemias
As alluded in the introduction, homozygotes or compound heterozygotes for β-thalassemia usually result in the phenotype of transfusion-dependent thalassemia major. However, in a consistent proportion of the cases, these intermediate genotypes are associated with milder forms, which are referred to as thalassemia intermedia. These forms of β-thalassemias show a marked phenotypic heterogeneity which ranges from the severe thalassemia major to the asymptomatic β-thalassemia carrier state.1–4 Genetic determinants capable of ameliorating the phenotype of β-thalassemia, so far defined (Figure 1), exert their effect by reducing the α/nonα chain imbalance, thereby resulting in a lesser degree of α-chain precipitation, which is the critical factor in the pathophysiology of these inherited anemias.5,6
Figure 1.
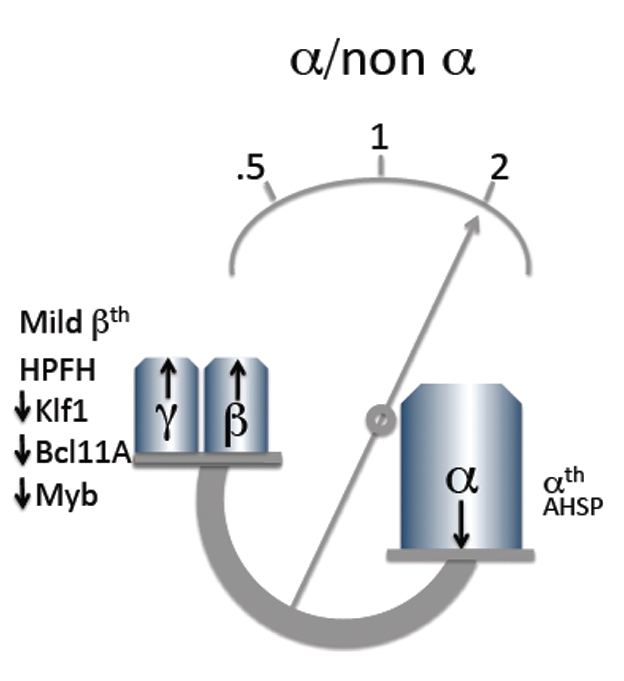
Genetic modifying factors in β-thalassemia. Modifying factors generally act by reducing or increasing the altered globin biosynthetic ratio typical of thalassemia.
A well-recognized mechanism for the production of a milder phenotype is the inheritance of a mild or silent β-thalassemia mutation, in homozygosity or in a compound heterozygous state with a severe mutation. Mild and silent mutations are molecular defects of the β-globin gene associated with a consistent residual output of β-globin chains from the affected β-locus.
A second characterized molecular mechanism is the co-inheritance with homozygous β-thalassemia of mutations of the α-globin gene associated with α-thalassemia which by reducing the output of α-chains decrease the α/nonα chain imbalance.
A third mechanism is related to the co-inheritance of genetic determinants able to substain a persistent production of γ-chain in the adult life, which again act by reducing the α/nonα globin chain imbalance.
Rarely the increase of γ-chain output depends on the type of thalassemia mutation per se as it occurs in δβ-thalassemias caused by deletions of variable extent in the β-globin gene cluster or in deletions removing the HBB promoter.
Most commonly the persistence of HbF production depends on the co-transmission of a specific determinant of hereditary persistence of fetal hemoglobin (HPFH). The inherited persistent production of HbF in adult life belongs to two different categories according to the distribution of HbF within the red blood cells, which can be pancellular (homogeneous distribution) or heterocellular (heterogeneous distribution). Pancellular HPFH are caused either by deletion at variable extent of the β-globin cluster or by point mutation in the Gγor Aγ promoter.
Heterocellular HPFH are associated with quantitative trait loci linked or unlinked to the β-globin cluster.7 These heterocellular HPFH determinants were recently shown to play a major role in determining the phenotype of β-thalassemia (Table 1).
Table 1. Genetic conditions with increased HbF in adult life.
| Deletion |
| - δβ thalassemia |
| -HPFH |
| Non deletion |
| Linked to the β-cluster |
| Gγ promoter mutations |
| Aγ promoter mutations |
| Unlinked to the β-cluster |
| -6q23 |
| -2q15 |
HbF, fetal hemoglobin; HPFH, hereditary persistence of fetal hemoglobin.
Hemoglobin F regulation
The residual amount of HbF in normal adults is distributed heterogeneously in the red blood cells of which those showing a measurable quantity are dubbed F cells.8 The level of HbF and the F cell count are strictly correlated and show a similar distribution. The majority of normal adult individuals have usually HbF levels <0.6% of the total Hb; however 10–15% show higher levels ranging between 0.8 and 5% of total Hb. As alluded before, these subjects are considered carriers of heterocellular HPFH genetic determinants.9
Twin studies have clearly demonstrated that HbF is a highly heritable trait. Genetic factors appear indeed responsible for 89% of the FC variance.9
Gγpromoter QTLs associated with heterocellular HbF
Since many years genetic determinants of high HbF production have been recognized in the β-globin gene cluster within the Aγ and Gγ promoter.10 In non-anemic subjects from North Europe the XmnI SNP (Single nucleotide polymorphysm) at position −158 upstream of the transcription start site of HBG2 (now denominated rs7482144) accounts for 13–32% of F-cell variance11 (Figure 2).
Figure 2.

HbF associated SNPs. Chromosome bands, associated genes and relative SNP positions are indicated. Variance of the HbF (H2) in the Sardinian population is shown in parenthesis for the SNPs with the highest association at each locus. SNP, Single nucleotide polymorphysm; HBF, fetal hemoglobin.
The effect of the −158 C→T determinant is, however not consistent, being associated also with normal HbF levels both in normal individuals as well as in β-thalassemia heterozygotes. In contrast in conditions of erythropoietic stress, as occurring in homozygous β-thalassemia or in sickle cell anemia,12 the Gγ −158 C→T mutation may lead to high production rate of HbF, thereby playing a role in the amelioration of the phenotype. For instance some β-globin mutations, i.e. codon 6 -A and codon 8 –AA, which are in linkage disequilibrium with the Gγ −158 C→T, are consistently associated with higher HbF production and evolution toward the clinical phenotype of thalassemia intermedia.
The role of the Gγ 158 C→T polymorphism as a quantitative trait locus for HbF has recently been confirmed by genomic association studies. By GWA (Genome wide association) studies the trait variance attributed to this locus was 10.2% in Northern Europeans.13–14 The mechanisms of the Gγ −158 C→T variance on the HbF level is not well defined, but may be related to an elevated Gγ gene expression.
Fine mapping of this region in African-Americans with sickle cell anemia found a SNP rs10128556 located downstream of HBG1, more strongly associated with HbF levels than rs7482144-XmnI indicating that at least in African-Americans the rs7482144-XmnI is not the causal variant.15 Similarly, in Sardinias a different SNP mapped in the HS3 (Hypersensitive site 3) of the locus control region of the β-globin gene cluster (rs4910742), which is not in linkage disequilibrium with the rs7482144-XmnI, accounts for 2.58% of the HbF variance.14
6q QTL locus
In addition to the β-globin gene cluster, positional cloning and successive genomewide linkage analysis of an Asian-Indian kindred family with β-thalassemia and an independently segregating high HbF trait led to map a quantitative HbF trait locus on chromosome 6q23–q24 (Figure 2). This trait appeared to be associated with the development of a mild β-thalassemia phenotype.16,17
Later on, high resolution association mapping in families of Northern European ancestry identified multiple genetic variants in the 1.5 Mb 6q23–q24 interval on chromosome 6 strongly associated with F cell levels (P=10–75). This variant maps in the intergenic region between the HBS1L gene and the MYB (myeloblastosis) oncogene and accounts for ∼19% of the population trait variance,13 in this population. In African-Americans with sickle cell anemia, haplotypes defined by three SNPs mapping at this locus explained more variation in HbF levels than the cumulative sum of the phenotypic variance explained by the SNPs individually (7.3% versus 6.8%, difference however not significant).15 Resequencing of the HBSA11L and MYB genes mapping in this region detected several missense variants in both genes. However, a significant difference for MYB was detected in carriers with high HbF versus non carriers, indicating that most likely MYB is causally involved in controlling HbF.15 The mechanism regulating the Hb F level of this locus has not been so far defined. Recent studies by chromatin immunoprecipitation with microarray analysis (ChIP-chip) in primary human erythroid precursors have shown within this region the presence of strong histone acetylation, GATA-1 recognition motifs, coincident with DNAse 1 sites and associated with intergenic transcription, suggesting the existence of a regulatory region controlling the MYB expression.18 The erythroid precursors of individuals with high F have low MYB expression. In addition to HbF level, this region influences also the erythrocyte count and volume and the platelet count. A further contribution of the role of MYB in controlling HbF variation comes from a study on the mechanism of delayed switching and persistence of HbF in human trisomy 13 patients.19 By using partial trisomy cases the HbF trait was mapped to chromosomal band 13q14. Examination of this region showed two microRNA (miR15a and −16.1), as candidate genes for elevated HbF levels. By a combination of genetic and functional studies in human cells, overexpression of miR15a/16.1 resulted in elevation of HbF expression, which may therefore explain the delayed switch and persistence of HbF in trisomy 13. This effect appears mediated through downregulation of the MYB transcription factor (a potent negative regulator of HbF expression), which is a direct target of miR15a and miR16.1.19 These results indicate that downregulation of MYB expression leads to delayed switch from HbF to HbA (Hemoglobin A) and persistent expression of HbF.
These findings indicate that the 6q23 locus controls the expression of MYB, which in turn influences the kinetics of erythroid differentiation and thereby HbF production. Previous studies have in fact demonstrated that the acceleration of erythropoiesis occurring in situation of erythropoietic stress is associated with high HbF production.20
The role of 6 QTLs (Quantitative trait loci) on the regulation of HbF have been later on confirmed in normal individuals and subjects affected by β-thalassemia and sickle cell anemia of different ancestry.14,2,22
Chromosome 2 QTLs
Genomic-wide association studies carried out in 4000 Sardinians, including normal individuals and carriers of β and α-thalassemia, confirmed the association of HbF levels with the β-locus (sr4910742 P <1.80×10−21) and the MYB/HBS1L locus (sr135468266 P<1.20×10−16), but also showed a strong association on chromosome 2, in intron 2 of the BCL11A gene (SNP sr11886868 P<6.70×10−35)14 (Figure 2). A similar result (P<1019) was obtained by genotyping a group of 179 individuals with extreme FC values (above the 95th and below the 5th percentile) selected from a cohort of 5184 individuals of Northern European ancestry. The BCL11A locus accounts for 15.1% of the FC variability in Northern European23 and for 7.66% of the HbF variance in Sardinians14 (Figure 2).
Fine mapping at the BCL11A locus in African-Americans with sickle cell anemia found three SNPs independently associated with HbF. Haplotype analysis (based on these SNPs), indicated that haplotypes at this locus explain more phenotypic variance than the cumulative sum of the three SNPs taken individually (18.1 versus 14.7%).15
These findings were later confirmed in normal population of different ancestry, in Chinese and Thai carriers of β-thalassemia24 and in patients with sickle cell anemia from USA, Brasil and UK.21
In our studies in 1412 individuals directly genotyped for rs11886868 (intron 2 of BCL11A) the genotype distribution was markedly different between individuals with high HbF levels (>0.8% HPFH) and those with normal levels (<0.8%). Genotype frequencies were 67% (T/T), 30% (T/C) and 3% (C/C) in individuals with normal HbF levels (Table 2). In subjects with HPFH these frequencies become 40%, 47% and 13% with a marked enrichment in the C allele and the C/C genotype. Furthermore, the C variant at rs11886868 in BCL11A locus was more frequent in β-thalassemia homozygotes with a mild phenotype (52 patients with thalassemia intermedia) as compared with those with a severe transfusion-dependent form (74 patients). These findings clearly indicate that the BCL11A gene is able to ameliorate the phenotype of homozygous β-thalassemia by increasing HbF levels (Table 2).
Table 2. Distribution of rs11886868 genotypes in thalassemic patients, HPFH individuals, and the general population from Ogliastra (Sardinia).
| N Individuals | C/C | C/T | T/T | |
|---|---|---|---|---|
| HPFH* | 66 | 0.227 | 0.546 | 0.227 |
| Thalassemia intermedia | 52 | 0.192 | 0.597 | 0.211 |
| Thalassemia major | 74 | 0.040 | 0.355 | 0.635 |
| Sardinia | 1,412 | 0.040 | .0316 | 0.644 |
HPFH, hereditary persistence of fetal hemoglobin.
HPFH was detected by population screening in the entire Sardinian population.
As in β-thalassemia, in patients with sickle cell disease from the Cooperative Study of Sickle cell disease (CSSCD), which includes African-American patients with homozygous sickle cell disease (SS) and sickle hemoglobin C disease (SC), the C allele at marker rs11886868 was strongly associated with increased HbF (P<10–20), accounting for 8.6% the variance in the trait14 and reduced pain crisis.21
Other loci
Recent studies have shown that polymorphisms in the olfactory receptor gene region mapping upstream of the β-globin gene cluster LCR, are associated with HbF concentration in sickle cell anemia.25 This region may modulate HbF levels by an effect on the chromatin structure of the β-globin cluster. On the other hand, other loci on Xp22.2–22.3 and 8q26–28 outside the β-globin gene cluster previously found to affect HbF, were not supported by GWA studies.13,14
Epistasis and relative contribution of the loci identified on HbF values
Epistasis among the various loci detected by GWA was limited to the interaction between pairs of SNPs on the β-globin locus and between rs4895441 on the MYB/HBS1L locus and rs1391619 on chromosome 11.14
In African-Americans the HbF phenotypic variation explained by the three loci on 11q, 2q and 6q following haplotype analysis increased from 23.5 to 30.1%.15
Recently our group has evaluated the relative contribution of BCL11A, HBS11/MYB loci and co-inheritance of α-thalassemia, on the clinical phenotype of homozygous β°-thalassemia.22 We confirmed that the α-thalassemia and allele C of SNP rs11886868 in BCL11A associated with high HbF were more frequent in thalassemia intermedia versus thalassemia major. Likewise the G at SNP rs9389268 in HBS1L-MYB region associated to elevated HbF was more represented in patients with thalassemia intermedia. The contribution of the three loci BCL11A, HBS11L-MYB and ケ-thalassemia expressed as OR were 5.15, 4.61 and 3.31 respectively (Table 3).
Table 3. Summary of the association with amelioration of β°-thalassemia.
| SNP/locus | Allele | Frequency thalassemia major | Frequency thalassemia intermedia | P | OR (95% confidence interval) |
|---|---|---|---|---|---|
| rs11896968/BCL11A | C | 0.21 | 0.48 | <.001 | 5.15 (2.46–12.06) |
| rs9389268/HBS1l-MYB | G | 0.15 | 0.35 | <.001 | 4.61 (2.18–10.76) |
| HbA* | - | 0.25 | 0.52 | <.001 | 3.32 (1.75–6.80) |
For each marker, we reported the minor allele with the correspondent frequency on the 2 groups of patients, the P value for allelic difference and the odds ratio.
For the HBA locus, genotypes have been coded as 0, 1 or 2 according to the number of mutated copies of the HBA gene. Because of small counts, persons with -α/αHpHα and -α/αNcoIα have been grouped with -α/-α, αNcoIα/αα with -α/αα HBA, human alpha-globin gene.
These three loci act in additive fashion, with each copy of the modulating allele at each locus contributing to the amelioration of the clinical phenotype. In complex these genetic factors account for 75% of the variation in phenotype severity.22
Our results are at variance with those observed in healthy Northern European population, where HBS1L-Myb locus has a stronger effect than BCL11A on the F cell number.13
Genotyping at these loci in neonatal period may have a practical utility, serving as a prognostic indicator for the severity of β-thalassemia and sickle cell anemia, thereby improving the genetic counseling. Furthermore, targeted manipulation of BCL11A in erythroid precursors may boost the production of HbF eventually leading to cure β-thalassemia and sickle cell anemia.
Accordingly, transduction of CD34 cells with lentiviral vector carrying a short hairpin RNA (shRNA) targeting the γ-globin gene repressor BCL11A, led to significant increase of HbF in differentiated erythroblasts, around 10% in cells derived from normal donors and from 33 to 45% in β-thalassemic cells.29
Function of BCL11A and implication for Hb switching
BCL11A (B cell lymphoma-leukemia 11A also dubbed EV19 (ecotropic viral integration site 19), CTIP1 (COUP-TF-interacting protein 1) is a zinc-finger transcriptional factor necessary for normal B and T lymphopoiesis.30 Recent GWA studies mentioned before have demonstrated an additional role in controlling HbF levels in peripheral blood.14,21,23
Further studies have demonstrated in human erythroid progenitors and in the knockout mouse that BCL11A functions as a regulator of γ-globin gene expression.31 BCL11A binds the HS3-LCR and the intergenic region between the Aγ-globin and δ-globin (Figure 3).32 The Aγ-δ intergenic region overlaps a previously reported deletion associated with high HbF levels.29 However, in these studies BCL11A does not bind to the γ-globin gene promoter. In partial agreement with Sankaran,31,33 Jawaid et al.,34 by ChIP-chip experiments, in mouse basophilic erythroblasts cultivated from peripheral blood, showed BCL11A binding to LCR-HS1, HS2, HS3 and HS7, 3′ end of HBE1, 3.4 Kb downstream of HBG1 and 1.5 Kb upstream of HBD, at OR52A1 gene, which is ∼75Kb downstream of HBB (a region which contains a 3′ enhancer as indicated by studies in HPFH deletion).35 Binding to LCR was usually coincident with GATA-1 binding. In contrast, EMSA studies in K562 cells found binding of BCL11A to a GGCCGG motif at position −56 to −51 of the HBG2 proximal promoter.36 However, expression of BCL11A in K562 cells has not been consistently detected.34
Figure 3.

Regions of the β-globin cluster bound by Bcl11A in ChIP and ChIP seq studies.
BCL11A shows abundant expression in primary adult human CD34 derived erythroid precursors, where its downregulation by siRNA or shRNA leads to marked induction of γ-chains and thereby increased HbF.31 Genetic variants at the BCL11A locus associated with high HbF levels show a reduced expression of BCL11mRNA. By proteomic approach, BCL11A was found to interact physically with the NURD (Nucleosome Remodelling Deacetylation) complex (which contains two histone deacetylase (HDAC1) and, the nuclear matrix component Matrin 3, the GATA1 and FOG1 erythroid transcriptional factors and the HMG-box protein SOX6, a chromatin associated protein [member of the SRY-related high mobility group (HMG) box transcription factors] that binds and induces a marked bending of DNA (Figure 4).31,33,37 In fetal liver definitive erythroid cells of the Bcl11a knockout mice the mouse embryonic globin genes are markedly upregulated, whereas in normal mice they are silent. Knockout of Bcl11a transgenic mouse, carrying the human β-globin locus, led to a failure of developmental silencing of the mouse embryonic globin and human γ-globin gene.38 These findings indicate the BCL11A in cooperation with SOX6 (see later) and other transcriptional factors is able to silence the γ-globin gene, thereby actively participating in the switching process.37
Figure 4.
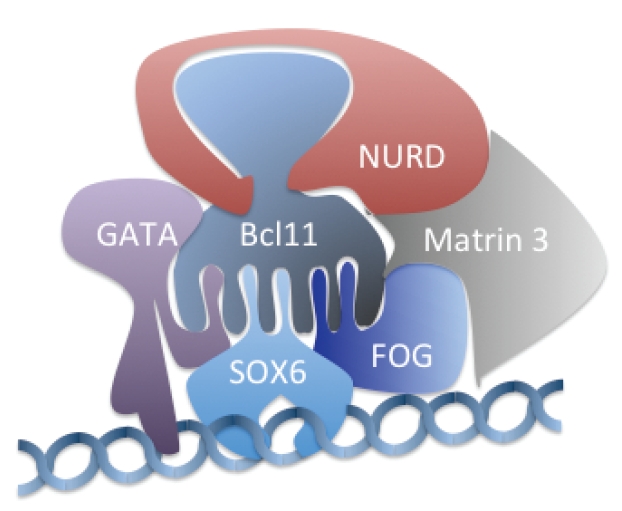
Schematic representation of the most important partners that interact with Bcl11A.
Downregulation of BCL11A expression may lead to a new approach for activation of HbF production in hemoglobin disorders.
Function of KLF1 (Krueppel-like factor1) in Hb switching
KLF1 (previously dubbed EKLF) is a zinc-finger erythroid transcriptional regulator that binds to the critical promoter elements (CACCC box) of the adult β-globin gene.39 Knockout of Klf1 in mice leads to embryonic lethality because of the development of severe β-thalassemia.40–41 Accordingly mutation of the CACCC box of the human β-globin gene results in the phenotype of the β+-thalassemia.42 Two new studies43–44 demonstrate that KLF1, in addition to activate the β-globin gene, down-regulates the BCL11A gene, thereby leading to increased γ-globin chain production. Zhou et al.,43 by studying human and mouse adult erythroid progenitors, have shown that knockdown of Klf1 by Klf1 shRNA lentiviral vectors leads to marked reduction of Bcl11a protein and Bcl11mRNA and to an increase in human γ-globin/β-globin expression ratio. Furthermore, by ChIP quantitative PCR (qPCR), they showed also that Klf1 binds the CACCC box of the mouse Bcl11a promoter in adult bone marrow erythroid cells, suggesting that Klf1 directly regulates the Bcl11a expression. Borg et al.44 studied a large family of Maltese origin with HFPH by genomic-wide SNP analysis followed by linkage analysis and identified a shared haplotype at 19p13.12,13 in all members of the family with HPFH. Sequencing of KLF1, mapping in this region, revealed a nonsense mutation (K288X) abrogating its DNA bindings domain and segregating with the HPFH phenotype in this family. Genomic-wide expression analysis from erythroid progenitors cultured from peripheral blood showed downregulation of KLF1 target genes, including BCL11A in all individuals with HPFH. Quantitative RT-PCR demonstrated, in addition, increased expression of the HBG1/HBG2 genes. KLF1 knockdown by specific lentiviral shRNA construct in human erythroid precursors from healthy donors, led to increase of HBG1/HBG2 expression and reduction in BCL11A expression. In adult erythroid progenitors, but not in fetal liver derived erythroid progenitors, KLF1 in addition to the HBB promoter binds also strongly to the BCL11A promoter. This study indicates that the HPFH phenotype may result from KLF1 haploinsufficiency and that KLF1 by regulating the BCL11A expression indirectly influences the γ-globin expression. In accordance with these studies in mouse and human, a transfusion-dependent congenital dyserythropoietic anemia associated with high HbF and resulting from a mutation in KLF1, was recently described.45 It is interesting to note that the causative mutation E325K maps on KLF1 at the equivalent site as the NAN mutation in the mouse which causes a severe congenital anemia in this species.46
All these studies led to propose a coherent mechanism of hemoglobin switching. In embryonic and fetal development or in KLF1 haploinsufficient adult Klf1 concentration is reduced resulting in low levels of β-globin and BCL11A and high levels of γ-globin. In normal adults with two copies of KLF1, the higher levels of Klf1 determines high expression of b-globin and BCL111A that represses γ-globin expression (Figure 5). Controlled pharmacological inhibition of KLF1 by downregulating BCL11A and augmenting γ-chain expression, could be explored as a strategy for ameliorating hemoglobin disorders such as β-thalassemia and sickle cell anemia.
Figure 5.

Current hemoglobin switching model. Globin gene activation is dependent upon concentration of Klf1. In the fetal stage of erythroid development or at low Klf1 concentration, weakly expressed Bcl11A can not repress γ-globin genes and Klf1 preferentially interacts with the γ-globin genes that are stimulated upon recruitment of LCR elements. In the adult stage or at high Klf1 concentration, overexpressed Bcl11A represses γ-globin genes and Klf1 binds preferentially the β-globin genes that are stimulated upon recruitment of the LCR (Inactive genes are grayed, active genes are in light green). LCR, Locus control region; KLF, Krueppel-like factor; Bcl11A: B cell lymphoma 11°.
However, exploration of a direct pharmacological activation on BCL11A could be preferable because this approach should avoid the eventual adverse effect of inhibition of KLF1 on β-globin chain synthesis.
Iron overload assessment and treatment
As a consequence of repeated long-life red blood cell transfusions, essential for survival, patients with thalassemia major accumulate over time iron in the body. Excess iron is toxic to many tissues, including the liver, endocrine organs and heart, leading to a series of complications which cause morbidity and mortality in these patients. Hypogonadism reported in more than 50% of the patients, hypothyroidism in 10% and hypoparathyroidism in 9%, are still the most common complications in thalassemia major.47 Diabetes, heart failure ad arrhythmias have been described in 6.4%, 6.8% and 5.7% respectively.47 Liver disease, resulting from infections with viruses that cause hepatitis B and C, affects up to 70% of the patients. Moreover, post-transfusional liver iron overload associated with viral chronic hepatitis increases the risk of cirrhosis and hepatocarcinoma.48 As the body has no effective means for removing iron, the only way to remove excess iron is to use iron binders (chelators), which allow iron excretion through the urine and/or stool. The first drug available for treatment of iron was deferoxamine (DFO), which has been introduced in clinical practice in the 70's. Its use has significantly reduced morbidity and improved survival in patients with thalassemia major. However, because of side effects and the inconvenient parenteral administration (see later), a consistent proportion of patients do not comply with deferoxamine treatment. This is probably the reason why iron overload related complications are still relatively frequent and the most common cause of death in patients with thalassemia major is cardiac disease. In fact, in different groups, death for cardiac disease has been reported in 60–70% of the patients.47,49,50 In the last years two major advances have substantially changed the prognosis of the patients with thalassemia major: the introduction in the clinical use of the oral iron chelators and the development of non-invasive methods for the assessment of specific organ iron overload.
Non-invasive assessment of organ iron overload
The accurate evaluation of iron overload is necessary to plan and to monitor iron chelation. Serum ferritin is an easy, worldwide available and cheap method to indirectly assess iron stores. In the absence of confounding factors such as infections, liver disease, neoplasia and vitamin C deficiency, serial measurements of serum ferritin allow longitudinal follow-up of patients and are useful for monitoring chelation therapy. However, serum ferritin does not accurately reflect hepatic and cardiac iron51,52 and since serious complications, limiting survival, may occur as a consequence of increased iron content in these organs, liver and heart iron assessment is recommended in patients with thalassemia major. Liver iron concentration (LIC) can be determined by liver biopsy, but the procedure is invasive and complications requiring hospitalization have been reported in about 0.5% of patients who undergo liver biopsy, even in experienced centres.53 Therefore, for the associated risks liver biopsy cannot be repeated for routine follow-up necessary to plan and adjust chelation therapy. Moreover, distribution of iron in the liver seems to be heterogeneous also in absence of cirrhosis.54,55 Magnetic bio-susceptometry, by the superconducting quantum interference device (SQUID) is a non-invasive method used to measure liver iron.56 The method has been validated, but has some disadvantages including underestimation of liver iron concentration, limited availability (only 4 functioning devices are at moment present in the world) and the incapability of assessing iron in the heart. Moreover, it has been shown that liver iron does not correlate with heart iron in a proportion of patients.51 Measuring cardiac iron is critical for thalassemia major patients because iron-related cardiac complications (i.e. cardiomyopathy, congestive heart failure and arrhythmias) are relatively common and severe and cardiac death is the most frequent cause of death in thalassemia.47,49,50 The introduction of magnetic resonance imaging (MRI) for the assessment of tissue iron in the early 2000, completely changed our understanding of iron overload and its management.
MRI has proven effective in detecting and quantifying iron in the heart, liver and other organs (Figure 6). Besides being non-invasive, MRI is inexpensive, widely available in developed and also in developing countries, robust and reproducible in the clinical relevant range.57,58 MRI does not image the iron directly but instead images water protons desynchronized by the iron deposits in the tissue of interest.59,60 Iron shortens T2* relaxation time, which has become the most sensitive, robust and therefore most used measure of cardiac iron. In 2001 Anderson et al.61 published a fundamental paper describing the relationship between cardiac T2* and left ventricular ejection fraction in patients with thalassemia major.61 Cardiac T2* greater than 20 milliseconds (ms) indicates normal iron content, while values between 10 and 20 ms or lower than 10 ms indicate moderate or severe iron overload respectively. As T2* decreases below 20 ms, increases the risk of having a decline in ejection fraction, but it should be pointed out that many patients with moderate or even severe cardiac iron may have normal cardiac function, suggesting that T2* is able to identify preclinical cardiac dysfunction. In a large retrospective study in UK, Kirk et al.62 evaluated the relevance of heart T2* as predictor of cardiac failure. Forty-seven percent of patients with T2* lower than 6 ms developed cardiac failure within 12 months from detection. Identifying at risk patients before cardiomyopathy develops, allows the use of targeted iron chelation treatment to reduce cardiac mortality.52 Prediction of arrhythmia was also significant but of lower magnitude.
Figure 6.
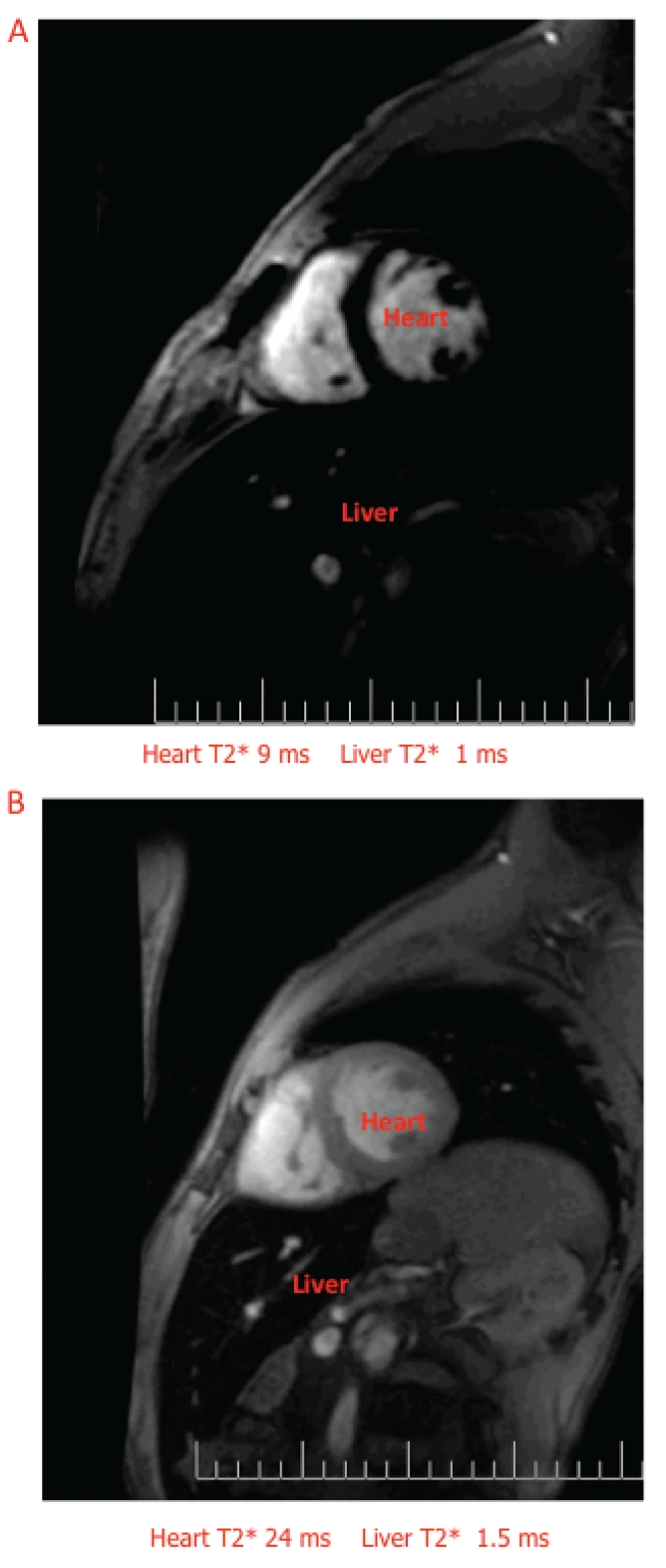
Cardiac MRI in patients with thalassemia major. A. Severe liver and heart iron. B. Severe liver iron and normal heart iron (courtesy of Dr. G. Matta).
Despite the limitation of the lack of correlation between LIC and heart iron in a proportion of patients, assessment of liver iron is important because iron is stored mainly in the liver and LIC accurately reflects total body iron stores.53 LIC can be used to monitor efficacy of iron chelation therapy. For MRI evaluation of liver siderosis different methods, including signal intensity ratio and relaxometry techniques, were introduced. At present T2*, R2 and R2* methods are the most widely used, R2 and R2* being the most accurate63 MRI methods have been calibrated with LIC obtained by liver biopsy.61,64,65 Although MRI is commonly used to assess liver and heart iron, it can also be used to estimate tissue iron overload in other organs including pituitary gland and pancreas. The pituitary gland is one of the most vulnerable target organ to early toxic effects from iron overload, leading to hypogonadotropic hypogonadism in a large proportion of patients.47,66 Pituitary R2 is closely correlated to liver and pancreatic iron, but not to heart iron. This suggests that kinetics of pituitary iron loading is relatively fast and starts already in childhood.67 Pancreatic MRI-T2*Iron overload is thought to produce oxidative stress in the pancreas beta cells and is frequently associated with diabetes and/or glucose intolerance. Au et al.68 studied pancreatic MRI-T2* in thalassemia major patients with and without diabetes. Pancreatic T2* signals were abnormal in 80% of patients in both groups, but diabetic patients had a smaller pancreas volume (P<0.0001). In normoglycemic patients, log-pancreatic T2* values correlated with homeostatic model assessment HOMA-B (beta cell reserve), HOMA-IR (insulin resistance) and fasting insulin/C-peptide levels. In a longitudinal study of 131 patients with thalassemia major pancreatic and cardiac R2* were correlated (r2=0.52), with significant pancreatic iron occurring earlier than cardiac iron.69 Based on these results, pancreatic R2* measurements can be considered a useful marker for future cardiac iron deposition allowing to plan an appropriate iron chelation therapy.
Iron deposition in other organs and tissues has not yet been extensively examined.
Oral iron chelators
Survival of patients with thalassemia major depends on long life red blood cell transfusions that inevitably lead to the accumulation of iron in the body. Excess iron is toxic because it generates free radicals and induces oxidative stress resulting in cell and organ damage. Excess iron from transfusions is distributed initially to macrophages and to the liver, then iron accumulates into the endocrine organs and heart causing, if not treated, the complications typically associated with iron overload, such as hypogonadism, hypothyroidism, hypoparathyroidism, diabetes, liver fibrosis and early death usually for cardiac failure. The aims of iron chelation are to reduce dangerous excess iron to safe iron levels, to maintain currently acceptable levels, to minimize exposure of tissues to the toxic free iron and possibly to prevent iron accumulation. Deferoxamine (DFO), the first iron chelator, was developed more than 40 years ago and has been shown to decrease morbidity from iron overload and to prolong life in patients who comply with therapy. Due to its high molecular weight ad its short half-life (approximately 20 min) DFO must be administered by parenterally route (usually subcutaneous), 8 to 12 hours/day, 5 to 7 days/week. This demanding regime results in poor adherence to therapy limiting the efficacy of this chelator in a significant proportion of patients. As a consequence the search for more practical orally available chelators has been a major challenge during the last few decades.
Deferiprone
Deferiprone (1,2 dimethyl-3-hydroxypyrid-4-one) (DFP) was synthesized in 1984 at the University of Essex in London and was first used in humans in 1987. The compound was initially screened in cell lines and subsequently tested in animal models of iron overload, proving that it was absorbed by gastrointestinal (GI) apparatus and caused excretion of excess iron. DFP is a bidentate chelator, therefore three DFP molecules are required to bind the six coordination sites of an iron atom (Fe+++ ) (Figure 7) DFP has strong iron-binding capacity particularly for ferric iron (pFe+++=19.6) and is rapidly absorbed after oral administration, reaching plasma level peak about 1 hour after giving medication.72
Figure 7.
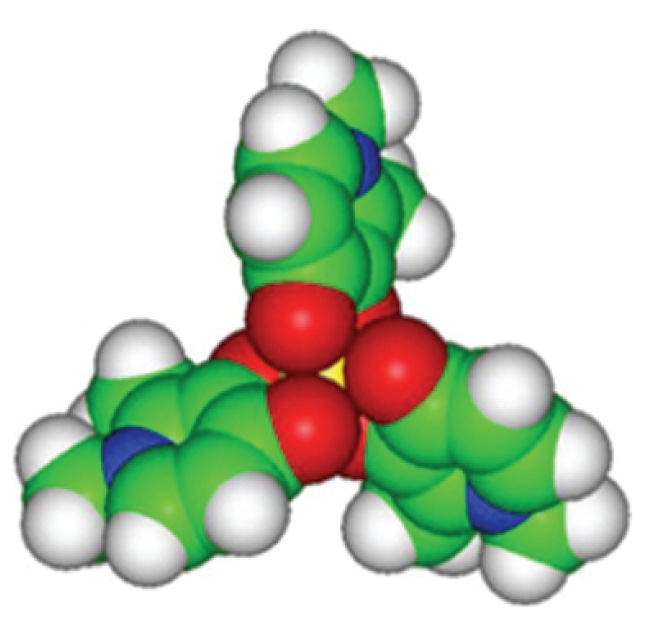
Structure of the deferiprone/iron complex. Three molecules of deferiprone bind one iron atom (yellow).
Most of the iron DFP complex is excreted in the urine and a minimal amount in the feces. Due to the relatively short half-life (160 min) and the rapid glucuronidation into an in active metabolite, DFP requires thrice-daily administration.73 Although the most used dose is 75 mg/kg/day, DFP can be given safely up to 100 mg/kg/day according to the severity of iron overload and to the patient's tolerability.
Deferiprone efficacy
The purpose of an effective iron chelation therapy is to prevent or reduce body iron accumulation. In regularly transfused patients the rate of iron accumulation, originated from destroyed senescent red cells, is 0.3–0.5 mg/kg/day. Therefore, a chelator should be able to remove at least this amount of iron to minimize the risk of iron-induced toxicity.
Several studies have shown that DFP, at comparable doses has an efficacy similar to that of DFO, and that iron excretion increases with the dose and with the degree of iron overload (Grady R. personal communication).73,74 DFP at appropriate doses is able to decrease or stabilize body iron as assessed by sequential serum ferritin or liver iron concentration, despite repeated RBC transfusions.75–77 Several independent studies have shown that DFP is more effective than DFO in removing cardiac iron, improving cardiac function and reducing mortality for cardiac disease.51,78–80 The most comprehensive results have been obtained in a large prospective trial, where 61 patients with moderate cardiac iron load (T2* between 10 to 20 ms), previously chelated with DFO where randomized to be maintained on DFO or switched to DFP.51 After 12 months of treatment a significant improvement in myocardial T2* was observed with both treatments, but the improvement was greater with DFP than with DFO [+27% vs 13% (P=0.023)]. Left ventricular ejection fraction also increased significantly, more in the DFP group [3.1% vs 0.3%(P=0.030)]. DFP treatment showed a significant greater cardiac protection as compared to DFO in a large natural history study on survival and causes of death in patients from seven Italian thalassemia centres.80 The study included cardiac events and survival for 359 patients on DFO, during 8 years of observation and for 157 patients switched from DFO to DFP. At the baseline, the two groups were comparable for age and sex, while serum ferritin was significantly higher in patients switched to DFP (mean serum ferritin 1860 vs 1460 µg/L, P<0.001). DFP- switched patients did not experience any cardiac event, whereas 52 cardiac events, including 10 cardiac deaths, occurred in patients on DFO. The greater efficacy of DFP in removing excess cardiac iron may be due to some pharmacochemical characteristics of DFP, such as low molecular weight, neutral charge and lipophilicity, which facilitate myocyte membrane crossing and chelation of intracellular iron.81
Deferiprone and Deferoxamine association
DFP and DFO can be given to the same patient with different regimes: in combination on the same day, either simultaneously (i.e. DFP given before breakfast, lunch and dinner and DFO infused during the day) or sequentially (i.e. DFP as above and DFO infused overnight), or as alternate treatment (i.e. one or the other chelator is given on different days). Combination therapy is considered an intensive chelation regimen and usually DFP is administered every day, while subcutaneous DFO is given 2 to 7 days/week, according to the severity of iron overload. In patients with heart failure, to reinforce chelation, DFO can be given intravenously 24 h/day. The potential advantages of the combined chelation are reported in Table 4. Several single case reports and prospective studies have shown that combined intensive chelation is effective in reducing cardiac siderosis and in improving cardiac function in patients with severe heart iron overload and heart failure.82–85 A randomized, placebo-controlled, double blind study suggested that in comparison to the standard chelation with DFO, combination therapy with DFO and DFP, was able reduce myocardial iron (ratio of change in cardiac T2* geometric means 1.5 in combined therapy vs 1.24 in DFO monotherapy, P=0.02) and improve LVEF (2.6% vs 0.6% P=0.05).52 In a study from Greece, reversal of endocrine complications, with very intensive combined chelation (DFP 75–100 mg/kg/day and DFO 20–60 mg/kg/day), has been reported.86 In particular, abnormal glucose metabolism was normalized in 17 (44%) of 39 patients. In 18 patients requiring thyroxine supplementation for hypothyroidism, 10 were able to reduce their thyroxine dose. In 14 hypogonadal males on testosterone therapy, 7 stopped treatment and quite remarkably, 6 of 19 females, who were hypogonadal on DFO monotherapy, were able to conceive. Combination therapy has been shown to improve survival in patients with thalassemia major.
Table 4. Potential advantages of combination therapy.
| Access to different iron pools |
| Greater efficacy |
| Dose decrease® toxicity decrease |
| Chelation of toxic free iron |
| Better tolerability and compliance |
In 1999 combination therapy was introduced in Cyprus because of the increased trend of deaths for cardiac disease, despite the use of intensive intravenous DFO in patients at increased risk of heart failure.49,87 The analysis of the survival trends from 1985 to 1999 and after 2000, showed a significant reversal of this trend in mortality for cardiac disease. None of the patient on combination therapy died for cardiac disease. Alternate chelation efficacy has been prospectively evaluated in a study where 60 thalassemia major patients, regularly chelated with DFO, were randomized to continue DFO alone or to receive 5 days/week DFP and 2 days/week DFO for 1 year.88 Comparable efficacy on serum ferritin and liver iron concentration was observed in both treatment groups. More recently, in a large long-term (5 years of follow-up), multicenter study, 213 patients were randomized to receive DFO for 4 days/week and DFP for the remaining 3 days, or DFP monotherapy for 7 days/week.89 In the alternating treatment group serum ferritin showed a significant reduction (P= 0.005) as compared to DFP group. DFO monotherapy versus DFP monotherapy, alternate or combined DFO-DFP regimes, have been evaluated in another long-term, multicenter, randomized trial including 265 patients.90 None of the patients on DFP alone or in combined treatment died, one death occurred with alternate treatment and 10 deaths with DFO treatment.
Deferiprone safety profile
Gastrointestinal symptoms (GI) (nausea, vomiting, abdominal pain) are the most commonly reported DFP-related adverse events. In a prospective long-term study 187 patients where monitored for 4 years.91 GI symptoms occurred in 33% subsequent years. In general these symptoms were mild or moderate and only a restricted number of patients discontinued therapy. Similar results have been reported in other studies.73,92 A lower incidence of GI adverse events (vomiting 6% of patients, abdominal pain 6% and nausea 1%) has been described in patients taking the new DFP liquid formulation.93 Arthropaty, particularly involving the knees, has been associated with the use of DFP in a variable number of patients (3.9–20%).91,92 A particularly high incidence of arthropaty, reported in Indian patients (up to 41%), has been attributed to the severe iron overload or undefined etnicHeading 2 genetic background.94 Unlike GI problems, which usually appear in the first weeks of treatment, the onset of joint symptoms may be late, even after years of treatment. The symptoms, usually mild, resolve in a few days with non-steroid anti-inflammatory drugs, or temporary discontinuation of DFP or dose reduction.95 Transient and mild to moderate increase in liver enzymes has been reported in about 7% of the patients, particularly in the first months of treatment. However, ALT monitoring at regular intervals is recommended and DFP interruption or dose reduction should be considered if ALT levels are significantly and persistently elevated.
The most serious side effect associated with DFP use is agranulocytosis [absolute neutrophil count (ANC) lower than 0.5×109/L]. In controlled studies it occurred in about 1% of the patients (0.2–0.3 episodes per 100 patient/years) usually within the first year of treatment.91,92,96 Agranulocytosis is reversible with interruption of DFP, but treatment with G-CSF may be needed for faster recovery. Deaths from severe infectious associated with DFP-related agranulocytosis have been reported in some patients.97 Weekly ANC monitoring is recommended to detect early signs of agranulocytosis. Moreover, patients should be counselled to stop DFP in case of fever and report to their physician any symptom indicative of infection. For the high risk of recurrence, rechallenge with DFP is not recommended in patients who developed agraulocytosis. Mild neutropenia (ANC between 0.5–1.5×109/L) occurs in 3.6–8.5% of the patients, more commonly in non-splenectomized patients and in association with viral infections.73,91,92,96 This condition is usually reversible with discontinuation of the drug.
Deferasirox
Deferasirox (DFX) is an oral iron chelator, administered as a suspension, that has been developed by computer modelling, after evaluation of more than 700 compounds from several chemical classes. It belongs to a new class of tridentate chelators, binding iron in a 2:1 ratio (Figure 8). DFX-iron complexes are primarily excreted with feces.98 DFX underwent extensive safety testing and clinical trials including several thousands of patients worldwide.99–105 The plasma half-life of 8 to 16 hours supports once-daily administration and scavenging of non-transferrin-bound labile plasma iron, the toxic form of iron responsible of tissue damage.106 DFX is currently approved in many countries for the treatment of chronic iron overload, associated with blood transfusions in patients older than 2 years. However, a small-scale study indicated that DFX may be suitable for controlling iron levels also in non-transfusion dependent patients with thalassemia intermedia.107
Figure 8.
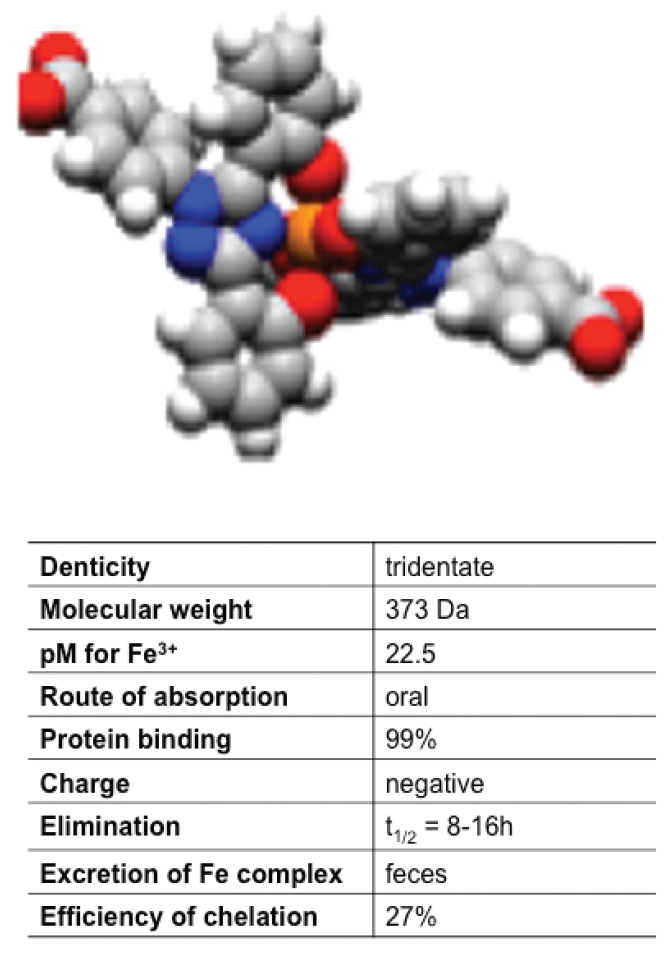
Structure of the deferasirox. Two molecules of deferasirox bind one atom of iron.
Deferasirox efficacy
Efficacy of DFX has been evaluated in large and long-term clinical studies in pediatric as well as adult patients.107–110 Serum ferritin and LIC monitoring in these studies have highlighted the importance to achieve the therapeutic goal of adjusting the dose of DFX on the basis of transfusional iron intake.93,111 In one-year studies, the proportion of patients in negative iron balance at 20 mg/kg/ day of DFX was 75% for those at low transfusional iron input (<0.3 mg/kg/day), falling to 55% and 47% respectively at intermediate (0.3 to 0.5 mg/kg/day) and high (>0.5 mg/kg/day) transfusional iron input. The highest recommended dose initially approved by health authorities in many countries was 30 mg/kg/day. A retrospective analysis of the extension phases of the core studies, investigated the efficacy (based on changes in serum ferritin levels) and safety of DFX doses higher than 30 mg/kg/day in adult and pediatric patients with different transfusion dependent anemias (i.e. thalassemia, sickle cell anemia, myelodisplasia).108 In the overall population there was a statistically significant median decrease in serum ferritin of 440 mg/L (P<0.0001) from pre-dose-escalation to the time-of-analysis, with no safety concerns (see below). Similar results have been obtained in two large prospective studies.107,109 DFX has been also evaluated long-term in pediatric patients with thalassemia major.112–114 In 24 out of 40 patients enrolled to receive DFX for 5 years, LIC was reduced in a dose-dependent manner.
Clinical data support the efficacy of DFX in removing cardiac iron and preventing myocardial siderosis in patients with thalassemia major.67,115,116 In a one year prospective multicenter trial (EPIC cardiac substudy), DFX significantly improved geometric mean myocardial T2* from baseline 11.2 to 12.9 ms (+16%, P<0.0001), with significant concomitant reduction in both serum ferritin and LIC.111 The two years extension of the same study showed continued improvement in myocardial T2* from the baseline 11.2 to 14.8 ms (P<0.001). In these studies the majority of the patients received doses of DFX higher than 30 mg/kg/day. In particular at the end of two years 53.5% of the patients were receiving 40 mg/kg/day, 9.9% were receiving 45 mg/kg/day and 2 were receiving 47 and 51 mg/kg/day, respectively.116 It should be pointed out that overall LVEF remained stable and in the normal range over the two years (67.5±5.7% at baseline; 67.7±5.1% at two years; P=0.72), but 5 patients developed LVEF less than 56% (the lowest normal value in thalassemia major patients) during the trial.116 As DFX is an oral, once-day easy to take iron chelator, compliance is expected to be high. Several studies, which have specifically assessed patient preference, demonstrated satisfaction and convenience with DFX therapy, which may translate into improvements in patient compliance and increased effectiveness of chelation therapy.117–119
Deferasirox and Deferoxamine association
Preliminary promising results have been obtained with iron balance studies, which evaluated the total (i.e. fecal and urinary) iron excretion, in three patients treated with DFX and DFO in combination.120 Total iron excretion was synergistic (i.e. higher than the sum of iron excretion obtained with each single drug) in two patients, but less than additive in the third patient, who responded the best to both drugs individually.
Alternating treatment with DFO and DFX resulted to be safe and effective in two retrospective studies including a limited number of patients.121,122
Safety
Gastrointestinal symptoms (i.e. nausea, vomiting, diarrhea, abdominal pain) and skin rash are the most common DFX-related adverse events, reported in the core pre-registration trials in 15% and 11% of the patients, respectively.123 The symptoms were mild and usually transient, though some severe cases required dose interruption and/or adjustments.124 (Exjade International website www.exjade.com) About one third of the patients have shown increases in serum creatinine, rising above the mean of the pre-treatment levels by >33% on two consecutive assessments.107 These increments were dose-dependent and non-progressive in study extensions up to 4.5 years.125 However, DFX should be interrupted if serum creatinine shows a progressive rise, beyond the age-appropriate upper limit of normal (Exjade International website www.exjade.com/exjade-safety/long-term-safety-profile.jsp) Elevations of liver transaminases have been reported in about 2% of patients and were not dose dependent. No worsening of renal or liver function has been observed following dose escalation (i.e. doses of DFX higher than 30 mg/kg/d) in a retrospective study including a large number of pediatric and adult patients with transfusion-dependent anemias.112 In the long-term (<5 years of treatment) trial conducted in pediatric patients, DFX was generally well tolerated.116 No signs of impaired growth or sexual development and no evidence of progressive renal, hepatic or bone marrow dysfunction have been reported. However, reports of renal tubulopathy (Fanconi syndrome) reversible with DFX cessation, have been recently described mainly in pediatric patients with low serum ferritin levels.126,127
Iron Chelation therapy has significantly improved in the recent years. This improvement is mainly due to greater chelation options and the possibility of accurately assess specific organ iron overload. The ability to recognize preclinical cardiac iron deposition by MRI and the availability of effective chelation regimes has significantly reduced cardiac deaths. DFP resulted to be particularly useful in reversing and preventing cardiac disease, alone and in combination with DFO. The cardiac profile of DFX is less well characterized, but the efficacy of this drug in reducing and controlling body iron overload has been clearly documented.
Despite both oral iron chelators showed a manageable safety profile, close monitoring for adverse events is recommended.
Gene therapy
Continuous improvements in the traditional care of β-thalassemia has ameliorated the quality of life and greatly improved the life expectancy.128–130 Despite these results, until recently a definitive cure could only be achieved with bone marrow transplantation (BMT) from related or unrelated donors. However, BMT is only available for a minority of patients and bears a significant risk of mortality and morbility, especially when the donor is unrelated131 or only aploidentical.132 In the search for a more general and definitive cure, hematologists have pursued alternative strategies aimed at correcting the defective β-globin gene by either gene transfer of a normal β-globin gene or substitution of the defective gene by homologous recombination. Although gene therapy would have been theoretically possible soon after the discovery and cloning of the human globin genes, for many years two main obstacles have hampered the progress in this field.133 The first obstacle has been the extremely complex regulation of the globin genes that has taken decades to at least partially unravel. The second and equally important obstacle has been the lack of an optimal vector for gene transfer into quiescent hematopoietic stem cells (HSC).
Traditional retroviral vectors
For at least two decades after the cloning of the globin genes, many different virus were explored as possible gene therapy vectors with very limited success. Moloney murine leukemia retrovirus based globin vectors were the only vectors able to infect HSCs, but they did it at a very low rate133–134 and, even worse, they were so unstable that the transduced globin gene only rarely integrated intact into the genome, thus losing any therapeutic efficacy. In fact, the regulatory sequences of the locus control region (LCR) needed to express high levels of globin, bore an intrinsic instability such that the traditional retrovirus globin vectors always displayed an inverse relationship between the amount of regulatory elements included in the vector and the final viral titer.135 Despite the failure in obtaining high level and persisting gene expression, those long frustrating years were not spent in vain as much more efforts were put to discover and assemble the optimal globin regulatory sequences.
Gene therapy with lentiviral globin vectors
Globin gene therapy perspectives changed abruptly close to the year 2000 when lentiviral based vectors were first introduced. The major advantages of these vectors were the ability to infect quiescent hematopoietic stem cells at higher rates and to export unspliced the vector RNA from nuclei, thereby ensuring the integrity of the expression cassette. In the year 2000, the group led by Sadelain published the first study in which a murine model of thalassemia intermedia was efficiently cured with a lentiviral globin vector.136 Soon after, therapeutic efficacy was demonstrated in a murine model of sickle cell disease137 and even in a murine model of thalassemia major developed by transplanting fetal livers from homozygous Th3/Th3 thalassemic mice into irradiated normal adult mice.138 In the following years different laboratories confirmed and extended initial results demonstrating efficacy of lentiviral gene therapy with xenotransplants of genetically corrected human thalassemic HCSs into recipient SCID mice.139 All these experiments used essentially the same lentiviral vector with only minor modifications. In the beginning the vector was a second generation lentivector, but, in view of the possible translation into clinical practice, to increase safety by reducing the chance to produce unwanted replication competent lentiviral particles, most of the laboratories replaced second140 with third generation lentiviral vectors141 as developed by Naldini et al. (Figure 9). The globin cassette of the vector is generally made of the beta-globin gene deleted of a destabilizing RsaI fragment contained in intron 2.142 The first gene therapy study used a long 600 bp β-globin promoter, whereas other studies preferred to use a shorter promoter version of approximately 270 bp. Person's group demonstrated good efficacy of a β-globin vector driven by the β-globin promoter and regulated by LCR sequences.143 All globin cassettes contained a small PstI DNA fragment thought to contain a 3′ β-globin enhancer. Several sequences around the DNAse I hypersensitive sites that make up the LCR are absolutely required to attain good levels of globin expression and are therefore included in all constructs for gene therapy. However, the number and the length of the DNAse I HS sites differ in all globin cassettes, reflecting the difficulty to localize the really important regulatory sequence within each HS, while omitting potentially destabilizing elements. In their ground breaking experiment, Sadelain's group provided evidences that the fragments larger than the core HS elements were needed to ensure elevated and long term gene expression of the transduced β-globin gene.144 Since then, most of the vectors included larger versions of the HSs 2–3–4, but recently Miccio et al. demonstrated good efficacy of a β-globin cassette bearing only HS2 and 3.145 On the other hand, even though human natural deletions that occurred in perfectly healthy subjects, suggest that HS1 could be omitted, recent studies showed that mice transplanted with globin cassette that included the HS1 did better than mice in which HS1 was omitted from the cassette.146 Finally, some of the vectors in use are flanked by insulators, such as the chicken HS4 insulator, to constrain the enhancing effect of the LCR to the globin transgene, while hampering the invasion of heterochromatin and the silencing of the transgene from the neighboring chromatin.147
Figure 9.

General scheme of 3rd generation globin lentiviral vectors. Vectors have deletions of all dispensable and potentially harmful HIV sequences (tat, vif, vpr, vpu, nef). The partial ORF of GAG that can not be removed because overlaps the packaging signal (Y) contains 2 in frame stop codons. The 400 bp deletion in the U3 region blocks provirus transcription after chromosomal integration (self inactivating vector). POL-GAG, REV and VSV-G envelopes are provided in trans to avoid recombination that could reconstitute wild type replication competent HIV particles. The b-globin cassette in the current vectors contains from 2 to 4 HSs, β-globin promoters of 260–600 bp, a stabilizing RsaI deletion of IVS2, variable length of the 3′ UTR and an enhancer region contained in a PstI fragment 3′ to the β-globin gene. As an additional safety feature some vector are insulated with the chicken HS4 or other equivalent insulator. Insulators that are inserted into the 3′ U3 region are duplicated and moved to the 5′ U3 region during viral reverse transcription achieving insulation of both sides of the vector. Insulation is expected to restrict the activity of the internal LCR enhancer to the globin cassette and to create a barrier to the invasion of adjacent heterochromatin that ensures persistent expression. ORF, Open reading frame; GAG, group-specific Antigen; 3′ UTR, 3′ untranslated region; LCR, locus control region.
Success in the first human gene therapy trial for β-thalassemia
After many animal experiments, in the year 2006, the first human gene therapy trial for β-thalassemia and sickle cell disease began in France. The first patient treated failed for technical reasons to reconstitute with the gene-corrected bone marrow and required back up of its own thalassemic bone marrow. The second patient, transplanted in June 2007 at age 18, has been recently reported to be free of transfusions now after 40 months of post-transplant follow up.148 The patient was a compound heterozygous HbE/β°-thalassemia with the phenotype of β-thalassemia major and regularly transfused since childhood. He lacked any available related or unrelated donor and met all criteria for inclusion in the gene therapy trial. The patient was prepared for gene transfer with myelo-suppressive doses of Busulfan (3.2mg/kg/da for 4 days)and transduced with a lentiviral vector already validated in the gene therapy of a murine model of sickle cell disease. The vector bore an antisickling β-globin(βA(T87Q)), a short 260 bp globin promoter, HSs 2, 3, 4 and 2 copies of the chicken core insulator flanking the globin cassette. The choice of a modified β-globin gene aimed at correcting anemia while antagonizing sickling and permitting discrimination of the globin produced by the vector from the endogenous derived β-globin chain.
In the months following gene transfer the patient required less and less transfusions until he became totally transfusion independent at 12 months post-transplant remaining so since then for over 30 months. Since the time he became transfusion independent, the patient was able to maintain a hemoglobin level between 9 and 10 gr/dL. Hemoglobin synthesis analysis at the steady state level showed that the vector produced 1/3rd (3.0 gr/dL) of the total hemoglobin, the remaining 2/3rd were accounted for by HbF and HbE in equal parts. Hence, in this patient a transfusion independent state was reached not only because of the supplementary 3 grams of exogenous hemoglobin, but also through a partial reactivation of the fetal γ-globin gene. Since fetal hemoglobin is genetically controlled, the increase observed in this patient may be an advantage restricted only to selected patients and not necessary related to the gene therapy procedure. Follow up analysis in the patient, discovered a dominant cell clone with integration site into the HMGA2 gene, a potential oncogene.149,150 In erythroblasts, clonal dominance reached a plateau when accounting for 77% of the integration sites at 20 months post-transplantation. The patient continues to be strictly monitored to anticipate a potential evolution into a leukemic state. However, studies suggest that isolated over expression of truncated HMGA2 is mainly associated with benign tumors or conditions such as lipomas or paroxysmal nocturnal hemoglobinuria. Although HMGA2 can also be associated with malignant tumors, the latter are tought to derive from a more complex dysregulation of several oncogenes (HMGA2, myc and ras) that are probably activated by deletions or inactivation of the let7 miR gene.151 In summary, the first gene transfer experiment in a β-thalassemia patient proves that gene therapy can succeed even in humans and represents a miliary stone in the road of gene therapy toward a definitive cure for all thalassemia patients. However, the parallel increase in fetal hemoglobin observed in the patient may suggest that other favorable modifying factors might have contributed to the therapeutic effect in this individual case. Furthermore, the emergence of a dominant HMGA2 positive cell clone, although so far harmless, casts a worrisome shadow, which only a more extended follow up should clear. Hence, larger number of patient are needed to confirm the initial positive results as well as to assure that the globin lentiviral vectors are sufficiently safe and offer to thalassemia patients a better option than the traditional transfusion and chelation therapy.
Therapeutic perspectives using iPS cells
In the last years, a new highly promising technology that allows derivation of patient specific embryonic–like stem cells from adult differentiated cells has been developed. These cells, produced by transduction with the transcription factors Sox2, Oct3/4, Myc and Klf4, are called induced pluripotent stem cells (iPS) because have the ability to differentiate into all embryonic tissues except extra-embryonic tissues. iPS can be easily obtained and have the great advantage of being expandable in large amounts and indefinitely. However the feature that makes them highly attractive in the field of gene therapy is the possibility of being amenable to gene correction by homologous recombination. A proof of principle of this use has been recently provided by employing iPS to correct a murine model of sickle cell anemia. In this experiment, iPS were derived from fibroblast harvested from a skin biopsy and induced to become iPS by transduction with retroviral vectors that encode four stem cell transcription factors. The sickle cell defect was then corrected by standard homologous recombination and, after in vitro differentiation of iPS into hematopoietic stem cells, were transplanted back into the original mouse donor, curing its SC disease. Another possible use of iPS is to overcome the risk of insertional mutagenesis by combining genetic and cell therapy.152 In this approach patient specific iPS cells are subjected to gene transfer and clones are selected based on the safety and the level of expression at the site of vector integration. Upon differentiation of such clones into hematopoietic stem cells, it should be possible to transplant back the autologous cells without the adverse effects linked to the random chromosomal integration of the vector.
Presently, iPS based techniques have the drawback of requiring potentially oncogenic retrovirus and oncogenic transcription factors to induce the iPS state. However the technology has being continuously improved in several respects mainly with the goal of reducing their oncogenic potential. Accordingly, iPS can be now derived with only transient expression of the reprogramming factors or by excision of the factors after reprogramming. It is however important to note that it is unknown whether the genomic integrity of the reprogrammed cells and reprogramming itself are consistent with safe and long-term hematopoietic reconstitution. The field is very exciting and rapidly developing and in the next future has the potential to completely revolutionize the field of genetic therapy and regenerative medicine.
References
- 1.Wheatherall DJ, Clegg JB. 4th Ed. Blackwell Science Ltd; 2001. The Thalassemia Syndromes. [Google Scholar]
- 2.Cao A, Galanello R. In: Beta-Thalassemia. Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington; Seattle: 1993-2000 [updated 2010] [Google Scholar]
- 3.Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2:245–55. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]
- 4.Thein SL. Genetic modifiers of the beta-haemoglobinopathies. Br J Haematol. 2008;141:357–66. doi: 10.1111/j.1365-2141.2008.07084.x. [DOI] [PubMed] [Google Scholar]
- 5.Cao A, Galanello R, Rosatelli MC. Genotype-phenotype correlations in beta-thalassemias. Blood Rev. 1994;8:1–12. doi: 10.1016/0268-960x(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 6.Thein SL. Genetic modifiers of beta-thalassemia. Haematologica. 2005;90:649–60. [PubMed] [Google Scholar]
- 7.Stamatoyannopoulos G. Molecular an cellular basis of haemoglobin switching. In: Steinberg MH, Forget B.G, Higgs D.R, Nagel R.L, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinica Management. Cambridge University Press; Cambridge UK: pp. 131–145. [Google Scholar]
- 8.Boyer SH, Belding TK, Margolet L, Noyes AN. Fetal hemoglobin restriction to a few erythrocytes (F cells) in normal human adults. Science. 1975;188:361–3. doi: 10.1126/science.804182. [DOI] [PubMed] [Google Scholar]
- 9.Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95:342–6. [PubMed] [Google Scholar]
- 10.Gilman JG, Huisman TH. DNA sequence variation associated with elevated fetal G gamma globin production. Blood. 1985;66:783–7. [PubMed] [Google Scholar]
- 11.Garner C, Tatu T, Game L, et al. A candidate gene study if F cell levels in sibling pairs using a joint linkage and association analysis. Gene Screen. 2000;1:9–14. [Google Scholar]
- 12.Labie D, Dunda-Belkhodja O, Rouabhi F, et al. The -158 site 5′ to the G gamma gene and G gamma expression. Blood. 1985;66:1463–5. [PubMed] [Google Scholar]
- 13.Thein SL, Menzel S, Peng X, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U S A. 2007;104:11346–51. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105:1620–5. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to effect fetal hemoglobin levels explains additional genetic variation. Nat Genetics. 2010;42:1049–51. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thein SL, Weatherall DJ. A non-deletion hereditary persistence of fetal hemoglobin (HPFH) determinant not linked to the beta-globin gene complex. Prog Clin Biol Res. 1989;316B:97–111. [PubMed] [Google Scholar]
- 17.Thein SL, Sampietro M, Rohde K, et al. Detection of a major gene for heterocellular hereditary persistence of fetal hemoglobin after accounting for genetic modifiers. Am J Hum Genet. 1994;54:214–28. [PMC free article] [PubMed] [Google Scholar]
- 18.Wahlberg K, Jiang J, Rooks H, et al. The HBS1L-MYB intergenic interval associated with elevated HbF levels shows characteristics of a distal regulatory region in erythroid cells. Blood. 2009;114:1254–62. doi: 10.1182/blood-2009-03-210146. [DOI] [PubMed] [Google Scholar]
- 19.Sankaran VG, Menne TF, Scepanovic D, et al. MicroRNA-15° and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci U S A. 2011 Jan 4; doi: 10.1073/pnas.1018384108. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18:R216–23. doi: 10.1093/hmg/ddp401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. 2008;105:11869–74. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galanello R, Sanna S, Perseu L, et al. Amelioration of Sardinian beta0 thalassemia by genetic modifiers. Blood. 2009;114:3935–7. doi: 10.1182/blood-2009-04-217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39:1197–9. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 24.Sedgewick AE, Timofeev N, Sebastiani P, et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis. 2008;41:255–8. doi: 10.1016/j.bcmd.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solovieff N, Milton JN, Hartley SW, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood. 2010;115:1815–22. doi: 10.1182/blood-2009-08-239517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dover GJ, Smith KD, Chang YC, et al. Fetal hemoglobin levels in sickle cell disease and normal individuals are partially controlled by an X-linked gene located at Xp22.2. Blood. 1992;80:816–24. [PubMed] [Google Scholar]
- 27.Miyoshi K, Kaneto Y, Kawai H, et al. X-linked dominant control of F-cells in normal adult life: characterization of the Swiss type as hereditary persistence of fetal hemoglobin regulated dominantly by gene(s) on X chromosome. Blood. 1988;72:1854–60. [PubMed] [Google Scholar]
- 28.Garner C, Silver N, Best S, et al. Quantitative trait locus on chromosome 8q influences the switch from fetal to adult hemoglobin. Blood. 2004;104:2184–6. doi: 10.1182/blood-2004-02-0527. [DOI] [PubMed] [Google Scholar]
- 29.Wilber A, Hargrove PW, Kim YS, et al. Therapeutic levels of fetal hemoglobin in erythroid progeny of {beta}-thalassemic CD34+ cells following lentiviral vector-mediated gene transfer. Blood. 2010 Dec 14; doi: 10.1182/blood-2010-08-300723. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu P, Keller JR, Ortiz M, et al. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4:525–32. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 31.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–42. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 32.Bank A. Regulation of human fetal hemoglobin: new players, new complexities. Blood. 2006;107:435–43. doi: 10.1182/blood-2005-05-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149:181–94. doi: 10.1111/j.1365-2141.2010.08105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jawaid K, Wahlberg K, Thein SL, Best S. Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells Mol Dis. 2010;45:140–6. doi: 10.1016/j.bcmd.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 35.Feingold EA, Forget BG. The breakpoint of a large deletion causing hereditary persistence of fetal hemoglobin occurs within an erythroid DNA domain remote from the beta-globin gene cluster. Blood. 1989;74:2178–86. [PubMed] [Google Scholar]
- 36.Chen Z, Luo HY, Steinberg MH, Chui DH. BCL11A represses HBG transcription in K562 cells. Blood Cells Mol Dis. 2009;42:144–9. doi: 10.1016/j.bcmd.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Sankaran VG, Ni M, et al. Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–98. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sankaran VG, Xu J, Ragoczy T, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–7. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bieker JJ. Probing the onset and regulation of erythroid cell-specific gene expression. Mt Sinai J Med. 2005;72:333–8. [PubMed] [Google Scholar]
- 40.Perkins AC, Sharpe AH, Orkin SH. Lethal β-Thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 41.Nuez B, Michalovic D, Bygrave A, et al. Defective haematopoiesis in fetal liver resulting from inactivation the EKLF gene. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 42.Moi P, Faà V, Marini MG, et al. A novel silent beta-thalassemia mutation in the distal CACCC box affects the binding and responsiveness to EKLF. Br J Haematol. 2004;126:881–4. doi: 10.1111/j.1365-2141.2004.05146.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhou D, Liu K, Sun CW, et al. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet. 2010;42:742–4. doi: 10.1038/ng.637. [DOI] [PubMed] [Google Scholar]
- 44.Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–5. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singleton BK, et al. A novel EKLF mutation in a patient with dyserythropoietic anemia: the first association of EKLF with disease in man. Blood. 2009;114:A162–A162. [Google Scholar]
- 46.Siatecka M, Sahr KE, Andersen SG, et al. Severe anemia in the Nan mutant mouse caused by sequence-selective disruption of erythroid Kruppel-like factor. Proc Natl Acad Sci U S A. 2010;107:15151–6. doi: 10.1073/pnas.1004996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematol. 2004;89:1187–93. [PubMed] [Google Scholar]
- 48.Restivo Pantalone G, Renda D, Valenza F, et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: clinical characteristics and outcome in a long term single centre experience. Br J Haematol. 2010;150:245–7. doi: 10.1111/j.1365-2141.2010.08180.x. [DOI] [PubMed] [Google Scholar]
- 49.Telfer PT, Warburton F, Christou S, et al. Improved survival in thalassemia major patients on switching from desferrioxamine to combined chelation therapy with desferrioxamine and deferiprone. Haematol. 2009;94:1777–8. doi: 10.3324/haematol.2009.009118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Modell B, Khan M, Darlison M, et al. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2008;10:42–42. doi: 10.1186/1532-429X-10-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pennell DJ, Berdoukas V, Karagiorga M, et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood. 2006;107:3738–44. doi: 10.1182/blood-2005-07-2948. [DOI] [PubMed] [Google Scholar]
- 52.Tanner MA, Galanello R, Dessi C, et al. A Randomized, Placebo-Controlled, Double-Blind Trial of the Effect of Combined Therapy With Deferoxamine and Deferiprone on Myocardial Iron in Thalassemia Major Using Cardiovascular Magnetic Resonance. Circulation. 2007;115:1876–84. doi: 10.1161/CIRCULATIONAHA.106.648790. [DOI] [PubMed] [Google Scholar]
- 53.Angelucci E, Baronciani D, Lucarelli G, et al. Needle liver biopsy in thalassaemia: analyses of diagnostic accuracy and safety in 1184 consecutive biopsies. Br J Haematol. 1995;89:757–61. doi: 10.1111/j.1365-2141.1995.tb08412.x. [DOI] [PubMed] [Google Scholar]
- 54.Ambu R, Crisponi G, Sciot R, et al. Uneven hepatic iron and phosphorus distribution in beta-thalassemia. J Hepatol. 1995;23:544–9. doi: 10.1016/0168-8278(95)80060-3. [DOI] [PubMed] [Google Scholar]
- 55.Emond MJ, Bronner MP, Carlson TH, et al. Quantitative study of the variability of hepatic iron concentrations. Clin Chem. 1999;45:340–6. [PubMed] [Google Scholar]
- 56.Fischer R, Longo F, Nielsen P, et al. Monitoring long-term efficacy of iron chelation therapy by deferiprone and desferrioxamine in patients with β-thalassaemia major: application of SQUID bio-magnetic liver susceptometry. Br J Haematol. 2003;121:938–48. doi: 10.1046/j.1365-2141.2003.04297.x. [DOI] [PubMed] [Google Scholar]
- 57.Westwood MA, Firmin DN, Gildo M, et al. Intercentre reproducibility of magnetic resonance T2* measurements of myocardial iron in thalassaemia. Int J Cardiovasc Imaging. 2005;21:531–8. doi: 10.1007/s10554-005-0651-2. [DOI] [PubMed] [Google Scholar]
- 58.Tanner MA, He T, Westwood MA, et al. Multi-center validation of the transferability of the magnetic resonance T2* technique for the quantification of tissue iron. Haematol. 2006;91:1388–91. [PubMed] [Google Scholar]
- 59.Ghugre NR, Coates TD, Nelson MD, Wood JC. Mechanisms of tissue-iron relaxivity: nuclear magnetic resonance studies of human liver biopsy specimens. Magn Reson Med. 2005;54:1185–93. doi: 10.1002/mrm.20697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wood JC, Fassler JD, Meade T. Mimicking liver iron overload using liposomal ferritin preparations. Magn Reson Med. 2004;51:607–11. doi: 10.1002/mrm.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson LJ, Holden S, Davis B, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171–9. doi: 10.1053/euhj.2001.2822. [DOI] [PubMed] [Google Scholar]
- 62.Kirk P, Smith GC, Roughton M, et al. Myocardial T2* is not affected by ageing, myocardial fibrosis, or impaired left ventricular function. J Magn Reson Imaging. 2010;32:1095–8. doi: 10.1002/jmri.22348. [DOI] [PubMed] [Google Scholar]
- 63.Fischer R, Harmatz PR. Non-invasive assessment of tissue iron overload. Hematology Am Soc Hematol Educ Program. 2009:215–21. doi: 10.1182/asheducation-2009.1.215. [DOI] [PubMed] [Google Scholar]
- 64.St Pierre TG, Clark PR, Chua-Anusorn W, et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105:855–61. doi: 10.1182/blood-2004-01-0177. [DOI] [PubMed] [Google Scholar]
- 65.Wood JC, Enriquez C, Ghugre N, et al. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood. 2005;106:1460–5. doi: 10.1182/blood-2004-10-3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogiatzi MG, Macklin EA, Trachtenberg FL, et al. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol. 2009;146:546–56. doi: 10.1111/j.1365-2141.2009.07793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wood JC, Kang BP, Thompson A, et al. The effect of deferasirox on cardiac iron in thalassemia major: impact of total body iron stores. Blood. 2010;116:537–43. doi: 10.1182/blood-2009-11-250308. [DOI] [PubMed] [Google Scholar]
- 68.Au WY, Lam WW, Chu W, et al. A T2* magnetic resonance imaging study of pancreatic iron overload in thalassemia major. Haematol. 2008;93:116–9. doi: 10.3324/haematol.11768. [DOI] [PubMed] [Google Scholar]
- 69.Noetzli LJ, Papudesi J, Coates TD, Wood JC. Pancreatic iron loading predicts cardiac iron loading in thalassemia major. Blood. 2009;114:4021–6. doi: 10.1182/blood-2009-06-225615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kontoghiorghes GJ, Aldouri MA, Sheppard L, Hoffbrand AV. 1,2-Dimethyl-3-hydroxypyrid-4-one, an orally active chelator for treatment of iron overload. Lancet. 1987;1:1294–5. doi: 10.1016/s0140-6736(87)90545-9. [DOI] [PubMed] [Google Scholar]
- 71.Hoffbrand AV. Oral iron chelators in the treatment of hematologic diseases. Clin Adv Hematol Oncol. 2005;3:536–8. [PubMed] [Google Scholar]
- 72.Matsui D, Klein J, Hermann C, et al. Relationship between the pharmacokinetics and iron excretion pharmacodynamics of the new oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one in patients with thalassemia. Clin Pharmacol Ther. 1991;50:294–8. doi: 10.1038/clpt.1991.139. [DOI] [PubMed] [Google Scholar]
- 73.al-Refaie FN, Sheppard LN, Nortey P, et al. Pharmacokinetics of the oral iron chelator deferiprone (L1) in patients with iron overload. Br J Haematol. 1995;89:403–8. doi: 10.1111/j.1365-2141.1995.tb03318.x. [DOI] [PubMed] [Google Scholar]
- 74.Collins AF, Fassos FF, Stobie S, et al. Iron-balance and dose-response studies of the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one (L1) in iron-loaded patients with sickle cell disease. Blood. 1994;83:2329–33. [PubMed] [Google Scholar]
- 75.Hoffbrand AV, Cohen A, Hershko C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood. 2003;102:17–24. doi: 10.1182/blood-2002-06-1867. [DOI] [PubMed] [Google Scholar]
- 76.Olivieri NF, Koren G, Hermann C, et al. Comparison of oral iron chelator L1 and desferrioxamine in iron-loaded patients. Lancet. 1990;336:1275–9. doi: 10.1016/0140-6736(90)92962-h. [DOI] [PubMed] [Google Scholar]
- 77.Maggio A, D'Amico G, Morabito A, et al. Deferiprone versus deferoxamine in patients with thalassemia major: a randomized clinical trial. Blood Cells Mol Dis. 2002;28:196–208. doi: 10.1006/bcmd.2002.0510. [DOI] [PubMed] [Google Scholar]
- 78.Anderson LJ, Wonke B, Prescott E, et al. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. The Lancet. 2002;360:516–20. doi: 10.1016/s0140-6736(02)09740-4. [DOI] [PubMed] [Google Scholar]
- 79.Piga A, Gaglioti C, Fogliacco E, Tricta F. Comparative effects of deferiprone and deferoxamine on survival and cardiac disease in patients with thalassemia major: a retrospective analysis. Haematol. 2003;88:489–96. [PubMed] [Google Scholar]
- 80.Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood. 2006;107:3733–7. doi: 10.1182/blood-2005-07-2933. [DOI] [PubMed] [Google Scholar]
- 81.Hider RC, Liu ZD. Emerging understanding of the advantage of small molecules such as hydroxypyridinones in the treatment of iron overload. Curr Med Chem. 2003;10:1051–64. doi: 10.2174/0929867033457629. [DOI] [PubMed] [Google Scholar]
- 82.Wu KH, Chang JS, Tsai CH, Peng CT. Combined therapy with deferiprone and desferrioxamine successfully regresses severe heart failure in patients with beta-thalassemia major. Ann Hematol. 2004;83:471–3. doi: 10.1007/s00277-003-0820-0. [DOI] [PubMed] [Google Scholar]
- 83.Porcu M, Landis N, Salis S, et al. Effects of combined deferiprone and desferrioxamine iron chelating therapy in beta-thalassemia major end-stage heart failure A case report. Europ J Heart Fail. 2007;9:320–2. doi: 10.1016/j.ejheart.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 84.Fabio G, Minonzio F, Delbini P, et al. Reversal of cardiac complications by deferiprone and deferoxamine combination therapy in a patient affected by a severe type of juvenile hemochromatosis (JH) Blood. 2007;109:362–4. doi: 10.1182/blood-2006-04-016949. [DOI] [PubMed] [Google Scholar]
- 85.Tanner MA, Galanello R, Dessi C, et al. Combined chelation therapy in thalassemia major for the treatment of severe myocardial siderosis with left ventricular dysfunction. J Cardiovasc Magn Reson. 2008;10:12–12. doi: 10.1186/1532-429X-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Farmaki K, Tzoumari I, Pappa C, et al. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. Br J Haematol. 2010;148:466–75. doi: 10.1111/j.1365-2141.2009.07970.x. [DOI] [PubMed] [Google Scholar]
- 87.Telfer P, Coen PG, Christou S, et al. Survival of medically treated thalassemia patients in Cyprus. Trends and risk factors over the period 1980–2004. Haematol. 2006;91:1187–92. [PubMed] [Google Scholar]
- 88.Galanello R, Kattamis CA, Piga A, et al. A prospective randomized controlled trial on the safety and efficacy of alternating deferoxamine and deferiprone in the treatment of iron overload in patients with thalassemia. Haematol. 2006;91:1241–3. [PubMed] [Google Scholar]
- 89.Maggio A, Vitrano A, Capra M, et al. Long-term sequential deferiprone-deferoxamine versus deferiprone alone for thalassaemia major patients: a randomized clinical trial. Br J Haematol. 2009;145:245–54. doi: 10.1111/j.1365-2141.2009.07609.x. [DOI] [PubMed] [Google Scholar]
- 90.Maggio A, Vitrano A, Capra M, et al. Improving survival with deferiprone treatment in patients with thalassemia major: a prospective multicenter randomised clinical trial under the auspices of the Italian Society for Thalassemia and Hemoglobinopathies. Blood Cells Mol Dis. 2009;42:247–51. doi: 10.1016/j.bcmd.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 91.Cohen AR, Galanello R, Piga A, et al. Safety and effectiveness of long-term therapy with the oral iron chelator deferiprone. Blood. 2003;102:1583–7. doi: 10.1182/blood-2002-10-3280. [DOI] [PubMed] [Google Scholar]
- 92.Ceci A, Baiardi P, Felisi M, et al. The safety and effectiveness of deferiprone in a large-scale, 3-year study in Italian patients. Br J Haematol. 2002;118:330–6. doi: 10.1046/j.1365-2141.2002.03554.x. [DOI] [PubMed] [Google Scholar]
- 93.El Alfy M, Sari TT, Lee CL, et al. The safety, tolerability, and efficacy of a liquid formulation of deferiprone in young children with transfusional iron overload. J Pediatr Hematol Oncol. 2010;32:601–5. doi: 10.1097/MPH.0b013e3181ec0f13. [DOI] [PubMed] [Google Scholar]
- 94.Choudhry VP, Pati HP, Saxena A, Malaviya AN. Deferiprone, efficacy and safety. Indian J Pediatr. 2004;71:213–6. doi: 10.1007/BF02724272. [DOI] [PubMed] [Google Scholar]
- 95.Porter JB. Optimizing iron chelation strategies in beta-thalassaemia major. Blood Rev. 2009;23(Suppl 1):S3–S7. doi: 10.1016/S0268-960X(09)70003-7. [DOI] [PubMed] [Google Scholar]
- 96.Cohen AR, Galanello R, Piga A, et al. Safety profile of the oral iron chelator deferiprone: a multicentre study. Br J Haematol. 2000;108:305–12. doi: 10.1046/j.1365-2141.2000.01866.x. [DOI] [PubMed] [Google Scholar]
- 97.PSUR. Toronto, Apotex: 2003. Apotex Research: Ferriprox pediodic safety update report. [Google Scholar]
- 98.Nick H, Acklin P, Lattmann R, et al. Development of tridentate iron chelators: from desferrithiocin to ICL670. Curr Med Chem. 2003;10:1065–76. doi: 10.2174/0929867033457610. [DOI] [PubMed] [Google Scholar]
- 99.Galanello R, Piga A, Alberti D, et al. Safety, Tolerability, and Pharmacokinetics of ICL670, a New Orally Active Iron-Chelating Agent in Patients with Transfusion-Dependent Iron Overload Due to β-Thalassemia. J Clin Pharmacol. 2003;43:565–72. [PubMed] [Google Scholar]
- 100.Nisbet-Brown E, Olivieri NF, Giardina PJ, et al. Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet. 2003;361:1597–602. doi: 10.1016/S0140-6736(03)13309-0. [DOI] [PubMed] [Google Scholar]
- 101.Porter JB, Vichinsky E, Rose C, et al. A phase II study with ICL670 (Exjade), a once-daily oral iron chelator, in patients with various transfusio-dependent anemias and iron overload. Blood. 2004;104:872A–872A. [Google Scholar]
- 102.Vichinsky E, Fischer R, Fung E, et al. A randomized, controlled phase II trial in sickle cell disease patients with chronic iron overload demontrates that the once-daily oral iron chelator deferasirox (Exjade ICL670) is well tolerated and reduces iron. Blood. 2005;106:95A–95A. [Google Scholar]
- 103.Piga A, Galanello R, Cappellini MO, et al. Phase II study of ICL670, an oral chelator, in adult thalassemia patients with trasfusional iron overload: Efficacy, safety, pharmaco kinetics (PK) and pharmacodyamics (PD) after 18 months of therapy. Blood. 2003;102:121A–121A. [Google Scholar]
- 104.Piga A, Galanello R, Foschini L, et al. Once-daily treatment with the oral iron chelator ICL670(Exjade): Results of a phase II study in pediatric patients with beta thalassemia major. Blood. 2004;104:983A–983A. [Google Scholar]
- 105.Cappellini MD, Cohen AR, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood. 2006;107:3455–62. doi: 10.1182/blood-2005-08-3430. [DOI] [PubMed] [Google Scholar]
- 106.Daar S, Pathare A, Nick H, et al. Reduction in labile plasma iron during treatment with deferasirox, a once-daily oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia. Eur J Haematol. 2009;82:454–7. doi: 10.1111/j.1600-0609.2008.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ladis V, Berdousi H, Gotsis E, Kattamis A. Deferasirox administration for the treatment of non-transfusional iron overload in patients with thalassaemia intermedia. Br J Haematol. 2010;151:504–8. doi: 10.1111/j.1365-2141.2010.08346.x. [DOI] [PubMed] [Google Scholar]
- 108.Piga A, Galanello R, Forni GL, et al. Randomized phase II trial of deferasirox (Exjade©, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematol. 2006;91:873–80. [PubMed] [Google Scholar]
- 109.Taher A, El-Beshlawy A, Elalfy MS, et al. Efficacy and safety of deferasirox, an oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia: the ESCALATOR study. Eur J Haematol. 2009;82:458–65. doi: 10.1111/j.1600-0609.2009.01228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taher A, Cappellini MD, Vichinsky E, et al. Efficacy and safety of deferasirox doses of >30 mg/kg per d in patients with transfusion-dependent anaemia and iron overload. Br J Haematol. 2009;147:752–9. doi: 10.1111/j.1365-2141.2009.07908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cappellini MD, Porter J, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematol. 2010;95:557–66. doi: 10.3324/haematol.2009.014696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Galanello R, Piga A, Forni GL, et al. Phase II clinical evaluation of deferasirox, a once-daily oral chelating agent, in pediatric patients with beta-thalassemia major. Haematol. 2006;91:1343–51. [PubMed] [Google Scholar]
- 113.Cohen AR, Glimm E, Porter JB. Effect of transfusional iron intake on response to chelation therapy in beta-thalassemia major. Blood. 2008;111:583–7. doi: 10.1182/blood-2007-08-109306. [DOI] [PubMed] [Google Scholar]
- 114.Piga A, Kebaili K, Galanello R. Cumulative efficacy and safety of 5-year deferasirox (Exjade) treatment in pediatric patients with thalassemia major: a phase II multicenter prospective trial. Blood. 2008;112:5413A–5413A. [Google Scholar]
- 115.Pennell DJ, Porter JB, Cappellini MD, et al. Efficacy of deferasirox in reducing and preventing cardiac iron overload in beta-thalassemia. Blood. 2010;115:2364–71. doi: 10.1182/blood-2009-04-217455. [DOI] [PubMed] [Google Scholar]
- 116.Pennell DJ, Porter JB, Cappellini MD, et al. Continued improvement in myocardial T2* over two years of deferasirox therapy in β-thalassemia major patients with cardiac iron overload. Haematol. 2011;96:48–54. doi: 10.3324/haematol.2010.031468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vichinsky E, Pakbaz Z, Onyekwere O, et al. Patient-reported outcomes of deferasirox (Exjade, ICL670) versus deferoxamine in sickle cell disease patients with transfusional hemosiderosis. Substudy of a randomized open-label phase II trial. Acta Haematol. 2008;119:133–41. doi: 10.1159/000125550. [DOI] [PubMed] [Google Scholar]
- 118.Cappellini MD, Bejaoui M, Agaoglu L, et al. Prospective evaluation of patient-reported outcomes during treatment with deferasirox or deferoxamine for iron overload in patients with beta-thalassemia. Clin Ther. 2007;29:909–17. doi: 10.1016/j.clinthera.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 119.Taher A, Al Jefri A, Elalfy MS, et al. Improved treatment satisfaction and convenience with deferasirox in iron-overloaded patients with beta-Thalassemia: Results from the ESCALATOR Trial. Acta Haematol. 2010;123:220–5. doi: 10.1159/000313447. [DOI] [PubMed] [Google Scholar]
- 120.Galanello R, Agus A, Campus S, et al. Combined iron chelation therapy. Ann N Y Acad Sci. 2010;1202:79–86. doi: 10.1111/j.1749-6632.2010.05591.x. [DOI] [PubMed] [Google Scholar]
- 121.Jetsrisuparb A, Komvilaisak P, Wiangnon S, Jetsrisuparb C. Retrospective study on the combination of desferrioxamine and deferasirox for treatment of iron-overloaded thalassemic patients: first evidence of more than 2 years. J Pediatr Hematol Oncol. 2010;32:400–3. doi: 10.1097/MPH.0b013e3181e015b0. [DOI] [PubMed] [Google Scholar]
- 122.Balocco M, Carrara P, Pinto V, Forni GL. Daily alternating deferasirox and deferiprone therapy for “hard-to-chelate” beta-thalassemia major patients. Am J Hematol. 2010;85:460–1. doi: 10.1002/ajh.21711. [DOI] [PubMed] [Google Scholar]
- 123.Porter JB. Deferasirox--current knowledge and future challenges. Ann N Y Acad Sci. 2010;1202:87–93. doi: 10.1111/j.1749-6632.2010.05582.x. [DOI] [PubMed] [Google Scholar]
- 124.Vichinsky E. Clinical application of deferasirox: practical patient management. Am J Hematol. 2008;83:398–402. doi: 10.1002/ajh.21119. [DOI] [PubMed] [Google Scholar]
- 125.Porter JB. Deferasirox: an update. Hemoglobin. 2009;33:S70–5. doi: 10.3109/03630260903347146. [DOI] [PubMed] [Google Scholar]
- 126.Yacobovich J, Stark P, Barzilai-Birenbaum S, et al. Acquired proximal renal tubular dysfunction in β-thalassemia patients treated with deferasirox. J Pediatr Hematol Oncol. 2010;32:564–7. doi: 10.1097/MPH.0b013e3181ec0c38. [DOI] [PubMed] [Google Scholar]
- 127.Rheault MN, Bechtel H, Neglia JP, Kashtan CE. Reversible Fanconi syndrome in a pediatric patient on deferasirox. Pediatr Blood Cancer. 2010 Dec; doi: 10.1002/pbc.22711. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 128.Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Survival and complications in thalassemia. Ann N Y Acad Sci. 2005;1054:40–7. doi: 10.1196/annals.1345.006. [DOI] [PubMed] [Google Scholar]
- 129.Cao A. Quality of life and survival of patients with beta-thalassemia major. Haematol. 2004;89:1157–9. [PubMed] [Google Scholar]
- 130.Lai ME, Grady RW, Vacquer S, et al. Increased survival and reversion of iron-induced cardiac disease in patients with thalassemia major receiving intensive combined chelation therapy as compared to desferoxamine alone. Blood Cells Mol Dis. 2010;45:136–9. doi: 10.1016/j.bcmd.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 131.La Nasa G, Argiolu F, Giardini C, et al. Unrelated bone marrow transplantation for beta-thalassemia patients: The experience of the Italian Bone Marrow Transplant Group. Ann N Y Acad Sci. 2005;1054:186–95. doi: 10.1196/annals.1345.023. [DOI] [PubMed] [Google Scholar]
- 132.Isgro A, Marziali M, Sodani P, et al. Immunohematologic reconstitution in pediatric patients after T cell-depleted HLA-haploidentical stem cell transplantation for thalassemia. Biol Blood Marrow Transplant. 2010;16:1557–66. doi: 10.1016/j.bbmt.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 133.Moi P, Sadelain M. Towards the genetic treatment of beta-thalassemia: new disease models, new vectors, new cells. Haematol. 2008;93:325–30. doi: 10.3324/haematol.12732. [DOI] [PubMed] [Google Scholar]
- 134.Case SS, Price MA, Jordan CT, et al. Stable transduction of quiescent CD34(+)CD38(−) human hematopoietic cells by HIV-1-based lentiviral vectors. Proc Natl Acad Sci U S A. 1999;96:2988–93. doi: 10.1073/pnas.96.6.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tisdale J, Sadelain M. Toward gene therapy for disorders of globin synthesis. Semin Hematol. 2001;38:382–92. doi: 10.1016/s0037-1963(01)90033-2. [DOI] [PubMed] [Google Scholar]
- 136.May C, Sadelain M. A promising genetic approach to the treatment of beta-thalassemia. Trends Cardiovasc Med. 2001;11:276–80. doi: 10.1016/s1050-1738(01)00125-6. [DOI] [PubMed] [Google Scholar]
- 137.Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–71. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- 138.Rivella S, May C, Chadburn A, et al. M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–9. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 139.Puthenveetil G, Scholes J, Carbonell D, et al. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–53. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- 140.Naldini L, Blomer U, Gage FH, et al. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A. 1996;93:11382–8. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gelinas R, Novak U. Retroviral vectors for the beta-globin gene that demonstrate improved titer and expression. Ann N Y Acad Sci. 1990;612:427–41. doi: 10.1111/j.1749-6632.1990.tb24330.x. [DOI] [PubMed] [Google Scholar]
- 143.Hanawa H, Hargrove PW, Kepes S, et al. Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassemia. Blood. 2004;104:2281–90. doi: 10.1182/blood-2004-03-0863. [DOI] [PubMed] [Google Scholar]
- 144.May C, Rivella S, Callegari J, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–6. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 145.Miccio A, Cesari R, Lotti F, et al. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105:10547–52. doi: 10.1073/pnas.0711666105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lisowski L, Sadelain M. Locus control region elements HS1 and HS4 enhance the therapeutic efficacy of globin gene transfer in beta-thalassemic mice. Blood. 2007;110:4175–8. doi: 10.1182/blood-2007-08-108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Emery DW, Yannaki E, Tubb J, et al. Development of virus vectors for gene therapy of beta chain hemoglobinopathies: flanking with a chromatin insulator reduces gamma-globin gene silencing in vivo. Blood. 2002;100:2012–9. doi: 10.1182/blood-2002-01-0219. [DOI] [PubMed] [Google Scholar]
- 148.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–22. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Italiano A, Bianchini L, Keslair F, et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int J Cancer. 2008;122:2233–41. doi: 10.1002/ijc.23380. [DOI] [PubMed] [Google Scholar]
- 150.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–30. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 153.Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–3. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 154.Papapetrou EP, Lee G, Malani N, et al. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol. 2011;29:73–8. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Si-Tayeb K, Noto FK, Sepac A, et al. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol. 2010;10:81–81. doi: 10.1186/1471-213X-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sommer CA, Sommer AG, Longmire TA, et al. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells. 2010;28:64–74. doi: 10.1002/stem.255. [DOI] [PMC free article] [PubMed] [Google Scholar]