

Abstract
ATP-sensitive potassium channels (KATP channels) are important sensors of cellular metabolic state that link metabolism and excitability in neuroendocrine cells, but their role in nonglucosensing central neurons is less well understood. To examine a possible role for KATP channels in modulating excitability in hippocampal circuits, we recorded the activity of single KATP channels in cell-attached patches of granule cells in the mouse dentate gyrus during bursts of action potentials generated by antidromic stimulation of the mossy fibers. Ensemble averages of the open probability (popen) of single KATP channels over repeated trials of stimulated spike activity showed a transient increase in popen in response to action potential firing. Channel currents were identified as KATP channels through blockade with glibenclamide and by comparison with recordings from Kir6.2 knock-out mice. The transient elevation in KATP popen may arise from submembrane ATP depletion by the Na+-K+ ATPase, as the pump blocker strophanthidin reduced the magnitude of the elevation. Both the steady-state and stimulus-elevated popen of the recorded channels were higher in the presence of the ketone body R-β-hydroxybutyrate, consistent with earlier findings that ketone bodies can affect KATP activity. Using perforated-patch recording, we also found that KATP channels contribute to the slow afterhyperpolarization following an evoked burst of action potentials. We propose that activity-dependent opening of KATP channels may help granule cells act as a seizure gate in the hippocampus and that ketone-body-mediated augmentation of the activity-dependent opening could in part explain the effect of the ketogenic diet in reducing epileptic seizures.
Introduction
ATP-sensitive K+ channels, or KATP channels, are inward-rectifier potassium channels that are inhibited by intracellular ATP. Their role of linking metabolism to electrical activity is best understood in pancreatic beta cells, where their modest open probability between meals is sufficient to maintain a hyperpolarized state; upon ingestion of a sugary meal, the channels close, allowing action potentials to fire and produce Ca2+ influx and insulin release (Aguilar-Bryan and Bryan, 1999; Ashcroft and Gribble, 1999; Seino and Miki, 2003; Nichols, 2006). KATP channels can play a similar role in central glucosensing neurons (Ashford et al., 1990; Miki et al., 2001).
KATP channels are also present in a large number of central neurons that do not have a primary glucosensing role (Karschin et al., 1997; Dunn-Meynell et al., 1998; Zawar et al., 1999); in these neurons, as in the heart (Benndorf et al., 1991), the channels are mainly thought to remain closed except under conditions of severe metabolic deprivation, such as anoxia or ischemia. Inhibitors of KATP channels can sometimes affect neuronal firing under less pathological circumstances (Pierrefiche et al., 1996; Allen and Brown, 2004), implying that some basal activity of KATP channels can affect neuronal excitability in nonglucosensing neurons. But it remains largely undetermined if and how normal levels of neuronal firing, via the consequent metabolic load, can lead to KATP channel opening. This possibility of activity-dependent KATP channel opening is suggested by results from brainstem inspiratory neurons (Haller et al., 2001), where bursts of single KATP channel opening are seen in synchrony with the respiratory firing rhythm; channel open probability (popen) is increased by ∼60% after a strong burst of action potentials, compared with immediately before. These changes are smaller with blockade of the Na+-K+ ATPase, suggesting that ATP consumption in response to the Na+ influx from action potentials contributes to the KATP channel opening.
We examined the ability of action potentials to induce KATP channel opening in the dentate granule neurons (DGNs) of the mouse hippocampus. These cells express a particularly high density of KATP channels (Karschin et al., 1997; Zawar et al., 1999), such that single KATP channels can be recorded in ∼50% of cell-attached patch recordings (Pelletier et al., 2000). Because these cells have a very low rate of spontaneous firing in superfused hippocampal slices, we induced action potential firing by electrical stimulation of the DGN axons in the dentate hilus. We found that modest firing activity, a burst of five action potentials at 20 Hz, was adequate to increase the activity of single KATP channels for 0.5–1 s. This effect on KATP largely disappeared in the presence of strophanthidin, a blocker of the Na+-K+ ATPase. It appears that even moderate action potential firing can evoke KATP channel opening via Na+ influx and ATP depletion. Both the level of firing-induced KATP opening and the steady-state level of KATP opening were higher in the presence of the ketone body R-β-hydroxybutyrate (R-βHB), consistent with a previous study (Ma et al., 2007) showing that ketone bodies can alter neuronal electrical activity by opening KATP channels.
Materials and Methods
Hippocampal slice preparation.
Acute hippocampal slices 400 μm thick were cut in the transverse plane from the brains of 12- to 16-d-old male mice (C57BL/6; Charles River Laboratories) or Kir6.2−/− mice (in a C57BL/6 background) (Miki et al., 1998), using a vibrating tissue slicer (Vibratome). All procedures involving animals were approved by the Harvard Medical Area Standing Committee on Animals. Mice were anesthetized by halothane or isoflurane inhalation and decapitated into an ice-cold slurry containing the following (in mm): 87 NaCl, 25 NaHCO3, 25 d-glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2 (osmolarity ∼340 mmol/kg; pH 7.4 with NaOH).
Slices were incubated at 37°C for 20 min in the slicing solution and then for 40 min in recording solution, then stored at room temperature for 0–3 h before use. During recording, ACSF was delivered to the bath using a peristaltic pump at a flow rate of ∼0.9 ml/min. All ACSF solutions for single-channel recordings contained the following (in mm): 26 NaHCO3, 10 d-glucose, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, and 1 MgSO4 (osmolarity ∼290 mmol/kg; pH 7.4). Standard single-channel ACSF also contained 120 mm NaCl; R-βHB solution contained 118 mm NaCl, 2 mm NaOH, and 2 mm R-β-hydroxybutyric acid (Kanto Chemical). For whole-cell recordings, ACSF contained the following (in mm): 119 NaCl, 26 NaHCO3, 1 NaH2PO4,10 d-glucose, 2.5 KCl, 2.5 CaCl2, and 1.3 MgSO4 (osmolarity ∼300 mmol/kg; pH 7.4). In addition, all recording solutions contained 100 μm picrotoxin and 1 mm kynurenic acid to block fast synaptic transmission (all chemicals from Sigma-Aldrich unless otherwise noted). All bath solutions were bubbled continuously with 95% O2 and 5% CO2.
Electrophysiological recordings.
Cell-attached patch recordings from individual DGNs identified under infrared differential interference contrast visual guidance were made in voltage-clamp mode following establishment of high-resistance (multi-GΩ) seals. Data were collected with an Axopatch 200B integrating patch-clamp amplifier (MDS Analytical Technologies) in whole-cell mode (β = 1), sampled at 10–14 kHz, and filtered online at 1–2 kHz. Pipette potential was held at 0 mV (Vm = Vrest) for most experiments. All recordings were obtained at room temperature.
Patch pipettes were pulled from borosilicate glass capillary tubes (VWR Scientific) on a horizontal heated-filament puller (P-97; Sutter Instrument) and were wrapped about the shank close to the tip with stretched Parafilm to reduce capacitance. Pipettes with tip resistances of 3–6 MΩ were used for cell-attached recordings and tip resistances of 2–4 MΩ were used for whole-cell recordings. Pipette tips were not fire-polished.
For cell-attached recordings, the pipette solution contained the following (in mm): 140 potassium methanesulfonate (KMeS), 10 HEPES, 2 CaCl2, and 10 TEA-Cl, 2 CsCl, and 1 4-aminopyridine (4-AP) (for blockade of non-KATP potassium channels); osmolarity ∼310 mmol/kg; pH to 7.4 with KOH). For experiments (except those summarized in Fig. 1B), the patch pipette also contained additional peptide blockers of non-KATP potassium channels: 100 nm charybdotoxin (in 0.001% BSA; Alomone Labs) to block BK channels and 100 nm apamin to block SK channels (including 40 μm acetic acid; Calbiochem).
Figure 1.

Single KATP channels in unstimulated dentate gyrus granule cells. A, Single-channel currents recorded from a DGN cell-attached patch at various apparent membrane voltages (Vm). Vm is based on the applied pipette potential and an estimated membrane potential of Vrest = −80 mV (Vm = Vrest − Vcommand). These current traces were selected to show bursts of channel openings and are thus not representative of typical open probability. Arrows indicate the zero current level. B, Plot of single-channel current (mean ± SEM) against apparent membrane voltage for eight patches in which a response to NN414 was observed. Linear fit corresponds to a single-channel conductance of 25 pS. C, Continuous records of single-channel currents from a cell-attached patch recorded with no applied voltage in the absence (left) or presence (right) of the KATP opener NN414 (5 μm). Single-channel openings in this and all subsequent figures are represented by upward deflections of the current trace.
DGNs were stimulated antidromically with a glass electrode placed in the hilus, proximal to the recording site. The stimulating electrode contained a HEPES-buffered ACSF and was driven by a D/A converter (Digidata 1321A; MDS Analytical Technologies) via an A360 stimulus isolation unit (WPI). Stimulation intensity was maintained at the minimum level required to reliably evoke spike activity. Stimulation was delivered in trains of five or 10 short (80 μs) pulses at a frequency of 20 Hz.
For paired experiments to assess glibenclamide effects, experiments were performed only on patches with relatively high initial channel activity. For this reason, the control segments of these paired experiments were not included in the averages of control activity that were used for unpaired comparisons (vs Kir6.2−/− animals or vs ketone body application), and the average peak popen was higher for these series of experiments. For the paired experiments, an approximately equal number of experiments were performed using vehicle (DMSO) without the experimental drug; the experiments were performed and analyzed by an investigator blind to the identity of the solution applied.
For whole-cell recordings, the pipette solution contained the following (in mm): 140 KMeS, 10 NaCl, 10 HEPES, 1 MgCl2, 0.1 EGTA (osmolarity ∼295 mmol/kg; pH 7.4), supplemented with 4 mm MgATP and 0.3 mm Na3GTP. A voltage ramp protocol was used to measure input resistance: the holding potential was set at −90 mV, followed by a 300 ms step to −70 mV, then a 200 ms step back to −90 mV (for assessment of resting leak) and another 100 ms step to −110 mV to allow the membrane potential to settle before initiation of a 35 mV/s ramp from −110 to −20 mV, followed by a return to the −90 mV holding potential. The voltage-ramp protocol was applied every 5 s to provide a running assessment of whole-cell conductance.
For perforated patch recordings, the pipette solution contained the following (in mm): 140 KCl, 10 NaCl, 10 HEPES, 1 MgCl2 (pH 7.3). The pipette solution also contained 50–100 μg/ml gramicidin D (Sigma). Before experiments, fresh gramicidin stock solution (5 mg/ml in DMSO) was prepared and diluted in the pipette solution. Additionally, Alexa 488 (10 μm; Invitrogen) was added to the pipette solution to monitor the integrity of the perforated cell membrane. The pipette tip was filled with gramicidin-free solution and then back-filled with the gramicidin-containing solution. Perforation generally occurred within 30 min of forming a seal. Current-clamp recordings were performed on cells held near −60 mV, which allowed for consistent observation of slow afterhyperpolarizations (Podlogar and Dietrich, 2006). Trains of action potentials were triggered by five current pulses (5 ms, 100 pA) at 20 Hz.
Drugs and solutions.
All drugs used were bath-applied in ACSF. Glibenclamide was diluted from a 1 mm stock solution in DMSO down to 100 nm in ACSF. DMSO as a vehicle control for glibenclamide was added to ACSF at 0.01%. NN414 was diluted from a 250 mm stock in DMSO to a concentration of 5–10 μm in ACSF. Strophanthidin was made as a 0.5 m stock in DMSO diluted down to 500 μm in ACSF. DMSO as a vehicle control for strophanthidin was added to ACSF at 0.1%. NN414 was a gift from Novo Nordisk.
Data analysis.
Most experiments were performed and analyzed by an experimenter blind to genotype or treatment condition. Data were analyzed offline with Clampfit, Microsoft Excel (Microsoft), MATLAB (Mathworks), and Microcal Origin (OriginLab).
In-house software written in MATLAB was used for primary offline analysis of raw data. For analysis, data were filtered to 500 Hz. For binary idealization of channel openings, stimulus artifacts and capacitive action currents were blanked from the filtered data traces, which were then subjected to a baseline subtraction where the mean current level at each time point was subtracted from all traces. The mean current for subtraction was calculated after exclusion of traces in the top quartile (for data with few channel openings) or in the top 67% (for data with large numbers of peristimulus openings).
Open and closed levels for channel activity were determined from an all-points histogram of the filtered data, and channel openings were detected using a 50% threshold criterion applied to filtered (500 Hz) raw data. Raw datasets featuring excessive baseline shift, sufficient high- or low-frequency noise to preclude effective idealization of channel openings, insufficient entrainment of spikes to stimulus, or too few data traces (<36; 3 min of recording) were generally discarded without idealization or further analysis. In the case of data recorded in the absence of stimulation, either for comparison with records where spiking was stimulated, or for channel identification with KATP opening drugs, trials containing <36 traces were occasionally included for analysis.
The amplitude of the slow afterhyperpolarization (sAHP) was calculated as the minimum of a 250 ms moving average voltage, minus the median voltage of the baseline. The post-sAHP spike latency was measured as the first time point to exceed −10 mV after the last evoked action potential. For each experiment, we calculated the 80% trimmed mean (excluding the high and low 10%), then calculated mean and SEM across experiments. Significance by a nonparametric (Mann–Whitney) test was <0.05 for both latency and sAHP voltage.
Results
We performed cell-attached single channel recordings on DGNs using a high-[K+] pipette solution with a mixture of ionic channel blockers (TEA, 4-AP, and Cs+) designed to isolate KATP channels (see Materials and Methods, above), as previously done by Pelletier et al. (2000). With this solution, we observed channels in nine of 14 patches (∼64%) that had characteristics of DGN KATP channels (Fig. 1). Single-channel currents reversed at a pipette potential close to the cell resting potential (∼−80 mV, corresponding to a transmembrane potential of ∼0 mV), as expected for a potassium-selective channel with 140 mm extracellular K+. The slope conductance of ∼25 pS was smaller than usually reported for Kir6.2-containing KATP channels (in the range of 50–80 pS) (Cook and Hales, 1984; Inagaki et al., 1995; Babenko et al., 1998) but very close to that reported for endogenous KATP channels recorded in rat DGNs (Pelletier et al., 2000). The single-channel current–voltage relationship showed little rectification up to Vrest + 50 mV (Fig. 1B), characteristic of the Kir6.x class of inward rectifier channels (Baukrowitz et al., 1999). Also, the channels recorded under these conditions showed increased popen in the presence of the SUR1-specific KATP-opening drug NN414 (5 μm) (Fig. 1C) (Dabrowski et al., 2003). For all subsequent experiments, the patch pipette also contained additional peptide blockers of non-KATP potassium channels: 100 nm charybdotoxin (in 0.001% BSA; Alomone Labs) to block BK channels and 100 nm apamin to block SK channels (including 40 μm acetic acid; Calbiochem).
Action potential firing elevates KATP channel open probability in cell-attached patches
We tested whether the firing of DGNs could bias KATP channels toward opening, perhaps because of the increased metabolic demand. We drove DGNs to fire using antidromic stimulation from the hilus, as this permitted us to record under conditions of synaptic blockade, thus further decreasing the already low rate of spontaneous DGN firing and eliminating some complications that might arise from synaptic transmission.
We stimulated 20 Hz bursts of five action potentials every 5 s and recorded dozens to hundreds of consecutive 5 s sweeps of voltage-clamped current from each cell. The cell-attached recordings simultaneously recorded openings and closings of individual KATP channels along with capacitive action currents fired in response to stimulation (Fig. 2A).
Figure 2.
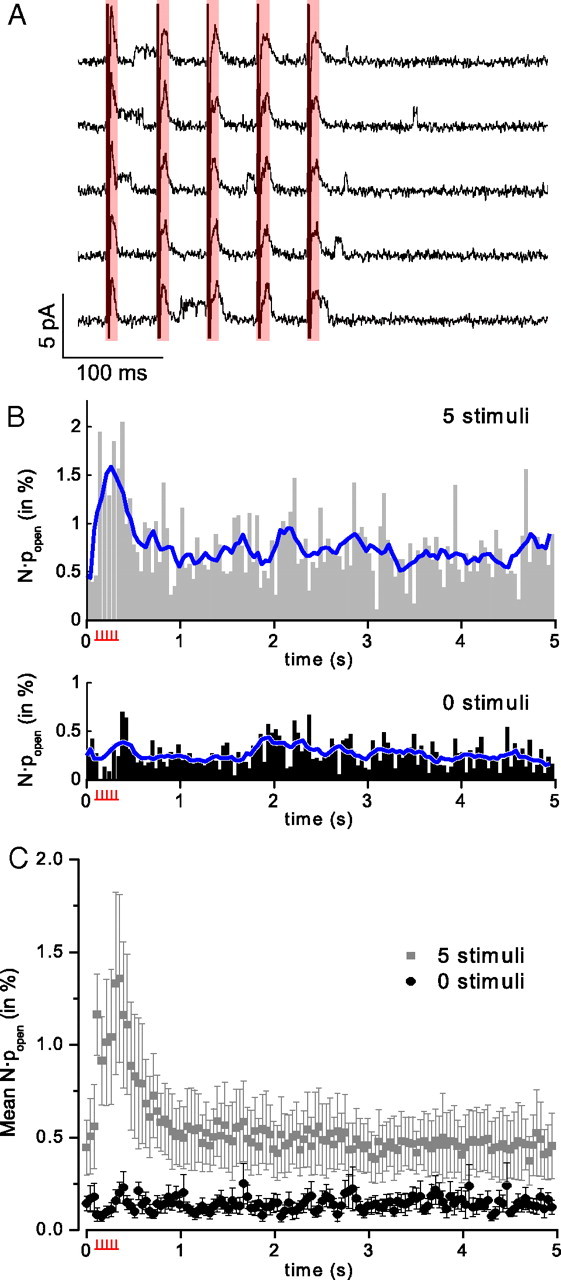
Stimulated spike activity increases channel popen. A, Five examples of cell-attached records from a DGN with five stimuli delivered at 20 Hz, showing channel openings just for the period during and after action potential stimulation. The red shading indicates the period of stimulus artifact and action potential. B, Top, Histogram of channel activity (ensemble average open probability) over all traces from a representative five-stimulus experiment. The blue line shows a 320 ms moving average value. A prominent elevation in channel popen can be observed during and after the stimulus period (indicated by the stimulus icon beneath the time axis). Bottom, A second histogram for the same patch for traces during which no stimuli were applied. Channel activity for all experiments is reported as N·popen to acknowledge the possibility that more than one channel is active in a given patch. C, Summary for all trials where cells were stimulated to fire five spikes at 20 Hz versus all trials where no stimulus was delivered. The peak stimulus-period popens for five stimuli are significantly different from those for the equivalent time period in trials where no spikes were stimulated to fire (p < 0.01; Mann–Whitney U test). Gray squares, Mean N·popen for all five-stimulus trials (n = 35); black circles, mean N·popen for all trials conducted without stimulation (n = 13). Data points are presented as mean channel popen ± SEM.
Time-course histograms of the average KATP channel open probability, obtained from analysis of a full set of sweeps, typically showed a marked elevation in channel popen during and immediately following the stimulus interval (Fig. 2B). On average, across all experiments recorded in ACSF with this stimulation paradigm, the elevation in channel popen lasted the duration of the stimulus period and attained a mean value of 1.09 ± 0.32% (n = 35) for the entire intrastimulus interval; this transient increase in popen persisted for ∼0.5–1 s beyond the end of the stimulation period (Fig. 2C). This increase in channel activity reached a level more than double that of the basal popen exhibited during the nonspiking quiescent period (taken as the last second of the 5 s recording epoch), where the open probability typically dropped to a very low level: <0.1% in some cases, and to 0.46 ± 0.17% on average.
Qualitatively, this observation suggests that KATP channels open preferentially in response to stimulated firing in DGNs. The effect depended on action potential firing; when the stimulus was turned off, the activation profile of the channels in the ensemble averages of the zero-spike condition showed no elevation (Fig. 2B,C). For the unstimulated condition, the peak open probability during the equivalent time period where spikes were stimulated to fire for the five-stimulus condition was 0.13 ± 0.03%, compared with a mean basal popen during the last second of the recording period of 0.14 ± 0.04% (n = 13). While there was no significant difference between the basal open probabilities over the last second of the recording epoch for the two conditions (5 vs 0 stimuli; p = 0.24, Mann–Whitney U test), the spike-elevated peak popen recorded with five stimuli delivered was significantly greater than that recorded during the equivalent time period in the absence of stimulation (p < 0.01). Comparable effects were seen with 10 stimuli, also administered at 20 Hz: mean popen was 0.85 ± 0.15% during the stimulus period and 0.38 ± 0.09% at steady-state (n = 40; p < 0.001). The decay of the popen back to a steady-state level after the end of the stimulus period was fitted with a single exponential, giving time constants of τ = 0.32 ± 0.02 s for five spikes and τ = 0.46 ± 0.06 s for 10 spikes.
Confirmation of KATP channel identity
Although our pipette recording solution contained a mixture of channel blockers (TEA, 4-aminopyridine, CsCl, charybdotoxin, and apamin) designed to isolate KATP channels (see Materials and Methods, above), we used both pharmacological and genetic tests to confirm this identification. To pharmacologically identify KATP channels, we recorded stimulus-dependent channel activity first in ACSF and then after 15 min perfusion with 100 nm glibenclamide, a potent blocker of KATP channels.
Glibenclamide substantially reduced the peak popen during the stimulus period (Fig. 3A). To analyze these paired experiments, we plotted Δpopen (glibenclamide − ACSF) as a function of the initial popen in ACSF, using a peak-weighted average measurement of popen (explained in Fig. 3's legend). The expected result is a line with a negative slope corresponding to the fractional reduction in single-channel activity (Fig. 3B). For peak channel-opening responses, the fitted slope corresponded to an 81% (±2%) fractional reduction in activity with glibenclamide (significantly different from zero by F-distribution test; p < 0.01). For measurement of channel opening over the entire trace, the fitted slope corresponded to an 87% (±2%) fractional reduction in channel current with glibenclamide (p < 0.01). In interleaved control experiments (performed and analyzed blind to the condition) where the 0.01% DMSO vehicle without glibenclamide was washed in, there was a slight downward trend for both the peak and the full-trace popen (Fig. 3B), but any apparent DMSO effect was much smaller than that for glibenclamide. Also, there was no overlap between the confidence bands for the fitted slopes of the drug and vehicle control conditions (Fig. 3B), indicating that the effect of glibenclamide treatment is significantly greater than the effect of DMSO treatment or any rundown that may have occurred.
Figure 3.

Both the firing-induced and basal channel openings are mainly KATP channels. A, Average effect of glibenclamide wash-in on channel open probability after recording of baseline activity in ACSF. The KATP blocking drug glibenclamide (100 nm) attenuates the spike-elevated opening of channels compared with paired five-stimulus trials recorded in ACSF alone. Mean ± SEM; n = 6. B, Estimate of the fractional blockade by glibenclamide. Δpopen (final − initial) is plotted against the initial popen (in ACSF), with each symbol representing a separate experiment with glibenclamide (black squares) or DMSO (gray circles) wash-in. Linear fits through the origin, for each dataset, represent a fixed fractional reduction in current produced by the blocker. The two graphs use different time samples: full-trace effects (bottom) simply used the full-trace average popen, while peak-weighted effects (top) were weighted with a function that was constant during the stimulus interval and decayed with a time constant of 0.3 s thereafter. Dotted lines mark the 95% confidence limits for the linear fits. For glibenclamide, the fitted slopes were (peak) −0.81 ± 0.02 and (full) −0.87 ± 0.02, consistent with ∼80–90% inhibition. For DMSO, the fitted slopes were (peak) −0.33 ± 0.06 and (full) −0.31 ± 0.08 (n = 7). C, Channel activity is reduced in DGNs from Kir6.2 knock-out mice. Summary for experiments with five-stimuli in wild-type animals (gray symbols, mean ± SEM, n = 35), and Kir6.2−/− (black symbols, mean ± SEM, n = 11). Most experiments were performed and analyzed blind to genotype.
As a genetic test of channel identity, we used brain slices from knock-out mice containing a disrupted Kir6.2 gene (Miki et al., 1998). These experiments showed a much smaller open probability and stimulus-induced elevation than seen in wild-type animals (Fig. 3C). There was still a small amount of channel activity in the knock-out animals, which may indicate that our recording pipette mixture does not completely block all other K+ channel types, or that there are some KATP channels formed by the alternative pore-forming Kir6.1 subunits (also capable of forming KATP channels).
Nevertheless, comparison of the results from the two genotypes studied showed that the prominent activity-dependent increase in channel popen seen in wild-type animals was largely attenuated or absent during the stimulus period in Kir6.2−/− knock-out animals. Over half (6 of 11; 55%) of the trials from knock-out animals showed little to no channel activity (<0.05% steady-state basal open probability) compared with only eight of 35 (23%) trials from wild-type animals exhibiting such low levels of activity, as recorded and analyzed under the same conditions (Fig. 3C). Of the five trials from knock-outs that did show some appreciable level of channel activity, only one exhibited any apparent stimulus-period elevation in popen at all. Overall, the peak open probability during the stimulus period was significantly lower in knock-out animals than in wild-type (p < 0.05, Mann–Whitney U test). The absence of a spike-induced elevation in the majority of slices from Kir6.2 knock-out animals again suggests that the activity observed in wild-type animals is largely due to KATP channels.
The Na+-K+ pump blocker strophanthidin attenuates the spike-dependent effect
KATP channel opening is primarily controlled by the intracellular ATP and ADP concentrations. The main metabolic consequence of action potential firing arises from the restoration of ion channel gradients by ATP-dependent pumps, principally the Na+-K+ ATPase. To test for a role of the Na+-K+ ATPase in the activity-induced opening of KATP channels, we performed a set of experiments in which we bath-applied the pump inhibitor strophanthidin following a control recording in ACSF. In experiments using 10 stimuli, bath application of strophanthidin often resulted in a marked decrement in both stimulus-elevated and steady-state popen compared with paired ACSF controls (Fig. 4). Control application of DMSO in these experiments resulted in a higher average popen than that recorded in ACSF-only conditions.
Figure 4.

The Na+-K+ pump blocker strophanthidin blocks both the firing-induced and basal channel opening. A, B, Channel activity was recorded first in ACSF, then in strophanthidin; histograms are shown for a representative experiment (A) and for the ensemble average (B, n = 13). Experiments were performed and analyzed with the experimenters blind to the identity of the washed-in solution. C, The effects of strophanthidin (or 0.1% DMSO) are summarized (as in Fig. 3B). For strophanthidin (Stroph), the fitted slopes were (peak) −0.71 ± 0.15 and (full) −0.75 ± 0.14, consistent with ∼70–75% inhibition. For DMSO, the fitted slopes were (peak) 0.96 ± 0.06 and (full) 1.35 ± 0.31 (n = 11).
There was a clear effect of strophanthidin in these experiments, decreasing popen during both the stimulus-elevated peak (71 ± 15% fractional reduction in peak-weighted open probability, p < 0.001, F-distribution test) and over the course of the full trace (75 ± 14% fractional reduction in open probability for the whole trace, p < 0.001, F-distribution test) (Fig. 4B). In several cases, strophanthidin eliminated the spike-dependent increase in channel popen completely (Fig. 4A). In control experiments where the 0.1% DMSO vehicle alone was washed in, comparison of Δpopen (vehicle − ACSF) versus ACSF popen showed an upward trend for both the peak and the full-trace popen. Again, as in the case of the glibenclamide–DMSO comparisons, there was no overlap between the confidence bands for the fitted slopes of the drug and vehicle control conditions (Fig. 4C).
We also analyzed these experiments by categorizing the results of individual experiments as increased, decreased, or no change, using an arbitrary threshold of ±0.25% change in the peak-weighted popen. By this criterion, strophanthidin reduced the stimulus-evoked increase in channel popen in six of 13 experiments, produced no change in six, and produced an increase in one. By the same criterion, DMSO produced a decrease in one of 11 experiments, no change in four, and an increase in six. A χ2 analysis of the contingency table confirmed a significant effect of strophanthidin versus vehicle controls (p < 0.01).
Ketone bodies increase both steady-state and stimulus-elevated popen
The strophanthidin experiments show that the metabolic load produced even by a short burst of action potentials could result in the opening of KATP channels. This result led us to investigate additional ways to manipulate KATP channel activity by altering metabolic conditions. It has been shown that KATP channels in DGNs can be opened under conditions of joint hypoxia and hypoglycemia (Pelletier et al., 2000), which underscores the metabolic sensitivity of these channels. In previous studies of a different population of neurons, the GABAergic neurons of substantia nigra pars reticulata (SNr), we found that ketone bodies, which circulate at high levels in the blood of patients and animals on an anticonvulsant ketogenic diet, can slow spontaneous neuronal firing by opening KATP channels (Ma et al., 2007). We therefore looked for a similar effect on KATP channels in DGNs.
Using the five stimulus paradigm, we recorded single-channel activity from DGNs in wild-type slices preincubated in ACSF containing 10 mm glucose (as before) and supplemented with 2 mm of the ketone body R-βHB. We found that 2 mm R-βHB increased not only the steady-state open probability of the channel, but also the stimulus-elevated popen (Fig. 5). For the experiments with R-βHB, the mean peak popen during the stimulus interval was nearly threefold higher than that recorded in ACSF (3.14 ± 0.90%, n = 8 in R-βHB, vs 1.09 ± 0.32%, n = 35 in ACSF; different with p < 0.05 by Mann–Whitney U test). The mean basal popen in R-βHB was also increased over that seen in ACSF (1.71 ± 0.64% in R-βHB vs 0.46 ± 0.17% in ACSF; different with p < 0.01). The decay time constant after stimulation was 0.30 ± 0.05 s, very similar to that determined for ACSF alone (τ = 0.32 ± 0.02 s).
Figure 5.

Channel open probability is elevated with the ketone body R-β-hydroxybutyrate. A, Top, Set of five current traces recorded in ACSF supplemented with 2 mm R-βHB. Bottom, Set of five current traces recorded in ACSF alone. Left, First second of recording period. Right, Last second. B, Summary of all experiments in R-βHB (black diamonds; n = 8) versus all in ACSF (gray squares; n = 35). Note that the y-axis is on a larger scale than in previous sets of summary data.
KATP channels contribute to the slow afterhyperpolarization following action potential firing
We tested whether the elevated KATP channel open probability after action potential bursts produced a hyperpolarization of the membrane potential. To avoid disturbing intracellular ATP levels, we used the gramicidin perforated patch technique (Kyrozis and Reichling, 1995) to record the membrane potential after evoking spike bursts. After perforation, cells were held in current clamp near −60 mV and, following a 20 Hz train of five action potentials evoked by brief current pulses, we observed an sAHP (Fig. 6). KATP channels contribute to this sAHP, as application of 100 nm glibenclamide reduced the amplitude of the sAHP (Fig. 6). The mean sAHP was −8.8 ± 1.1 mV, and was significantly smaller in the presence of glibenclamide, −5.7 ± 0.7 mV (n = 5, p < 0.05). We also observed that spontaneous action potential firing stopped during the sAHP and resumed only after the resting potential returned to the baseline level. Glibenclamide shortened the latency to resumption of firing after the evoked train of action potentials and the AHP from a mean latency of 6.5 ± 0.7 s to 4.1 ± 0.6 s (n = 5, p < 0.05).
Figure 6.

A glibenclamide-sensitive, long, slow AHP following a burst of evoked action potentials. Representative perforated patch voltage recordings from DGNs, with a burst of five action potentials evoked by five brief current pulses just before the indicated t = 0. A total of eight traces are overlaid, with a single trace shown in black. In recordings with glibenclamide, AHPs were smaller (ΔV = −5.7 ± 0.7 mV vs −8.8 ± 1.1 mV for control; n = 5, p < 0.05) and spontaneous action potential firing resumed after a shorter latency (4.1 ± 0.6 s vs 6.5 ± 0.7 s for control; n = 5, p < 0.05).
Discussion
Spike activity in granule cells opens up KATP channels via Na+-K+ pump activity
In our recordings of single KATP channels in membrane patches of hippocampal dentate granule neurons, we have identified a link between spike-burst activity in the DGN and the opening of these metabolically sensitive channels. With a brief burst of action potentials, it was possible to systematically increase KATP channel open probability above the usually very low levels of activity that these channels exhibit in the resting state. This spike-dependent increase in KATP channel popen was attenuated or abolished in the presence of strophanthidin, a blocker of the Na+-K+ ATPase. This result suggests that an increase in cellular firing activity imposes a metabolic stress on the cell that, in the absence of pump blockade, is sufficient to open KATP channels above their low steady-state resting levels, most likely via ATP depletion (Kabakov, 1998).
We included a number of channel blockers in our experiments to eliminate most K+ channels other than KATP channels, including the ionic blockers 4-AP, cesium, and TEA (to broadly block Kv and Kir channels), as well as two peptide blockers, charybdotoxin (to block BK channels) and apamin (to block SK channels). The channel activity observed under these conditions was blocked by glibenclamide and nearly eliminated in knock-out mice lacking the major pore-forming subunit of neuronal KATP channels, Kir6.2 (Fig. 3). However, some small activity did remain. We cannot rule out some contribution in our experiments from apamin-insensitive SK channels, or from Kir6.1-containing KATP channels, but it does appear that the majority of the stimulus-evoked channel opening that we observed can be accounted for by glibenclamide-sensitive, Kir6.2-containing KATP channels.
We also found that action potential bursts evoked in individual DGNs recorded using a perforated patch approach produced an sAHP that was sensitive to glibenclamide. Our finding from our cell-attached recordings that KATP channel open probability is elevated after bursts of action potentials is consistent with a role of KATP channels in the sAHP. The long duration of the sAHP would require a contribution not only from the transient elevation in open probability induced by firing, but also from the chronic elevation apparent in the steady-state open probability [which was also dependent on regular stimulation, as seen from its disappearance when stimulation was turned off (Fig. 2)]. KATP channels account for only a fraction of the sAHP; another component of the sAHP in hippocampal neurons has been attributed to KCNQ channels (Tzingounis and Nicoll, 2008). Loss of KCNQ channels also eliminates only a portion of the sAHP. Our finding suggests that KATP channels may contribute the sAHP in DGNs and possibly function to reduce excitability in conditions of elevated neuronal firing.
The likely size of these KATP effects at a whole-cell level
To gain an additional sense for how this spike-induced opening of KATP channels might functionally affect overall cellular excitability, we can estimate the fractional change in input conductance. In whole-cell recordings with a high-ATP pipette solution, we found that the KATP channel opener NN414 produced a whole-cell KATP conductance of ∼0.8 nS (n = 2). In cell-attached single-channel recordings, we observed a mean popen in NN414 of 4.7 ± 1.7%, compared with a resting popen (in ACSF alone) of 0.19 ± 0.02%. The peak popen responses in our five-stimulus experiments of ∼1.1%—approximately one-fifth of the NN-414-induced popen—may thus correspond to a total conductance on the order of ∼160 pS, which would represent a substantial (∼30%) increase over the resting conductance of ∼400–500 pS observed at this age (and in our own whole-cell experiments). A similar calculation applied to the R-βHB treatment yields an even more substantial (∼100%) increase in conductance.
The intrinsic membrane properties (Liu et al., 2000) and synaptic plasticity (Schmidt-Hieber et al., 2004) of young or newly generated dentate granule cells change significantly as they age. A key difference between cells of different ages is their input resistance: ∼2.5 GΩ for young neurons (corresponding to 0.4 nS conductance) and ∼250 MΩ for older neurons (corresponding to 4 nS conductance). This difference suggests that the potential effects of a spike-induced elevation of channel popen may be more pronounced in younger animals.
Could KATP channel opening represent one mechanism for tempering seizure propagation in the temporal lobe?
The hippocampal formation is one of the most widely studied regions of the brain where KATP channels are known to be broadly expressed. To date, however, no clear understanding has emerged of what role these channels might serve in the hippocampus beyond neuroprotection during ischemia or ischemia-induced seizures (Krnjević and Ben-Ari, 1989; Ben-Ari et al., 1990; Yamada et al., 2001). We have shown that externally driven spiking in hippocampal DGNs, imposed at relatively high rates, leads to the sustained opening of KATP channels in these cells.
This result agrees with and extends studies of KATP channels in rat respiratory neurons (Haller et al., 2001). In the respiratory neurons, which have a spontaneous population rhythm, the clearest change in popen was the closure of KATP channels seen immediately before burst cycle; opening of channels occurred fairly gradually after each burst. The slow response may reflect the asynchrony of action potential firing in different respiratory cells, so that the increase in KATP channel opening is dispersed in time from burst to burst. In our preparation, with precisely controlled timing, we observe KATP channels opening up promptly following stimulated spiking—an occurrence that could serve to temper any further excitation in the wake of an unusually high-frequency spike burst.
In the basic trisynaptic hippocampal circuit, DGNs are the first point of input from the entorhinal cortex into the hippocampus. This privileged positioning of DGNs in the circuit may permit them to act as a seizure gate in temporal lobe epilepsy (Heinemann et al., 1992; Lothman et al., 1992; Coulter and Carlson, 2007). While DGNs are typically reported to fire at very low rates (Jung and McNaughton, 1993), the spike frequencies we delivered are not out of the range of possibility for these cells. Indeed, these cells have been reported to fire at frequencies of up to 25 Hz or more at the onset of seizures in rats during pilocarpine-induced status epilepticus (Bower and Buckmaster, 2008). Therefore, our chosen rates of stimulation are likely to be visited during epileptic-like states.
The opening of KATP channels in DGNs could serve to limit the passage of excitation to more distal parts of the circuit in cases where the firing rate of these gating neurons is vastly increased by excessive excitatory synaptic input, such as might occur during seizure episodes. Indeed, it has been demonstrated in paired recordings that single spikes in DGNs generally do not induce spiking in CA3 pyramidal cells, but that high-frequency trains of DGN spikes can do so consistently (Henze et al., 2002). This result suggests that mechanisms for tempering high-frequency excitation in DGNs can have significant effects on downstream target neurons.
We also found that applying ketone bodies to hippocampal slices augments both the basal channel popen and the stimulus-elevated open probability above that observed in ACSF alone. This result supports the role of KATP channels as a link between metabolism and neuronal excitability in central neurons, and supplements our earlier findings that ketogenic diet metabolites can slow the spontaneous firing of neurons in the SNr via opening of KATP channels (Ma et al., 2007). Like the dentate gyrus, the SNr is closely associated with the gating of epileptic seizures in the brain (Iadarola and Gale, 1982; Garant and Gale, 1983; McNamara et al., 1983, 1984; Depaulis et al., 1994). The effects seen here of ketone bodies in DGNs may suggest that the opening of KATP channels represent a general mechanism—under metabolic control—for tempering excitability during high-activity, epileptic-like states.
Footnotes
This work was supported by NIH/NINDS Grants NS029693 and NS055031 to G.Y. We also received support for biostatistics consultation from Harvard Catalyst–The Harvard Clinical and Translational Science Center (NIH Grant RR025758 and financial contributions from Harvard University and its affiliated academic health care centers). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Center for Research Resources, or the National Institutes of Health. We thank members of the Yellen laboratory for valuable discussions and comments on the manuscript. We also thank Drs. Bruce Bean, Rachel Wilson, and Bernardo Sabatini for assistance and advice, and Dr. E. Andres Houseman for discussing statistical methods. NN414 was a gift from Novo Nordisk. Kir6.2 knock-out mice were generously provided by Drs. S. Seino and C. G. Nichols; we thank Tatiana Abramson for expert technical assistance in genotyping the mice and Drs. James Berg, Weiyuan Ma, and Mathew Tantama for assistance in colony maintenance.
References
- Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- Allen TG, Brown DA. Modulation of the excitability of cholinergic basal forebrain neurones by KATP channels. J Physiol. 2004;554:353–370. doi: 10.1113/jphysiol.2003.055889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- Ashford ML, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflügers Arch. 1990;415:479–483. doi: 10.1007/BF00373626. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Gonzalez G, Aguilar-Bryan L, Bryan J. Reconstituted human cardiac KATP channels: functional identity with the native channels from the sarcolemma of human ventricular cells. Circ Res. 1998;83:1132–1143. doi: 10.1161/01.res.83.11.1132. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Tucker SJ, Schulte U, Benndorf K, Ruppersberg JP, Fakler B. Inward rectification in KATP channels: a pH switch in the pore. EMBO J. 1999;18:847–853. doi: 10.1093/emboj/18.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Krnjević K, Crépel V. Activators of ATP-sensitive K+ channels reduce anoxic depolarization in CA3 hippocampal neurons. Neuroscience. 1990;37:55–60. doi: 10.1016/0306-4522(90)90191-6. [DOI] [PubMed] [Google Scholar]
- Benndorf K, Friedrich M, Hirche H. Anoxia opens ATP regulated K+ channels in isolated heart cells of the guinea pig. Pflugers Arch. 1991;419:108–110. doi: 10.1007/BF00373754. [DOI] [PubMed] [Google Scholar]
- Bower MR, Buckmaster PS. Changes in granule cell firing rates precede locally recorded spontaneous seizures by minutes in an animal model of temporal lobe epilepsy. J Neurophysiol. 2008;99:2431–2442. doi: 10.1152/jn.01369.2007. [DOI] [PubMed] [Google Scholar]
- Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311:271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Carlson GC. Functional regulation of the dentate gyrus by GABA-mediated inhibition. Prog Brain Res. 2007;163:235–243. doi: 10.1016/S0079-6123(07)63014-3. [DOI] [PubMed] [Google Scholar]
- Dabrowski M, Larsen T, Ashcroft FM, Bondo Hansen J, Wahl P. Potent and selective activation of the pancreatic beta-cell type KATP channel by two novel diazoxide analogues. Diabetologia. 2003;46:1375–1382. doi: 10.1007/s00125-003-1198-1. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Vergnes M, Marescaux C. Endogenous control of epilepsy: the nigral inhibitory system. Prog Neurobiol. 1994;42:33–52. doi: 10.1016/0301-0082(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. 1998;814:41–54. doi: 10.1016/s0006-8993(98)00956-1. [DOI] [PubMed] [Google Scholar]
- Garant DS, Gale K. Lesions of substantia nigra protect against experimentally induced seizures. Brain Res. 1983;273:156–161. doi: 10.1016/0006-8993(83)91105-8. [DOI] [PubMed] [Google Scholar]
- Haller M, Mironov SL, Karschin A, Richter DW. Dynamic activation of KATP channels in rhythmically active neurons. J Physiol. 2001;537:69–81. doi: 10.1111/j.1469-7793.2001.0069k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Iadarola MJ, Gale K. Substantia nigra: site of anticonvulsant activity mediated by gamma-aminobutyric acid. Science. 1982;218:1237–1240. doi: 10.1126/science.7146907. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Horie M, Seino Y, Mizuta M, Seino S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem. 1995;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- Jung MW, McNaughton BL. Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus. 1993;3:165–182. doi: 10.1002/hipo.450030209. [DOI] [PubMed] [Google Scholar]
- Kabakov AY. Activation of KATP channels by Na/K pump in isolated cardiac myocytes and giant membrane patches. Biophys J. 1998;75:2858–2867. doi: 10.1016/S0006-3495(98)77728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin C, Ecke C, Ashcroft FM, Karschin A. Overlapping distribution of KATP channel-forming Kir6.2 subunit and the sulfonylurea receptor SUR1 in rodent brain. FEBS Lett. 1997;401:59–64. doi: 10.1016/s0014-5793(96)01438-x. [DOI] [PubMed] [Google Scholar]
- Krnjević K, Ben-Ari Y. Anoxic changes in dentate granule cells. Neurosci Lett. 1989;107:89–93. doi: 10.1016/0304-3940(89)90796-9. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- Liu X, Tilwalli S, Ye G, Lio PA, Pasternak JF, Trommer BL. Morphologic and electrophysiologic maturation in developing dentate gyrus granule cells. Brain Res. 2000;856:202–212. doi: 10.1016/s0006-8993(99)02421-x. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Rigsbee LC, Galloway MT. Evidence that substantia nigra is crucial to neural network of kindled seizures. Eur J Pharmacol. 1983;86:485–486. doi: 10.1016/0014-2999(83)90202-9. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Galloway MT, Rigsbee LC, Shin C. Evidence implicating substantia nigra in regulation of kindled seizure threshold. J Neurosci. 1984;4:2410–2417. doi: 10.1523/JNEUROSCI.04-09-02410.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A. 1998;95:10402–10406. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Pelletier MR, Pahapill PA, Pennefather PS, Carlen PL. Analysis of single KATP channels in mammalian dentate gyrus granule cells. J Neurophysiol. 2000;84:2291–2301. doi: 10.1152/jn.2000.84.5.2291. [DOI] [PubMed] [Google Scholar]
- Pierrefiche O, Bischoff AM, Richter DW. ATP-sensitive K+ channels are functional in expiratory neurones of normoxic cats. J Physiol. 1996;494:399–409. doi: 10.1113/jphysiol.1996.sp021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlogar M, Dietrich D. Firing pattern of rat hippocampal neurons: a perforated patch clamp study. Brain Res. 2006;1085:95–101. doi: 10.1016/j.brainres.2006.02.050. [DOI] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, Shimizu T, Seino S, Inagaki N. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science. 2001;292:1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
- Zawar C, Plant TD, Schirra C, Konnerth A, Neumcke B. Cell-type specific expression of ATP-sensitive potassium channels in the rat hippocampus. J Physiol. 1999;514:327–341. doi: 10.1111/j.1469-7793.1999.315ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]